1. Introduction
Antisense oligonucleotides (ASOs) bind complementary mRNA and modulate its function to yield a pharmacological response [
1,
2,
3]. Second-generation ASOs are typically 16–20 nucleotides in length connected by phosphorothioate linkages and contain a DNA nucleotide “gap” subtended by 2′-
O-methoxyethyl (MOE) modified RNA “wings”. Kynamro, a second generation ASO targeting Apolipoprotein B-100 mRNA, was recently approved by the FDA for the treatment of homozygous familial hypercholesterolemia [
4]. There are more than 100 ASOs advancing in the clinic for a variety of indications [
5], many of which target mRNA expressed primarily in the hepatocytes in the liver. Relatively recently, conjugation of ASOs to triantennary
N-acetyl galactosamine (GalNAc) ligands has been shown to improve potency in hepatocytes [
6,
7]. GalNAc conjugation on both the 3′ and 5′-termini of the oligonucleotide has been evaluated with the former having slightly enhanced potency in cells and in animals [
8].
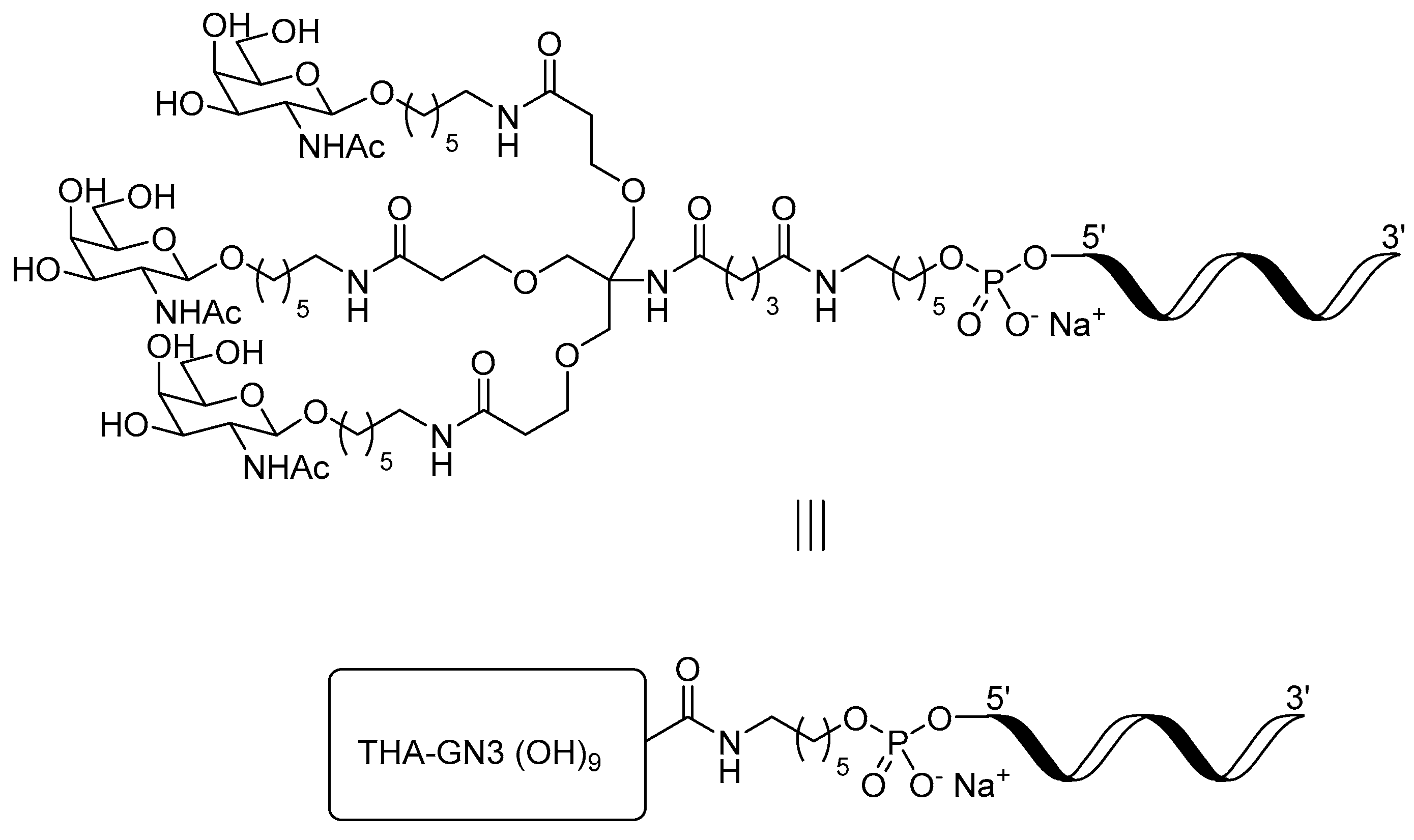
The structure of our optimized 5′-GalNAc-ASO conjugate is shown in
Figure 1. In the structure, the trishexylamino (THA) GalNAc ligand is connected to the ASO 5′-terminus through an aminohexanol linker. The linker is attached to the ligand via an amide bond and to the ASO via a phosphate diester. Synthetic routes to the GalNAc-ASO conjugate differ in the method and order of formation of both the amide and phosphate diester links.
