1. Introduction
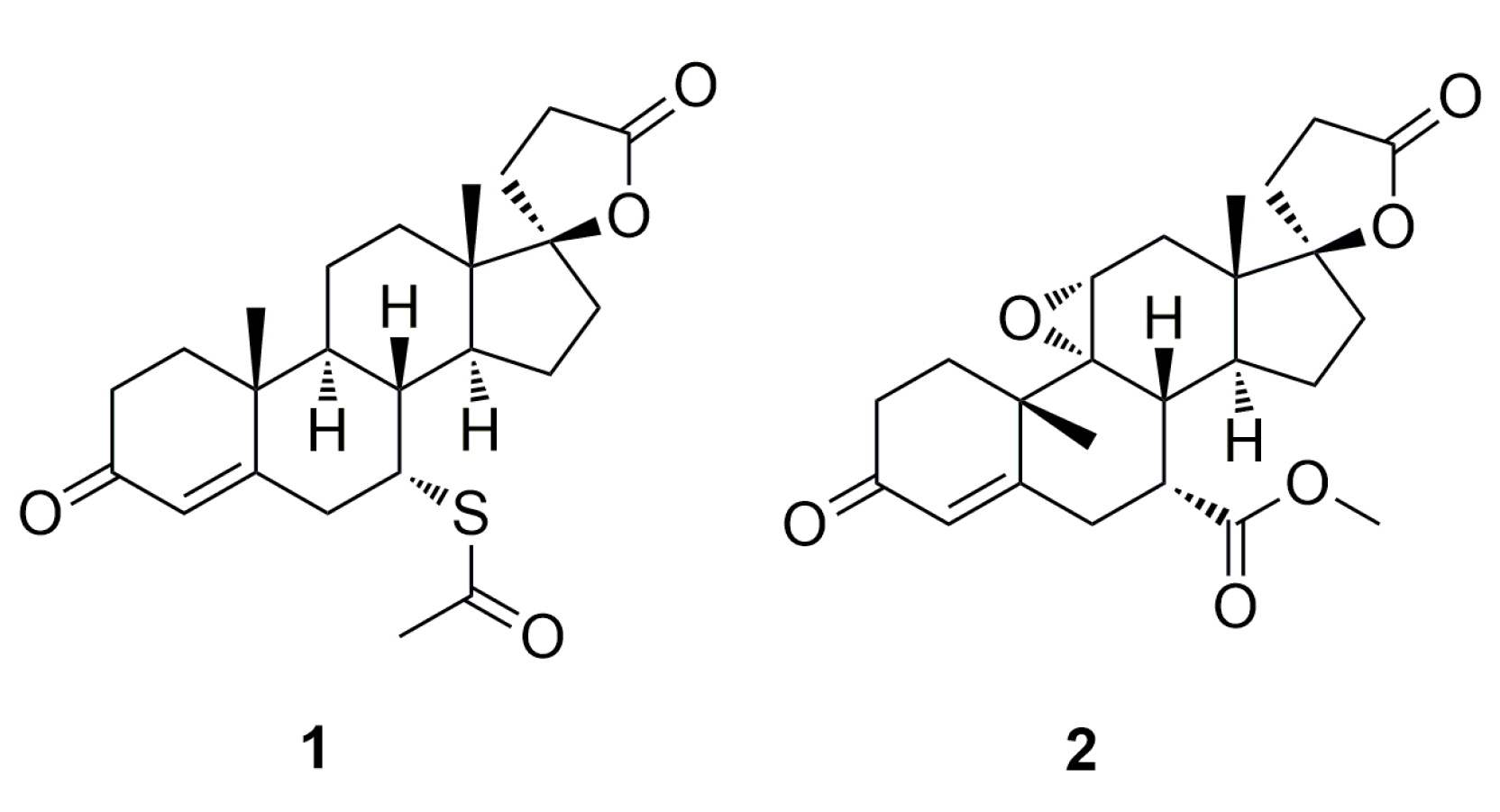
Eplerenone (
2,
Figure 1) is a cardiovascular drug indicated for the treatment of essential hypertension and congestive heart failure that, in contrast to its predecessor spironolactone
1 (
Figure 1) demonstrates a high degree of selectivity for the aldosterone receptor and a low-binding affinity for progesterone and androgen receptors [
1,
2,
3,
4,
5,
6,
7,
8]. As a result of the presence of a 9,11-epoxide group in the eplerenone structure, its selectivity for the aldosterone receptor is enhanced and the drug minimizes the risk of adverse hormonal effects and provides important clinical benefits not previously available with spironolactone
1. Treatment with eplerenone is associated with reductions in blood pressure and improved survival (15% reduction in total mortality) for patients with heart failure who are in stable condition after a myocardial infarction. The product was originally developed by scientists at Ciba-Geigy AG (Basel, Switzerland) and launched in the US in 2003 by Pharmacia (Sandwich, Kent, UK) under the trade name Inspra
®.
Eplerenone contains sensitive 9,11α-epoxide, 17α-γ-lactone and 7α-carbomethoxy moieties that, depending on the conditions, may hydrolyze or epimerize to afford degradation or epimerization products [
9,
10,
11,
12,
13]. The SCXRD structure of eplerenone confirms the relative
cis configuration of the 9α,11α-epoxide ring and the 7α-carbomethoxy substituent [
14,
15,
16,
17]. Manufacture of eplerenone is always accompanied by side reactions leading to the unwanted impurities that vary with the different starting materials and reaction conditions used. Most of the patents and literature information dealing with the preparation of eplerenone are based on the use of canrenone derivatives. As stereogenic centers in eplerenone precursors give rise to various process-related impurities, including diastereo- and regioisomers of starting materials, intermediates, by-products and the final drug substance, the manufacture of eplerenone with the required stereochemistry and pharmaceutical grade purity is a significant challenge. In designing a synthesis of eplerenone from canrenone derivatives, the principal challenges are the stereoselective introduction of the carbomethoxy substituent at the C-7α position of the steroid skeleton and the regioselective dehydration of the 11α-hydroxy group [
1,
9,
16,
17,
18,
19,
20,
21].
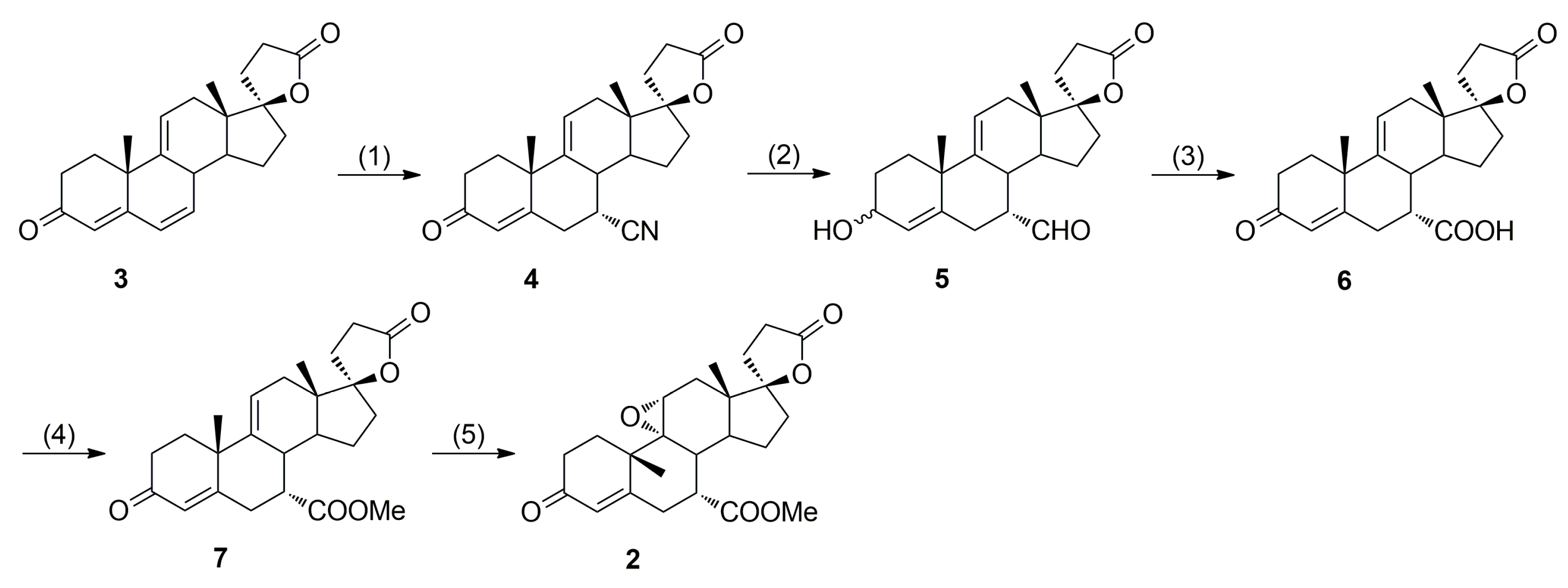
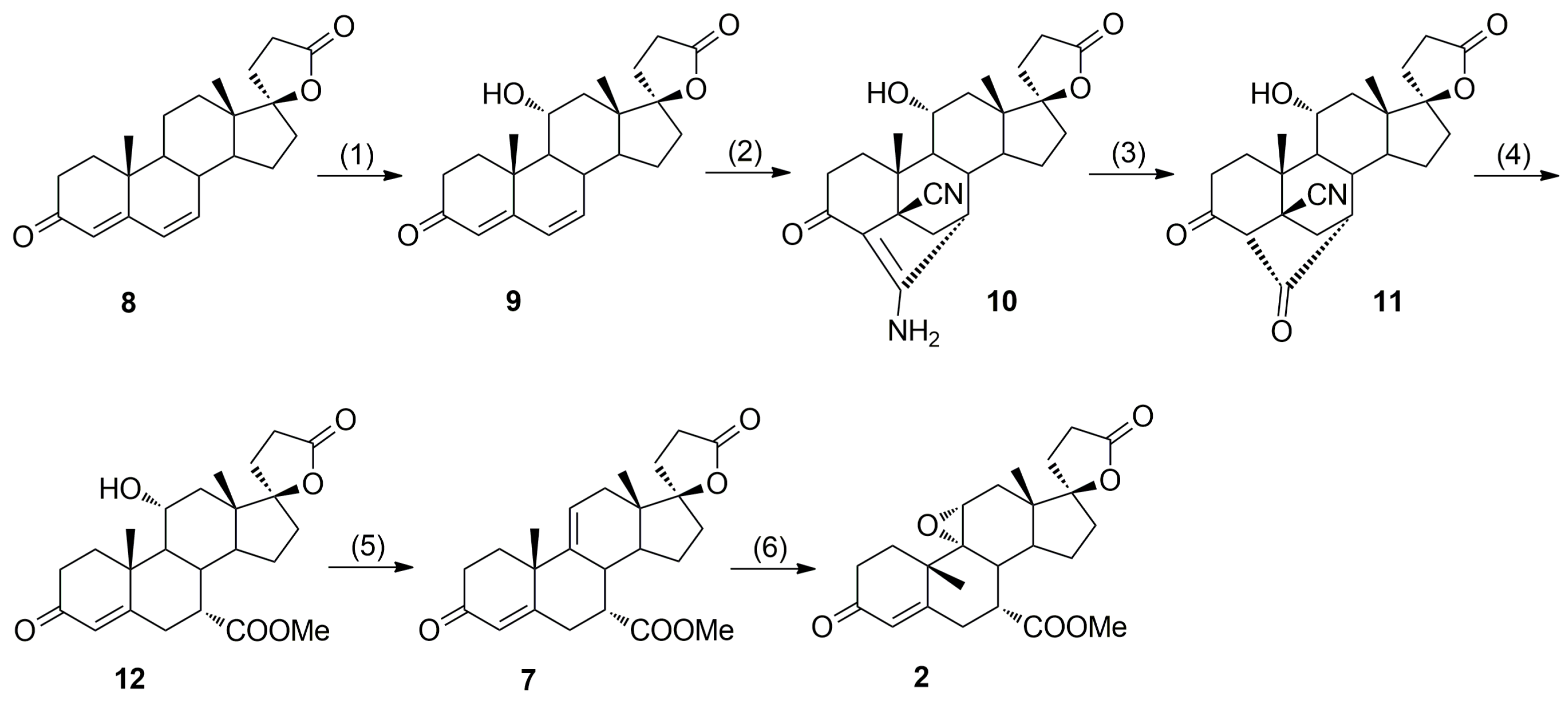
In 1984, Grob et al. [
17,
18] from Ciba-Geigy AG accomplished the first synthesis of eplerenone by employing Nagata hydrocyanation of Δ
9(11)-canrenone (
3) as the key step (
Scheme 1), but with moderate stereoselectivity in the 7α-cyano derivative
4 formation (7α/7β ≈ 4:1) necessitating tedious column chromatographic separations. The original EP 122232 B1 patent does not disclose any information with respect to the purity of the material obtained, its purification to pharmaceutical-grade or the removal of impurities.