ASOs are commonly synthesized on a solid support using the phosphoramidite method [
9]. The method, which has been highly optimized over the years, involves assembly of the ASO one nucleotide at a time on the support using protected nucleoside phosphoramidite building blocks. The oligonucleotide is typically synthesized in the 3′ ➔ 5′ direction. Numerous synthetic methods have been developed to conjugate molecules to ASOs [
10], with conjugation approaches generally falling within two categories: solution-phase and solid-phase. We evaluated each type to synthesize our conjugate structure and report the results herein.
Our initial synthesis route (
Figure 2) is an example of a solution-phase approach, consisting of solid-phase synthesis and purification of a 5′-aminohexyl modified ASO, followed by solution-phase conjugation with THA-GalNAc glutarate activated as the pentafluorophenyl (PFP) ester [
8]. There are several advantages to this synthetic route. First, the aminohexyl-ASO intermediate can be synthesized, purified, and isolated in high yield and purity using standard oligonucleotide manufacturing techniques. Second, the conjugation reaction is reliable, efficient, and selective. Third, the protocol is scalable, having been successfully used to produce multi-kilogram quantities of conjugated drug substance. The solution-phase strategy, however, is lengthy, involving numerous unit operations, and notably two purification steps. We, therefore, became interested in developing alternative approaches.
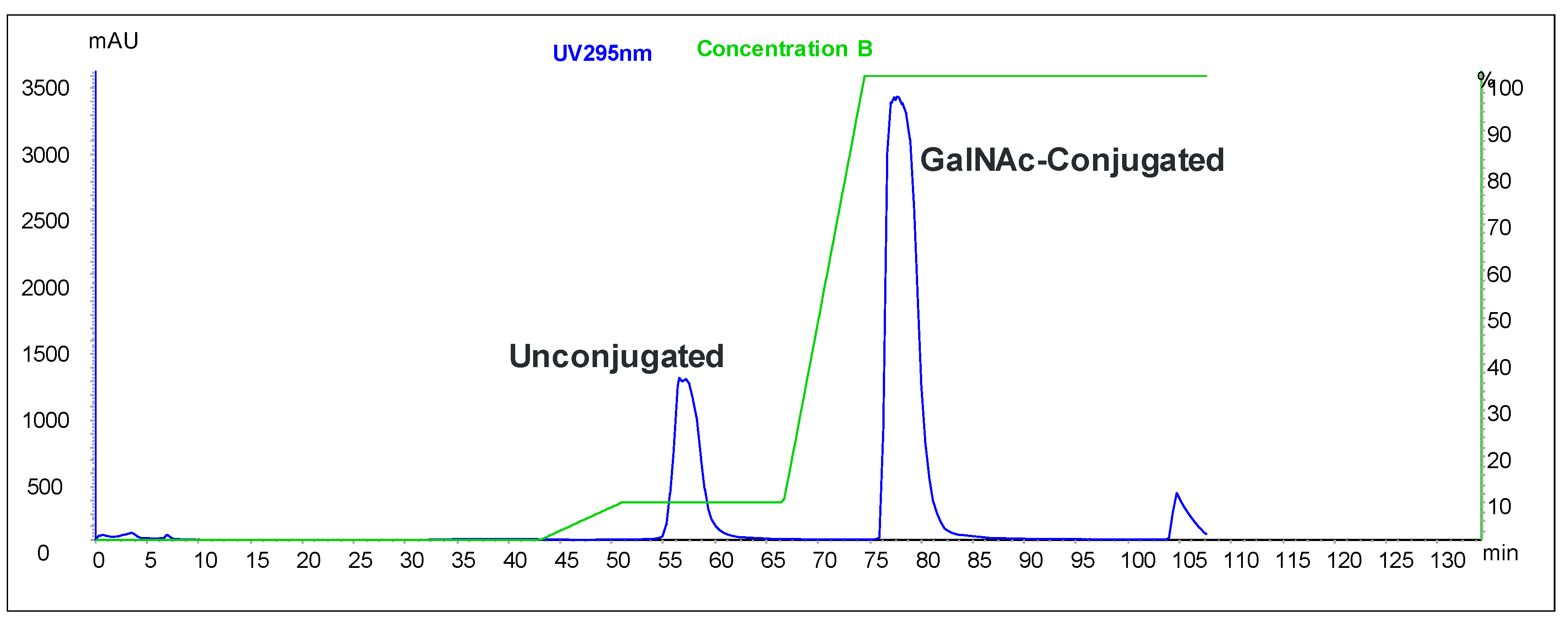
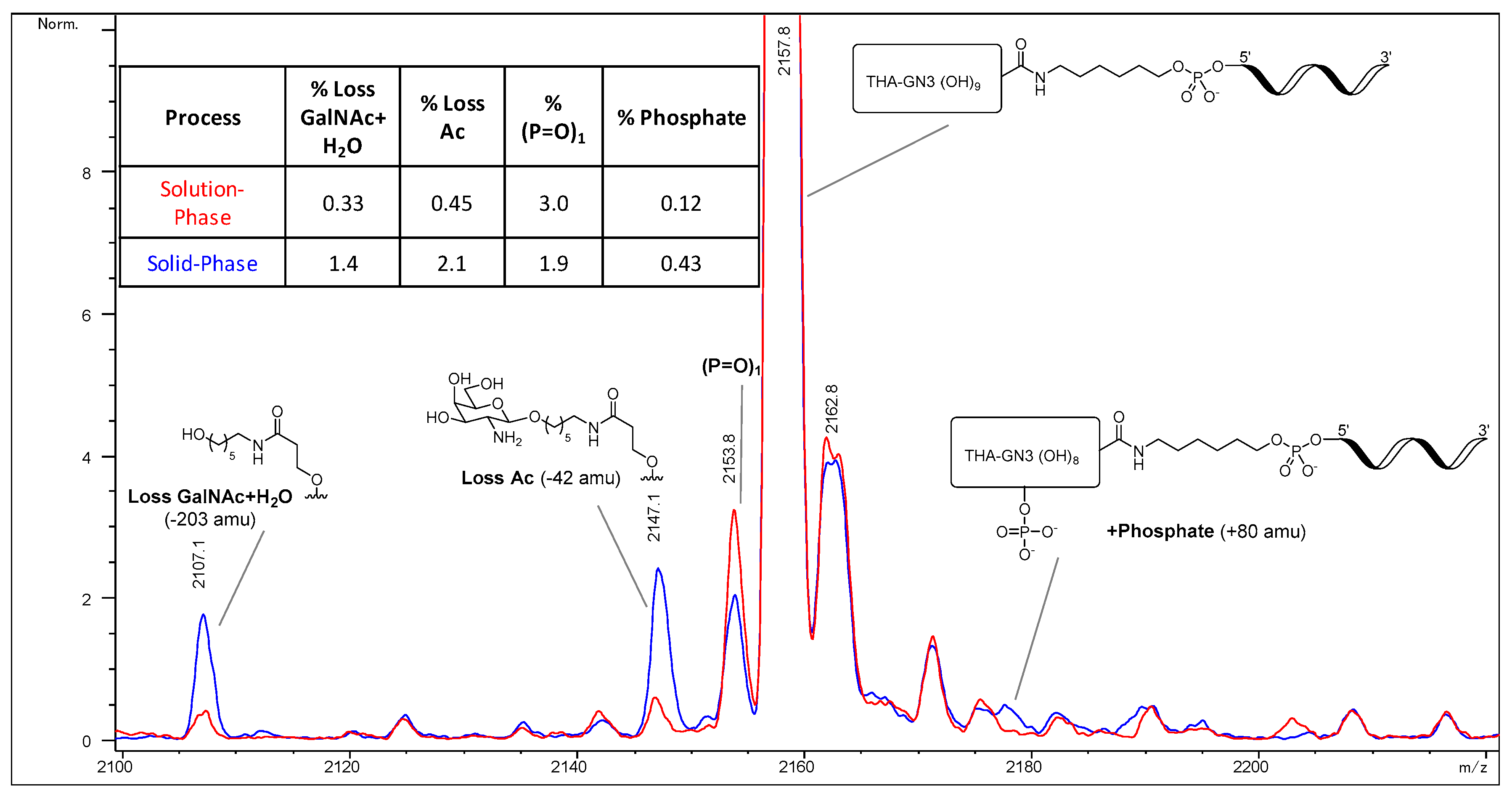
We developed an alternative solid-phase conjugation strategy consisting of the coupling of THA-GalNAc-aminohexyl phosphoramidite to support-bound ASO (
Figure 2). The process requires fewer unit operations and is considerably shorter than the solution-phase approach. Since there is no trityl-protecting group, a purification protocol capable of discriminating GalNAc-conjugated from unconjugated ASOs was developed.