In alternative eplerenone synthesis by Ng et al. [
9], the 7α-carbomethoxy group was introduced stereoselectively via 4,6-bishydrocyanation of 11α-hydroxycanrenone (
9,
Scheme 2), but regioselective dehydration of the intermediate (7α,11α,17α)-11-hydroxyester
12 was problematic giving the regioisomeric (7α,17α)-11(12)-enester
7b apart from the main (7α,17α)-9(11)-enester
7 product (
Table 1). The 7α/7β diastereoisomeric purity of (7α,11α,17α)-
12 obtained by the cleavage of diketone
11 with sodium methoxide in refluxing methanol was not reported; however, the literature sources on syntheses of related 7α-carboalkoxy stereoidal spirolactones provide information on their 7β-carboalkoxy diastereoisomers resulting from basic epimerization of the steroid C-7α asymmetric center under the same conditions as used for diketone
11 cleavage [
22].
When the authors repeated the preparation taught in WO 9825948 A2 it was observed that the purity of the resulting eplerenone was low and attempts to purify the resulting product to a level meeting the requisite specifications for use as a pharmaceutical active were unsuccessful. Although the epimeric 11-hydroxyester (7β,11α,17α)-
12a was not reported by Ng et al. [
9], or any other literature sources, it contaminated the main (7α,17α)-9(11)-enester
7 product. In particular, the regioisomeric (7α,11α,12α,17α)-11,12-epoxyester
2b (Imp. E), the new (7α,11β,17α)-9,11-dichloro derivative
13 and the 7α,9:21,17-dicarbolactone
14 (Imp. A) were difficult to remove from the final product. All these contaminants resulted from side reactions taking place on the steroid ring C of the (7α,11α,17α)-11-hydroxy derivative (
12) and the key intermediate (7α,17α)-9(11)-enester
7, including epimerization of the C-7α asymmetric center, oxidation, dehydration, chlorination and lactonization.
Recently, we have developed an improved, scalable, cost-effective and environment-friendly technology for the industrial-scale synthesis of eplerenone (
2) from commercially available 11α-hydroxy-7α-(methoxycarbonyl)-3-oxo-17α-pregn-4-ene-21,17-carbolactone (
12), based on the last two steps of the general route described by Ng et al. (
Scheme 2) [
9]. During the process development, two new and seven known process-related impurities of eplerenone were observed, and/or synthesized and fully characterized, including four impurities A, B, C and E listed in the European Pharmacopoeia 8.4 [
23]. The new impurities were identified as 11α-hydroxy-7β-(methoxycarbonyl)-3-oxo-17α-pregn-4-ene-21,17-carbolactone (
12a) and 9,11β-dichloro-7α-(methoxycarbonyl)-3-oxo-17α-pregn-4-ene-21,17-carbolactone (
13). To the best of our knowledge, the 11-hydroxyester
12a and the 9,11-dichloro derivative
13 are new compounds that have never been identified before.
The syntheses of the starting (7α,11α,17α)-11-hydroxyester
12 and the key intermediate (7α,17α)-9(11)-enester
7 are broadly described in the literature sources [
9,
16,
17,
18,
19,
20,
21,
24]; however, their comprehensive structure elucidation and confirmation is still missing and SCXRD studies are reported here for the first time. As starting materials and intermediates in active pharmaceutical ingredient (API) synthesis often afford numerous impurities affecting the quality of the final drug product, their comprehensive structural elucidation and confirmation is essential for impurities identification and characterization. A complete physicochemical characterization, not only for an API, but also starting materials and key synthetic intermediates, became a requirement of both the U.S. Food and Drug Administration (FDA) and the European Medicine Agency (EMA). The five other impurities listed in
Table 1, i.e., the isomeric (7β,17α)-9(11)-enester
7a and (7α,17α)-11(12)-enester
7b, the isomeric (7β,11α,17α)-9,11-epoxyester
2a (Imp. E) and (7α,11α,12α,17α)-11,12-epoxyester
2b (Imp. B) and the 7α,9:21,17-dicarbolactone
14 (Imp. A), were mentioned elsewhere in the literature, mainly as part of HPLC studies; however, their syntheses, methods of removal from the final product and comprehensive structural elucidation and confirmation were not disclosed [
9,
12]. The determination of a drug substance impurity profile, including starting materials, by-products, intermediates and potential degradation products, is critical for the safety assessment of API and manufacturing process thereof. In any API, it is necessary to study the impurity profile, and control it during the preparation of the pharmaceutical. As indicated in the ICH guidelines, any impurity, formed at a level of ≥0.10% with respect to the API, should be identified, synthesized and characterized thoroughly [
25,
26]. Only two eplerenone impurities, i.e., 7α,9:21,17-dicarbolactone
14 (Imp. A) and 11,12-epoxy ester
2b (Imp. B), are accepted at a level greater than 0.10%, i.e., maximum 0.3%, in accordance with pharmacopoeial acceptance criteria [
22].
The present paper deals with synthesis, physicochemical characterization and comprehensive structural elucidation and confirmation of the process-related eplerenone impurities found in samples of the key intermediate (7α,17α)-9(11)-enester
7 and eplerenone bulk drug substance manufactured according to the modified and optimized procedure described in Searle Co. patent [
9] (
Scheme 2,
12 as starting material). Apart from the starting material
12, the key intermediate enester
7 and the final product
2, seven process-related eplerenone impurities
2a–
b,
7a–
b,
12a,
13 and
14 were fully characterized by IR, NMR and MS. The molecular structures of the epimeric 11-hydroxyesters (7α,11α,17α)-
12 and (7β,11α,17α)-
12a, the isomeric (7α,17α)-9(11)-enester
7, (7β,17α)-9(11)-enester
7a and (7α,17α)-11(12)-enester
7b, the isomeric (7β,11α,17α)-9,11-epoxyester
2a and (7α,11α,12α,17α)-11,12-epoxyester
2b, and the new (7α,11β,17α)-9,11-dichloro derivative
13, were solved and refined using SCXRD. The full identification and characterization of these compounds should be useful for the quality control and the validation of the analytical methods in the manufacture of eplerenone.
2. Results and Discussion
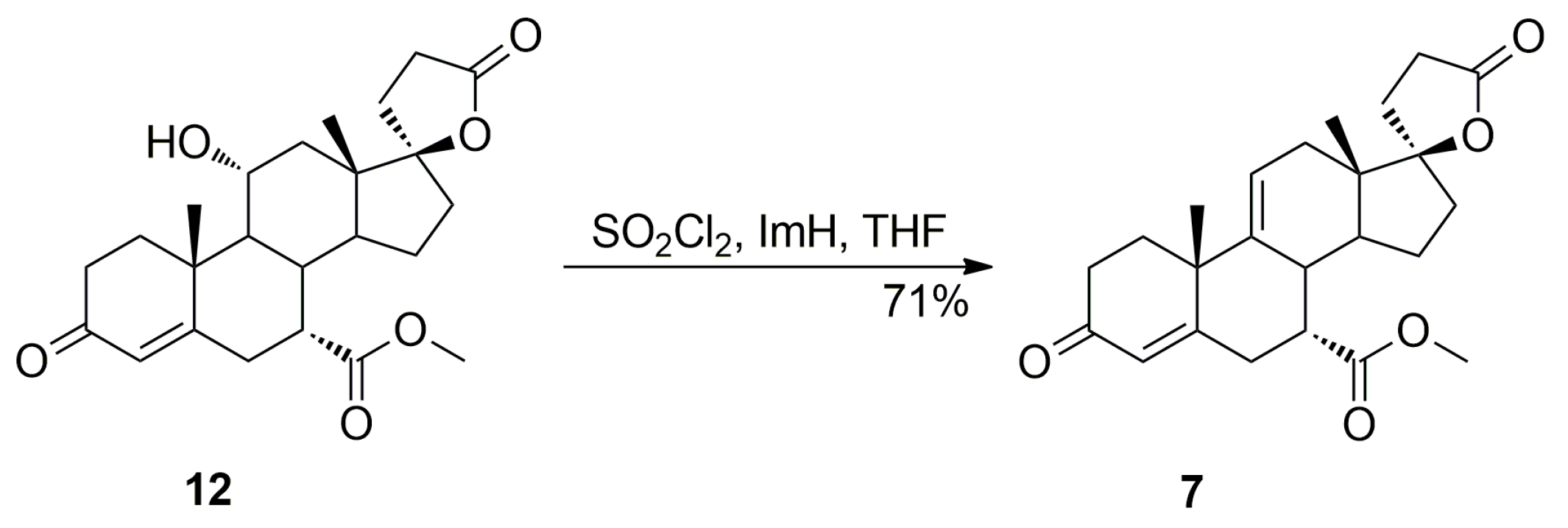
Dehydration of 11-hydroxy steroids is commonly used for the introduction of the 9,11-double bond into the steroid skeleton. In a preferred technological embodiment, the (7α,17α)-9(11)-enester 7 is synthesized directly by in situ replacement of the 11α-hydroxy group of the ester (7α,11α,17α)-12 with halogen followed by thermal 9,11-dehydrohalogenation. The nucleophilic substitution of the 11α-hydroxy group is effected by reaction with sulfuryl halide at about −70 °C, after which a hydrogen halide scavenger is added. In manufacturing technology of eplerenone developed by the authors, the 11α-hydroxyester 12 and imidazole were dissolved simultaneously in anhydrous tetrahydrofuran and cooled to −10 °C. Sulfuryl chloride was added slowly and the reaction mixture was allowed to warm to room temperature, and then stirred for a time sufficient to complete the elimination reaction, typically about 1 h. The key intermediate 7 was isolated in a crude form by dichloromethane extraction followed by evaporation of the solvent and recrystallized twice, from ethanol and a mixture of dichloromethane/diethyl ether respectively, to give the pure (7α,17α)-9(11)-enester 7 (71% yield) as white crystals.
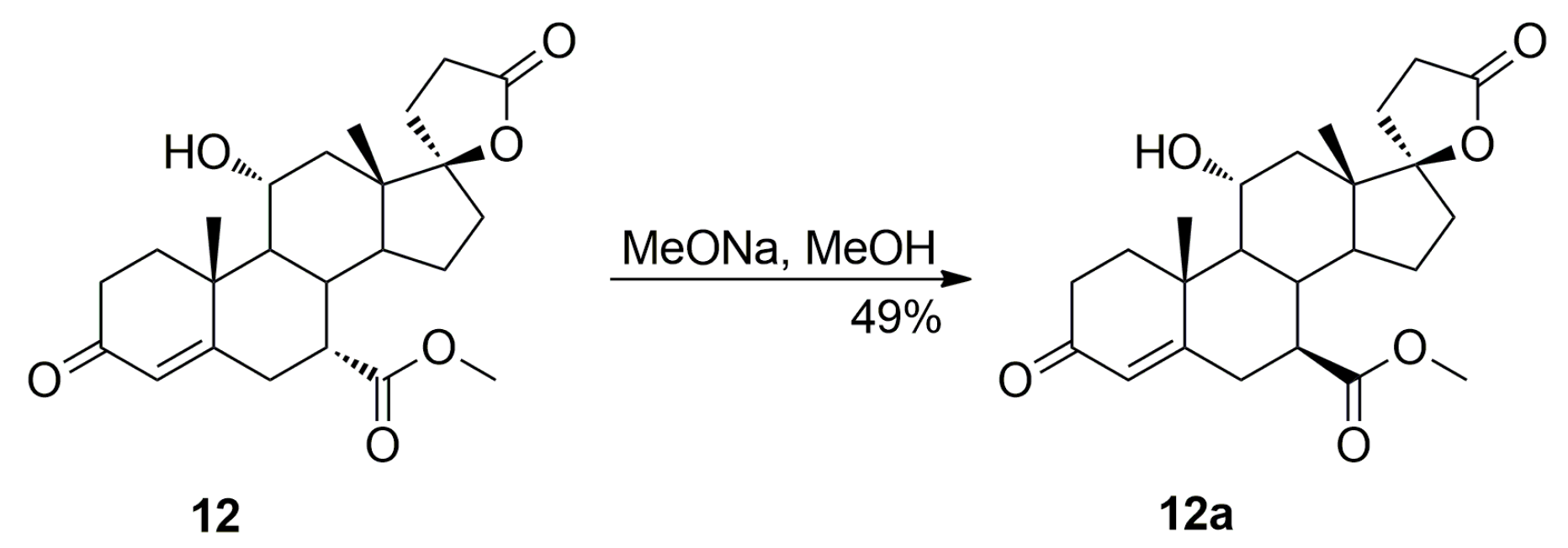
The dehydration step produced the three main side products, i.e., the regioisomeric (7β,17α)-11(12)-enester 7b, the new (7α,11β,17α)-9,11-dichloro derivative 13 and the 7α,9:21,17-dicarbolactone 14 (Imp. A), which were isolated chromatographically from the recrystallization mother liquors of 7 by elution with varying mixtures of ethyl acetate/dichloromethane. Further gradient elution with acetonitrile/toluene afforded the new 11-hydroxyester (7β,11α,17α)-12a, which was isolated in only a small yield, apart from the unreacted (7α,11α,17α)-12 isomer used as the starting material.
The commercial 11-hydroxyester (7α,11α,17α)-
12 was synthesized according to the procedure described by Ng et al. [
9] and the chemical purity declared by the manufacturer was 98%. The novel 11-hydroxyester (7β,11α,17α)-
12a was not previously detected by chromatographic or spectroscopic methods in its commercially available (7α,11α,17α)-
12 isomer. However, the literature data on syntheses of related 7α-carboalkoxy steroidal spirolactones [
22] allowed us to assume that the new 11-hydroxyester (7β,11α,17α)-
12a originates from the fourth step of the
Scheme 2 synthesis in which sodium methoxide reacts with diketone
11 to afford the (7α,11α,17α)-
12 isomer as the main product of the cleavage, which is contaminated with the (7β,11α,17α)-
12a epimer resulting from the competitive basic epimerization of
12. The 11-hydroxyester (7β,11α,17α)-
12a was synthesized independently by basic epimerization of the (7α,11α,17α)-
12 isomer. As expected, sodium methoxide in methanol solution converted the C-7α ester
12 into an epimeric mixture of the C-7α
12 and C-7β
12a esters. The pure C-7β ester
12a was isolated chromatographically from the crude mixture of epimers
12 and
12a by 10–50% acetonitrile/toluene gradient elution to give (7β,11α,17α)-
12a (49% yield) as a white solid. A sample of
12a was also isolated by column chromatography from recrystallization mother liquors of the commercial 11-hydroxyester (7α,11α,17α)-
12. Even a small amount of the newly detected (7β,11α,17α)-11-hydroxyester
12a is hardly separable from its (7α,11α,17α)-
12 isomer by large scale chromatography or recrystallization, therefore, the commercial 11-hydroxyester (7α,11α,17α)-
12 was used further without purification. As the two 11-hydroxyesters of the C-7α and C-7β series, respectively
12 and
12a, are crucial for impurities formation during multi-gram scale synthesis of eplerenone, they were subjected to comprehensive structural elucidation and confirmation.
The [M + Na]
+ values,
m/
z 439.2110 and 439.2080, obtained for both 11-hydroxyesters
12 and
12a correspond to C
24H
32O
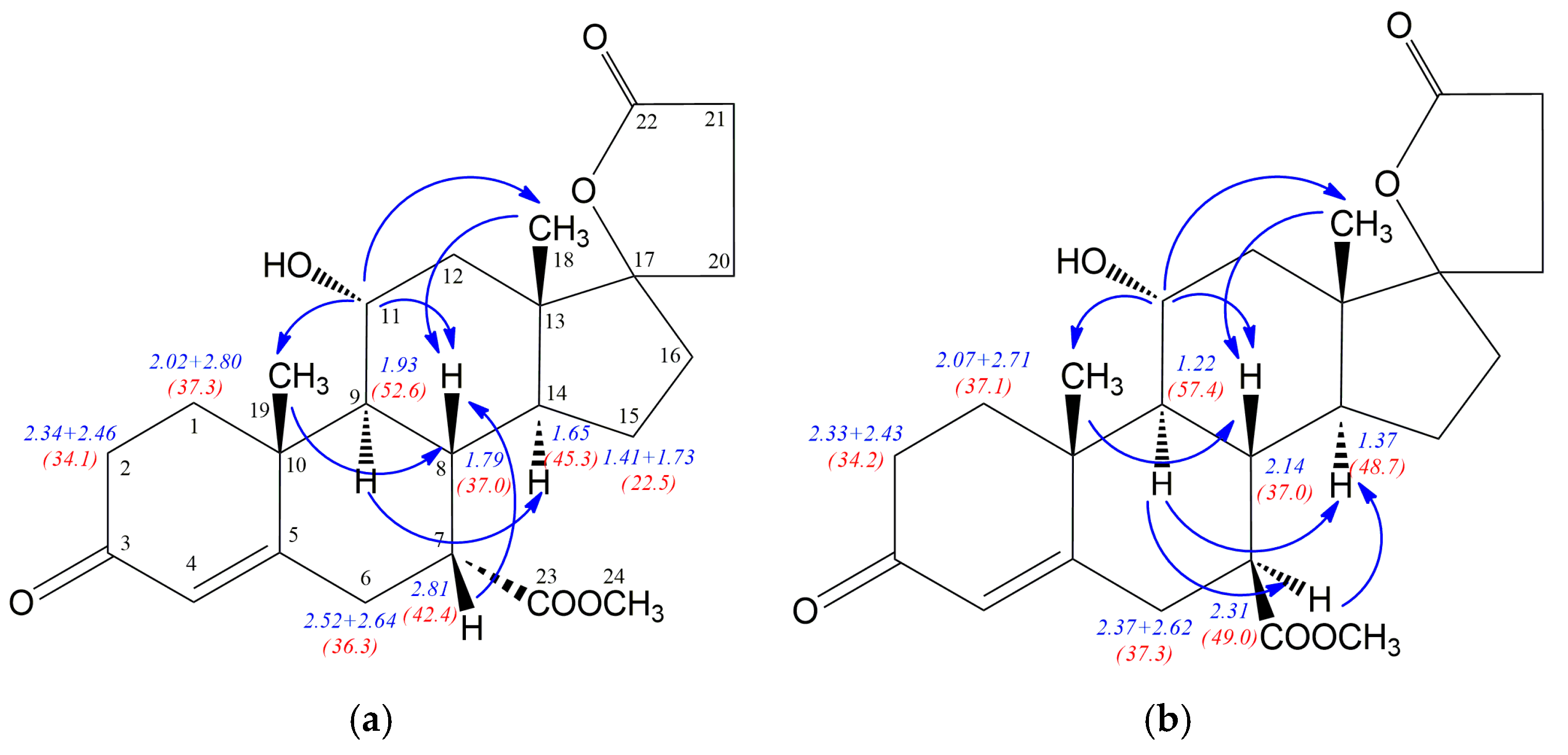
6Na. The NMR data for epimeric hydroxyesters
12 and
12a are given in
Table 2. The
1H-/
13C-NMR chemical shifts assignment was made after careful and precise 2D NMR spectra analysis and the data obtained for the known isomer
12 are in full compliance with those presented by Preisig et al. [
24]. The 2D NOESY experiments allowed discrimination between epimeric structures
12 and
12a and fully confirmed the stereochemistry at the C-7 atom in both isomers. Blue arrows in
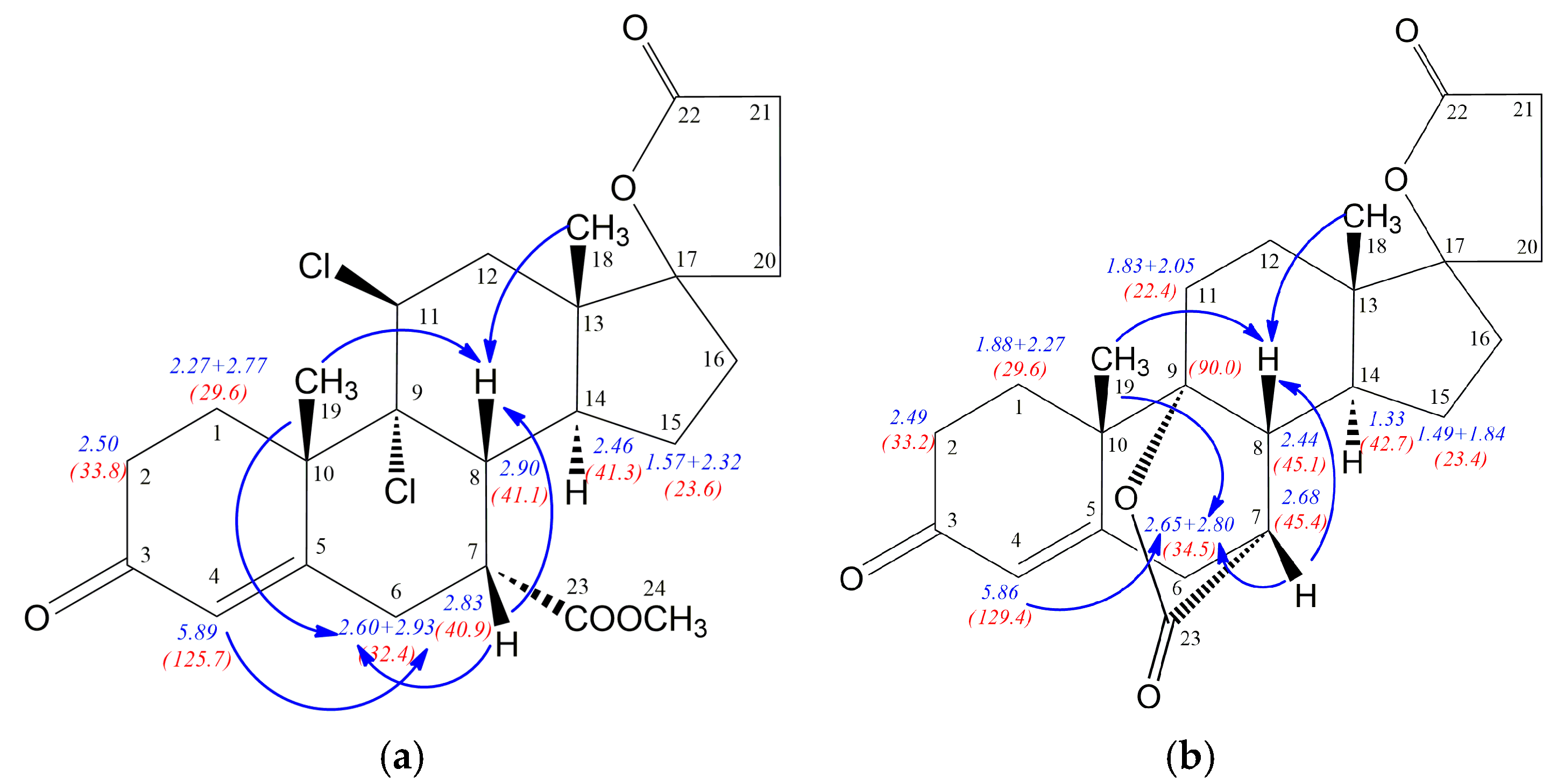
Figure 2 show the most important NOE effects involving H7 proton, simultaneously clearly indicating the 7α or 7β positioned carbomethoxy group. The strong H7-H8 NOE effect is observed for the compound
12 with β positioned H7 proton, whereas α situated H7 proton in
12a is involved in two significant H9-H7 and H7-H14 NOE effects. Additionally, the strong NOE effect between H9 and H14 protons for both
12 and
12a epimers is observed. Comparison of the NMR data for epimeric structures
12 and
12a revealed that some of the
1H/
13C nuclei shieldings within the steroid rings B, C and D are related to the α or β configuration of the C-7 atom. The
1H shielding increase of 0.5 ppm for H7, 0.71 ppm for H9 and 0.28 ppm for H14 nuclei is observed when passing from the compound
12 with 7α positioned carbomethoxy group to its 7β epimer
12a. Additionally, the change of configuration at the C7 is accompanied by H8 (0.35 ppm) shielding decrease. Simultaneously, both diasterotopic H15 protons of the epimer
12a with 7β positioned carbomethoxy group became equal having the same proton chemical shift (1.49 ppm). In the case of
13C-NMR data the opposite effect is observed, the transition from
12 to
12a leads to the shielding decrease of 6.6 ppm for C7, 4.8 ppm for C9 and 3.4 ppm for C14 nuclei. Surprisingly, the change of configuration from 7α in
12 to 7β in
12a is related with no change of the shielding of the C8 nucleus and decreasing of shielding by 2.7 ppm for C23 carbon of the carbomethoxy substituent.
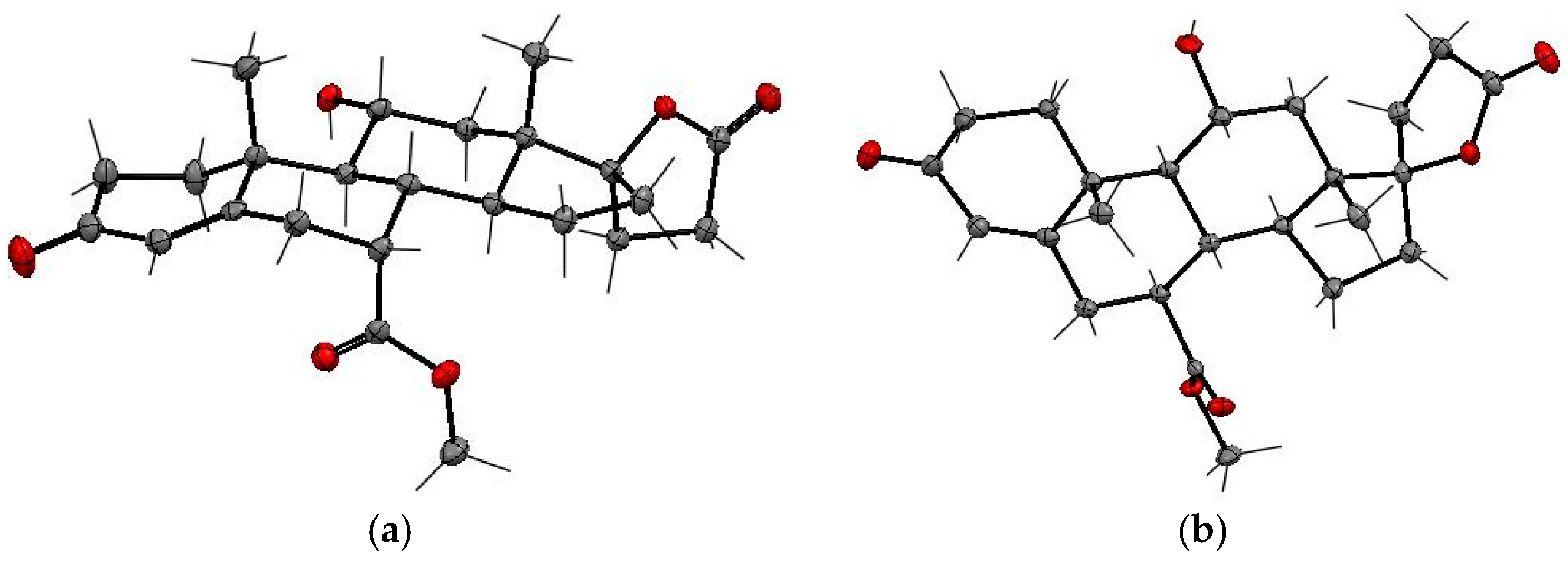
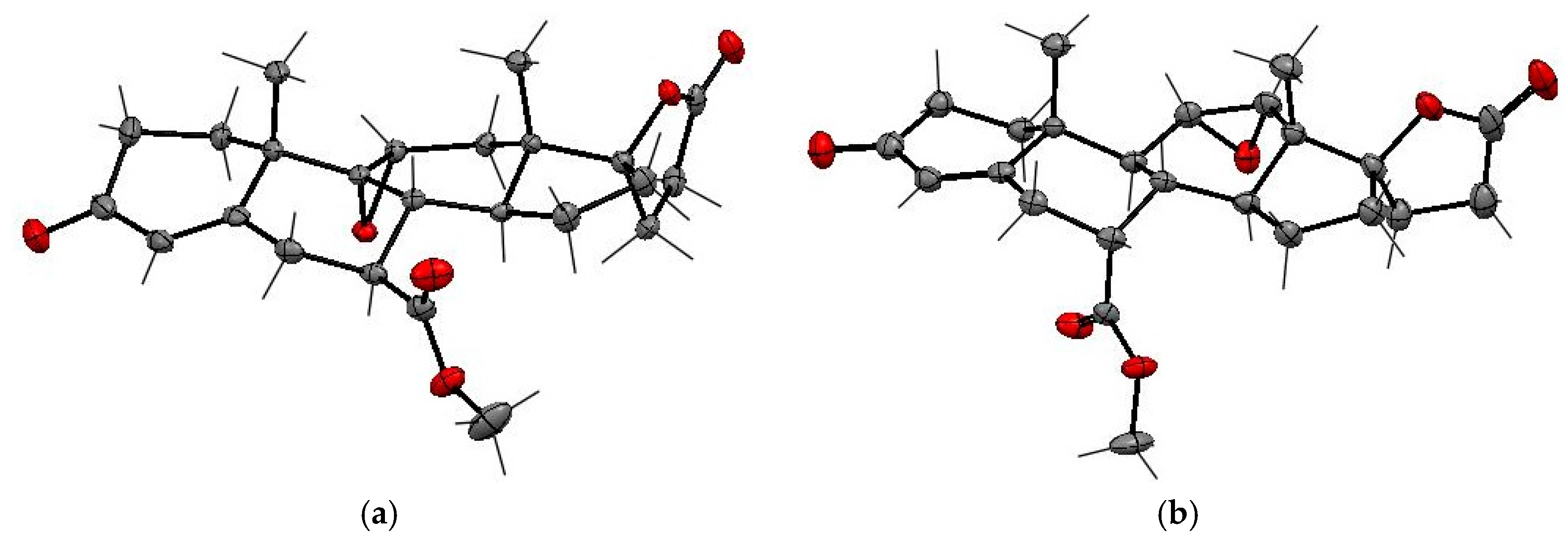
The NMR structure assignments of the isomeric 11-hydroxyesters (7α,11α,17α)-
12 and (7β,11α,17α)-
12a were confirmed by X-ray analysis (
Figure 3 and
Table 3). The SCXRD structures confirmed the relative
cis- and
trans-configurations of the 11α-hydroxy and 7α-carbomethoxy substituents in
12 and
12a, respectively. The carbomethoxy group is situated at the C-7α position in
12 and the C-7β position in
12a, whereas the hydroxy group adopts the C-11α position in both
12 and
12a isomers. The dehydration of 11α-hydroxy steroids leads predominantly to the formation of the double bond between C-9 and C-11 carbons of the steroid skeleton. As expected, the (7α,17α)-9(11)-enester
7 was the main product of the 11-hydroxyester
12 dehydration with sulfuryl chloride in the presence of imidazole; however, a considerable amount of the regioiosmeric (7α,17α)-11(12)-enester
7b, as a result of competitive 11,12-dehydration of
12, was also isolated from the recrystallization mother liquors of
7.
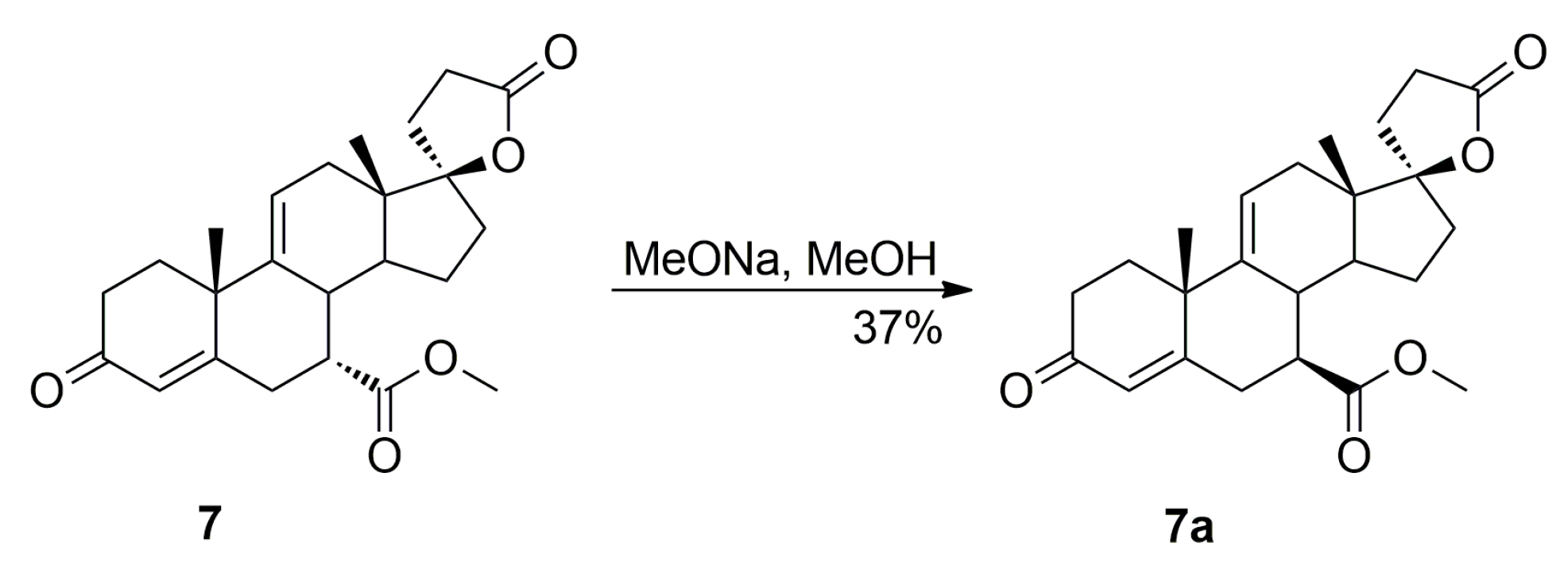
Additionally, the 9,11-dehydration of the residual 11-hydroxyester (7β,11α,17α)-12a contaminating the starting 12 isomer afforded the epimeric (7β,17α)-9(11)-enester 7a, which was also isolated chromatographically from recrystallization mother liquors of 7. It was synthesized independently by epimerization of the (7α,17α)-9(11)-enester 7 with sodium methoxide in methanol, or dehydration of the 11-hydroxyester (7β,11α,17α)-12a with sulfuryl chloride and imidazole, and then purified by column chromatography with 5–20% ethyl acetate/dichloromethane gradient elution to give the pure (7β,17α)-9(11)-enester 7a, with 37% or 85% yield respectively, as a white solid.
The [M + Na]
+ values,
m/
z 421.1986, 421.2006, and 421.1988, obtained for the free isomeric enesters
7,
7a and
7b correspond to C
24H
30O
5Na. The full NMR data confirming the structure of the known enester
7 were reported by Grob et al. [
17]; however several
1H and
13C signals were assigned reversibly. The proper
1H/
13C chemical shift assignment for isomeric enesters
7,
7a and
7b was based on the careful analysis of 2D NMR experiments and is given in
Table 4. Similarly to the epimeric hydroxyesters
12 and
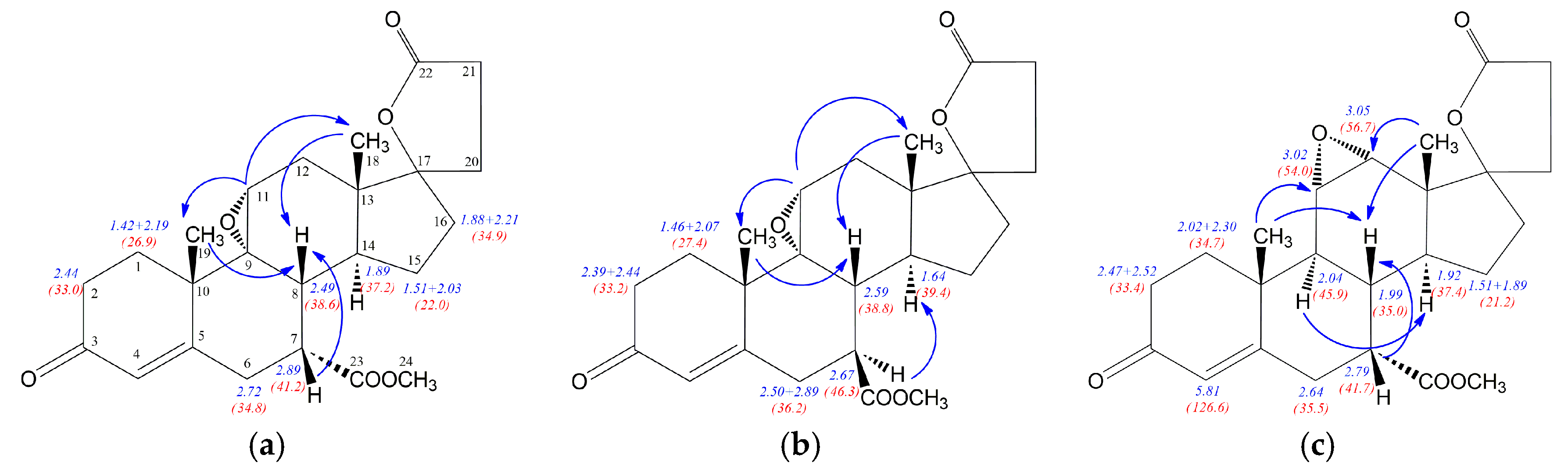
12a, the NOE effects involving H7 proton allowed distinction between 7α (
7 and
7b) and 7β (
7a) diastereoisomers (
Figure 4). The strong H7-H8 NOE effect was noted for isomers
7 and
7b with β positioned H7 proton, whereas α situated H7 in
7a is involved in H7-H14 NOE effect disturbed by H7-H15 interaction. Additionally, the significant H9-H14 NOE effect for
7b regioisomer was observed, while H8 proton of the epimers
7 and
7b is involved in H8-H6 interaction. Similarly to the epimeric hydroxyesters
12 and
12a, some of the
1H/
13C nuclei shieldings within the steroid rings B, C and D are related to the α or β configuration of the C-7 atom. The
1H shielding increase of 0.7 ppm for H7 and 0.22 ppm for H8 nuclei was observed when passing from the compound
7 with 7α positioned carbomethoxy group to its 7β epimer
7a. Similarly to
12a, both diasterotopic H15 protons of the epimer
7a with 7β positioned carbomethoxy group became equal having the same proton chemical shift (1.54 ppm). In the case of
13C-NMR the opposite effect is observed, the transition from
7 to
7a leads to the shielding decrease of 5.9 ppm for C7 and 2.4 ppm for C14 nuclei. The change of configuration from 7α in
7 to 7β in
7a is related with no change of the shielding of the C8 nucleus and decreasing of shielding by 2.3 ppm for C23 carbon of carbomethoxy substituent. The dehydration of the epimeric 11-hydroxyesters
12/
12a to the corresponding 9(11)-enesters
7/
7a caused the strong
1H shielding decrease of 89.7 ppm/84.7 ppm for C9 and 49.8 ppm/51.9 ppm for C11 carbons, respectively. The transition from
12/
12a to
7/
7a also resulted in medium shielding increase of 10.5/10.8 ppm for C12 and shielding decrease of 8.8/7.3 ppm for C19 and 3.4/3.4 ppm for C8, whereas for C10 (1.1/1.3 ppm), C13 (1.7/1.9 ppm) and C14 (2.2/3.2 ppm) a weak increasing effect was observed. The competitive dehydration of
12 to the regioisomeric 11,12-enester
7b resulted in the strong
1H shielding decrease of 57.3 ppm for C11 and 90.4 ppm for C12 nuclei. Similarly to the hydroxyesters
12 and
12a, the change of configuration from 7α in
7 to 7β in
7a is related with no change of the shielding of the C8 nucleus and decreasing of shielding by 2.3 ppm for C23 carbon of the carbomethoxy substituent. Additionally, the shielding increase of 4.5 ppm for C8 carbon was observed when passing from epimeric 9(11)-enesters
7 and
7a to regioisomeric 11-enester
7b.
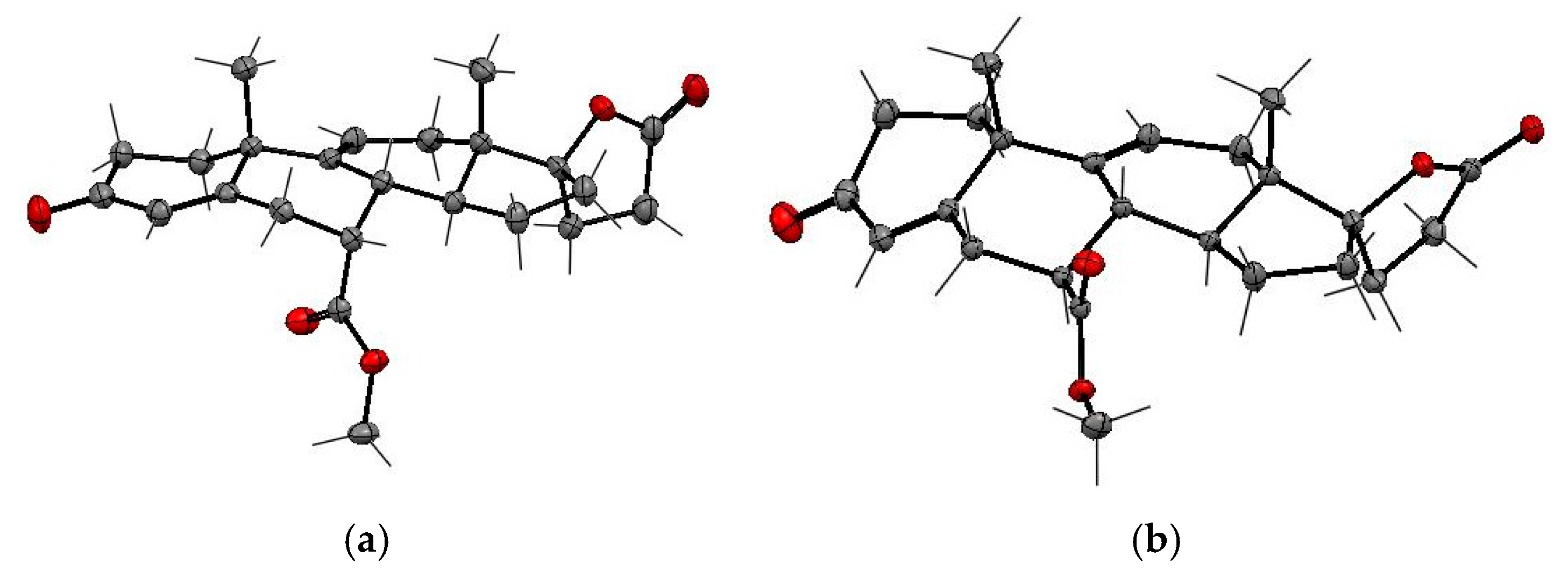
The NMR structure assignments of the isomeric (7α,17α)-9(11)-enester
7, (7β,17α)-9(11)-enester
7a and (7α,17α)-11(12)-enester
7b were confirmed by X-ray analysis (
Figure 5 and
Table 5 and
Table 6). The SCXRD structures confirmed the presence of the 9,11-double bond in
7 and
7a and the 11,12-double bond in
7b. The carbomethoxy group is situated at the C-7α position in
7 and
7b and at the C-7β position in
7a.
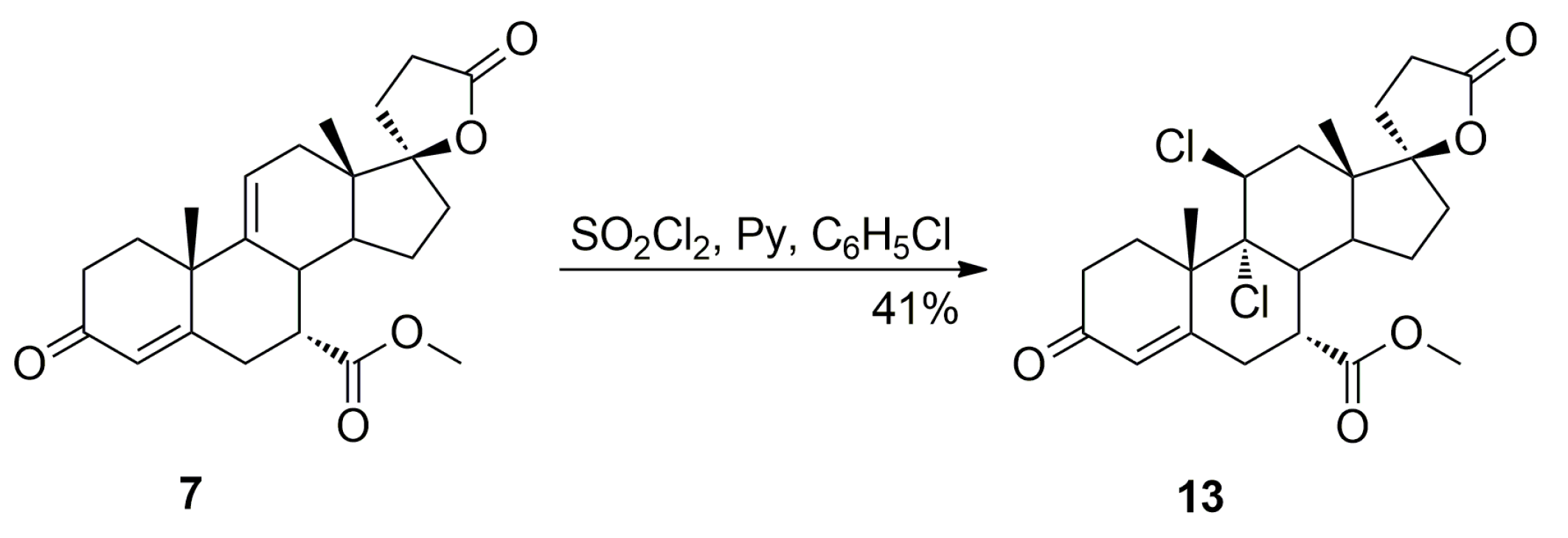
Sulfuryl chloride used for dehydration of 11α-hydroxy steroids is also known as a chlorinating agent and, under some conditions, applied for chlorination of steroid double bonds [
27,
28]. Indeed, the novel (7α,11β,17α)-9,11-dichloro derivative
13, formed as a result of competitive chlorine addition to the newly formed 9,11-double bond of the key intermediate (7α,17α)-9(11)-enester
7, was isolated chromatographically from the recrystallization mother liquors of
7. It was also synthesized by chlorination of the pure enester
7 with sulfuryl chloride in the presence of pyridine in chlorobenzene, and then purified by column chromatography with 5–30% EtOAc/CH
2Cl
2 gradient elution to give the pure (7α,11β,17α)-9,11-dichloro derivative
13 (41% yield) as a white solid. The novel 9,11-dichloro impurity
13 was formed by the nucleophilic attack of a chloride anion on the intermediate chloronium cation, a structural analogue of the eplerenone epoxide ring. No other isomers of
13 were obtained.
The [M + Na]
+ value,
m/
z 491.1386 obtained for the 9,11-dichloroderivative
13 corresponds to C
24H
30O
5NaCl
2. The stereochemistry at the C9 and C11 carbons of the dichloro derivative
13 was established on the basis of 1D/2D NOESY experiments, which indicated the relative
trans configuration of the C-9α and C-11β positioned chlorine atoms. The addition of chlorine to the 9,11-double bond of enester
7 caused significant changes in the
1H/
13C nuclei shieldings within the steroid rings A, B, C and D of the dichloro derivative
13 (
Table 7).
The strong shielding increase of 55.7 ppm for C9 and 59.5 ppm for C11 nuclei was observed when passing from
7 to
13. Minor shielding increase of 4.1 ppm for C1, 2.9 ppm for C5, 3.3 ppm for C6, 2.9 ppm for C7 and 1.8 ppm for C14 and shielding decrease of 6.6 ppm for C10 nuclei were also noted. The introduction of chlorine into C9 and C11 positions of
7 also resulted in shielding increase of 0.97 ppm for H11 and shielding decrease of 0.18 ppm for H4, 0.42 ppm for H8 and 0.97 for H14 nuclei in dichloro derivative
13. Noteworthy, the diasterotopic effect observed for H15 protons of
7 (0.4 ppm) increased to 0.75 ppm for the same protons in
13. Similarly to the other compounds with 7α-carbomethoxy group, the β positioned H7 proton in dichloro derivative
13 is involved in strong H7-H8 NOE effect (
Figure 6). The NMR structure assignment of the novel (7α,11β,17α)-9,11-dichloro derivative
13 was confirmed by X-ray analysis (
Figure 7 and
Table 5). The SCXRD structure confirmed the presence of the two chlorine atoms at the C-9α and C-11β positions of the steroid ring C in the relative
trans configuration and the C-7α positioned carbomethoxy substituent.
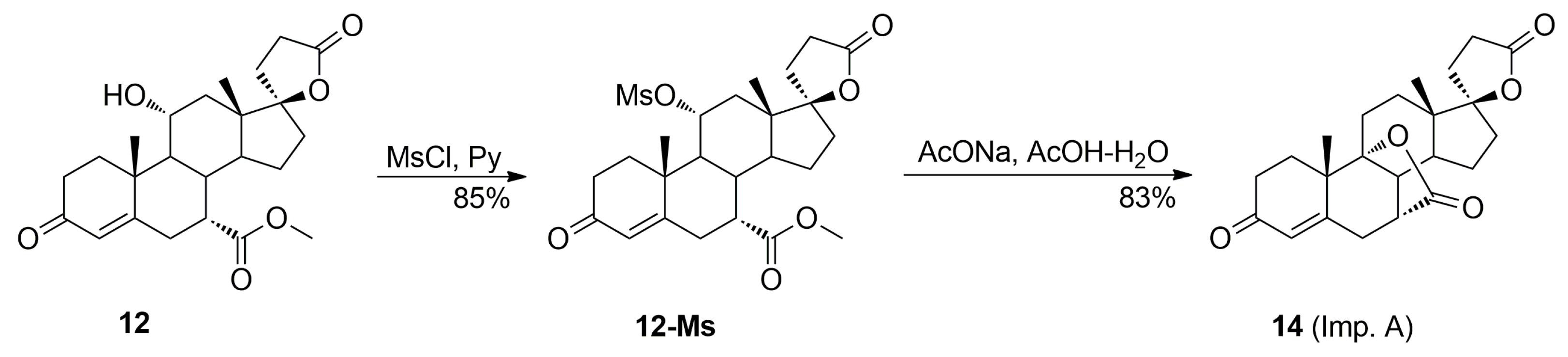
The acidic conditions applied for the dehydration of the 11α-hydroxy group in 12 resulted in competitive lactonization between the hydroxyl and carbomethoxy groups and lead to 7α,9:21,17-dicarbolactone 14 (Imp. A), which tends to be formed even in the presence of traces of free water. The dicarbolactone 14 was isolated chromatographically from the recrystallization mother liquors of 7. It was also synthesized independently from the mesylate of the starting 11α-hydroxyester 12 by reaction with acetic acid in the presence of sodium acetate, and then purified by column chromatography with 5–30% ethyl acetate/dichloromethane gradient elution to afford the pure 7α,9:21,17-dicarbolactone 14 (83% yield) as a white solid.
The [M + Na]
+ value,
m/
z 407.1847, obtained for the dicarbolactone
14 corresponds to C
23H
28O
5Na. The
1H-/
13C-NMR chemical shifts assignment for dilactone
14 is presented in
Table 7. The detailed analysis of 2D NMR spectra, especially
1H-
13C HMBC and NOESY experiments, unambiguously confirmed the structure consisting of the two γ-lactone rings. The second γ-lactone moiety results from internal lactonization between 9α-hydroxy and 7α-carbomethoxy groups of intermediate ester, formed by water addition to the 9,11-double bond of enester
7 during the dehydration step of
12. This significant change in the steroid structure entails numerous changes in the
1H/
13C nuclei shielding. The shielding increase of 46.8 ppm for C11 and 16.5 ppm for C12 and decrease of 37.4 ppm for C9 nuclei were observed when passing from hydroxyester
12 to the dicarbolactone
14. Minor shielding increase of 7.7 ppm for C1 and 6.2 ppm for C5 was also noted, whereas for C8 shielding decrease of 8.1 ppm was observed. Similarly to the other compounds of the series with 7α positioned carbomethoxy group, the H7 proton of the dicarbolactone
14 is involved in two strong H7-H8 and H6-H7 NOE effects (
Figure 6).
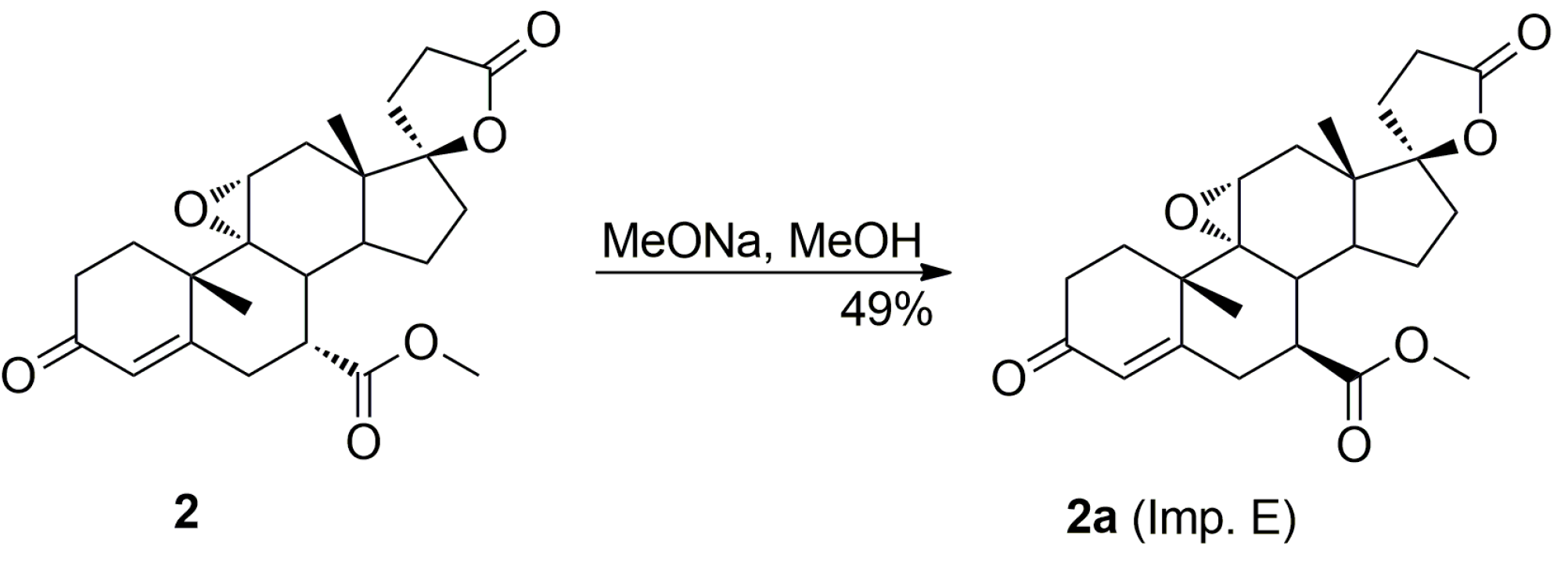
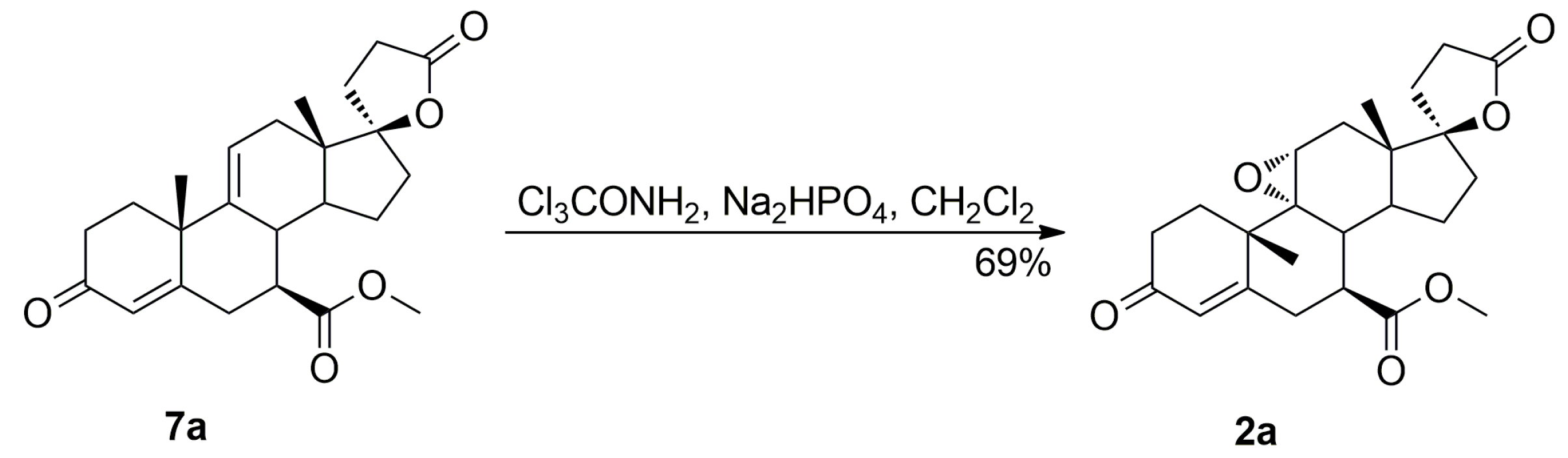
The purification of the key intermediate (7α,17α)-9(11)-enester 7 by large-scale recrystallization only partially removes the impurities formed during the dehydration of the starting 11α-hydroxyester 12. Thus, in a preferred technological embodiment, the key intermediate enester 7 was isolated in a crude form by dichloromethane extraction followed by evaporation of the solvent, and used directly in the following step of the process, which was the epoxidation of the 9,11-double bond to produce the eplerenone. Thus, the crude enester 7 was dissolved in dichloromethane and epoxidized with hydrogen peroxide in the presence of disodium hydrogen phosphate and trichloroacetamide. The crude product was recrystallized from ethanol and 2-butanone to give the pharmaceutical grade eplerenone. Apart from the (7α,11β,17α)-9,11-dichloro derivative 13 and the 7α,9:21,17-dicarbolactone 14 (Imp. A), which were present in the crude enester 7, the epoxidation step produced the two further impurities, i.e., the isomeric (7β,11α,17α)-9,11-epoxyester 2a and (7α,11α,12α,17α)-11,12-epoxyester 2b, which were isolated chromatographically from the recrystallization mother liquors of 2. The trichloroacetamide is known to preferentially epoxidize the more highly substituted double bond, e.g., the 9,11-olefin. In the case of the crude 9,11-enester 7 contaminated with the 11,12-enester 7b, the 9,11-epoxide should be formed with the minimal competitive epoxidation of the 7b isomer yielding the 11,12-epoxide as the by-product. As expected, the chromatography of the recrystallization mother liquors of 2 with 1–10% acetone/dichloromethane gradient elution gave the (7α,11α,12α,17α)-11,12-epoxyester 2b, which was undoubtedly formed via epoxidation of the (7α,17α)-9(11)-enester 7b. It was also synthesized independently by epoxidation of the enester 7b with hydrogen peroxide in the presence of disodium hydrogen phosphate and trichloroacetamide, and purified by column chromatography to afford the pure 11,12-epoxyester 2b (60% yield) as a white solid. Further gradient elution of the recrystallization mother liquors of 2 with 1–10% acetone/dichloromethane gave the isomeric (7β,11α,17α)-9,11-epoxyester 2a, which was formed by epoxidation of the (7β,17α)-9(11)-enester 7a contaminating the crude intermediate enester 7. It was also synthesized independently by epoxidation of the enester 7a with hydrogen peroxide in the presence of disodium hydrogen phosphate and trichloroacetamide, or basic epimerization of eplerenone (2), and then purified by column chromatography to give the pure (7β,11α,17α)-9,11-epoxyester 2a, with 69% or 49% yield respectively, as a white solid. The recrystallization of the crude eplerenone (2) from ethanol removes the new 9,11-dichloro derivative 13, the isomeric (7β,11α,17α)-9,11-epoxyester 2a and the residuals of the unreacted isomeric (7β,17α)-9(11)-enester 7a and (7α,17α)-9(11)-enester 7b. The recrystallization from 2-butanone removes the 7α,9:21,17-dicarbolactone 14 (Imp. A) and affords the pharmaceutical grade eplerenone (2).
The NMR data of eplerenone were presented in literature sources many times; however the full and correct
1H/
13C chemical shifts assignment confirming its structure has never been done. Although the complete assignment of
1H-/
13C-NMR signals was presented by Grob et al. [
17], some of the signals were assigned reversibly. The careful analysis of 2D NMR experiments, including HSQC, HMBC and NOESY measurements, allowed to assign signals to the corresponding protons and carbons of the isomeric epoxy esters
2,
2a and
2b unambiguously, and thus confirm the structure of eplerenone and its isomers (
Table 8). Blue arrows in
Figure 8 show the most important NOE effects involving H7 proton, simultaneously indicating the 7α or 7β positioned carbomethoxy group. The strong H7-H8 NOE effect is observed for the compounds
2 and
2b with β positioned H7 proton, whereas α situated H7 proton in
2a is involved in the strong interaction with H14. The epoxidation of the 9,11-double bond caused several shielding effects observed for
1H/
13C nuclei within the steroid rings A, B and C. The strong shielding increase of 77.1/75 ppm for C9, 67.6/66.8 ppm for C11 carbons and 2.5/2.4 ppm for H11 protons was observed when passing from enesters
7/
7a to epoxides
2/
2a, respectively. Minor shielding increase of 7/6.5 ppm for C1, 5.9/6.1 ppm for C14, 2.6/3.4 for C7, 1.8/1.6 ppm for C8, 2/2 ppm for C12 and 0.7/0.4 for C13 carbons was also noted. The transition from enesters
7/
7a to epoxides
2/
2a caused shielding increase of 0.76/0.66 ppm for one of the H1 protons and 0.36/0.31 ppm for one of the H12 protons, whereas for H-14 protons shielding decrease of 0.4/0.14 ppm was noted. Similarly to the epimeric hydroxyesters
12/
12a and enesters
7/
7a, some of the
1H/
13C nuclei shielding within the steroid rings B, C and D are related to the α or β configuration of the C7 atom. The
1H shielding increase of 0.22 ppm for H7 and 0.25 ppm for H14 nuclei is observed when passing from the compound
2 with 7α positioned carbomethoxy group to its 7β epimer
2a. Additionally, the change of configuration at the C7 is accompanied by weak shielding decrease of 0.1 ppm for H8. Simultaneously, both diasterotopic H15 protons of the epimer
2a with 7β positioned carbomethoxy group became equal having the same proton chemical shift (1.51 ppm). In the case of
13C-NMR data the opposite effect is observed, the transition from
2 to
2a leads to the shielding decrease of 5.1 ppm for C7, 1.9 ppm for C9 and 2.2 ppm for C14 nuclei. The change of configuration from 7α in
2 to 7β in
2a is also related with shielding decrease of 2.1 ppm for C23 carbon of the carbomethoxy substituent. The competitive epoxidation of the 11,12-enester
7b to the 11,12-epoxyester
2b resulted in the strong shielding increase of 76.1 ppm for C11 and 77.1 ppm for C12 nuclei, whereas for C9 (3.3 ppm), C18 (4.4 ppm) and C14 (6.7 ppm) medium effects were observed. The strong shielding increase of 2.57 and 2.84 ppm was noted for H11 and H12 protons, respectively. Minor changes in increasing of shielding for H7 (0.1 ppm), H8 (0.24 ppm) and H9 (0.4 ppm) protons were also observed. Similarly to the enesters
7,
7a and
7b, the shielding increase of 3.6 ppm and 3.8 ppm for C8 carbon was noted when passing from epimeric 9,11-epoxides
2 and
2a to regioisomeric 11,12-epoxyester
2b.
The [M + Na]
+ values,
m/
z 437.1941 and 437.1942, obtained for the two isomeric epoxyesters
2 and
2b correspond to C
24H
30O
6Na. The [M + H]
+ value,
m/
z 415.2113, obtained for the third isomeric epoxyester
2a corresponds to C
24H
31O
6. The NMR structure assignments of the isomeric (7β,11α,17α)-9,11-epoxyester
2a and (7α,11α,12α,17α)-11,12-epoxyester
2b were confirmed by X-ray analysis (
Figure 9 and
Table 9). The SCXRD structures confirmed the presence of the 9α,11α-epoxide ring in
2a and 11α,12α-epoxide ring in
2b. The carbomethoxy group is situated at the C-7α position in
2b and at the C-7β position in
2a.























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
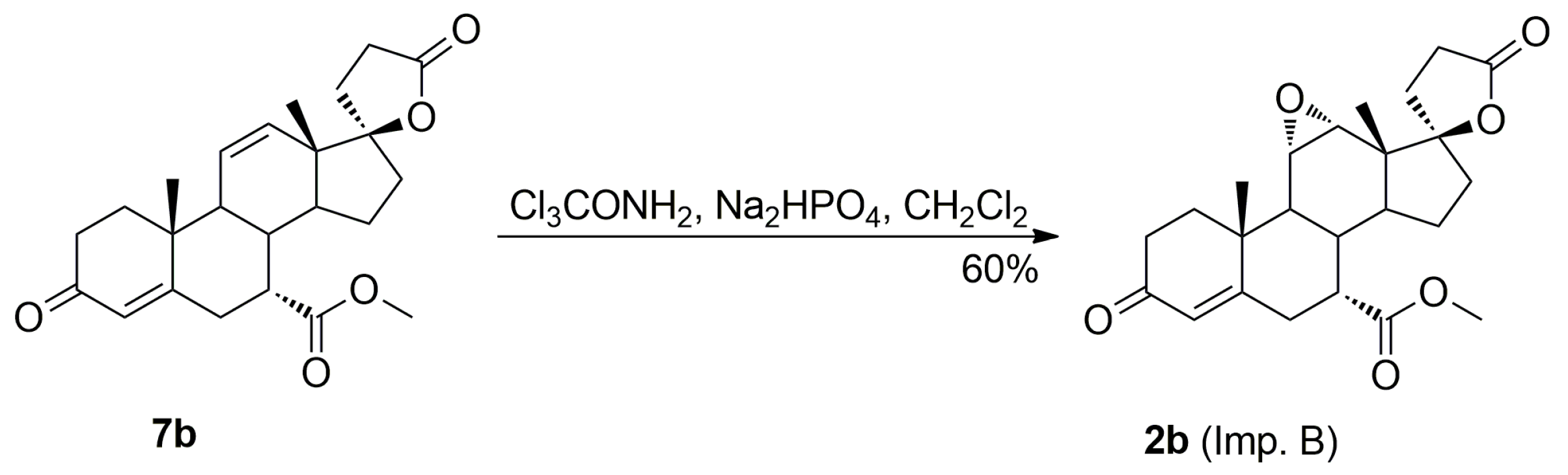{kind=link}
{kind=link}
{kind=link}
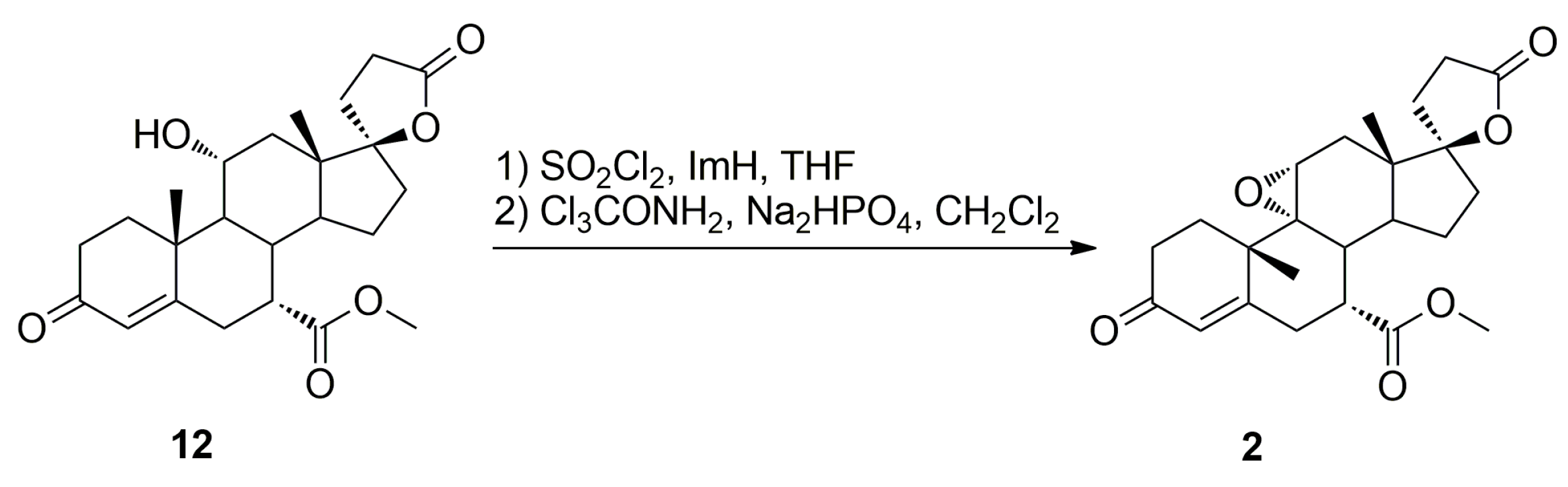{kind=link}