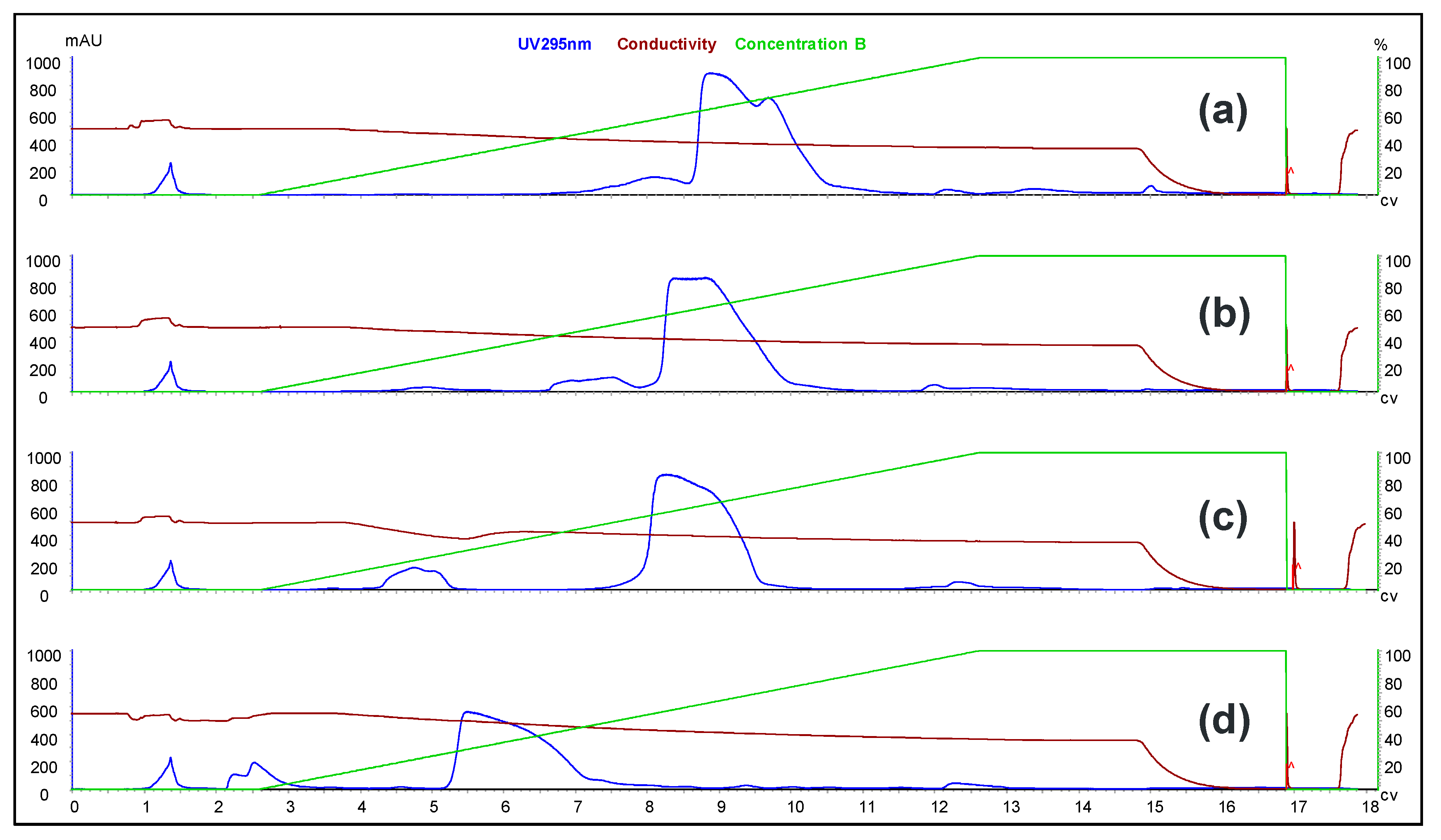
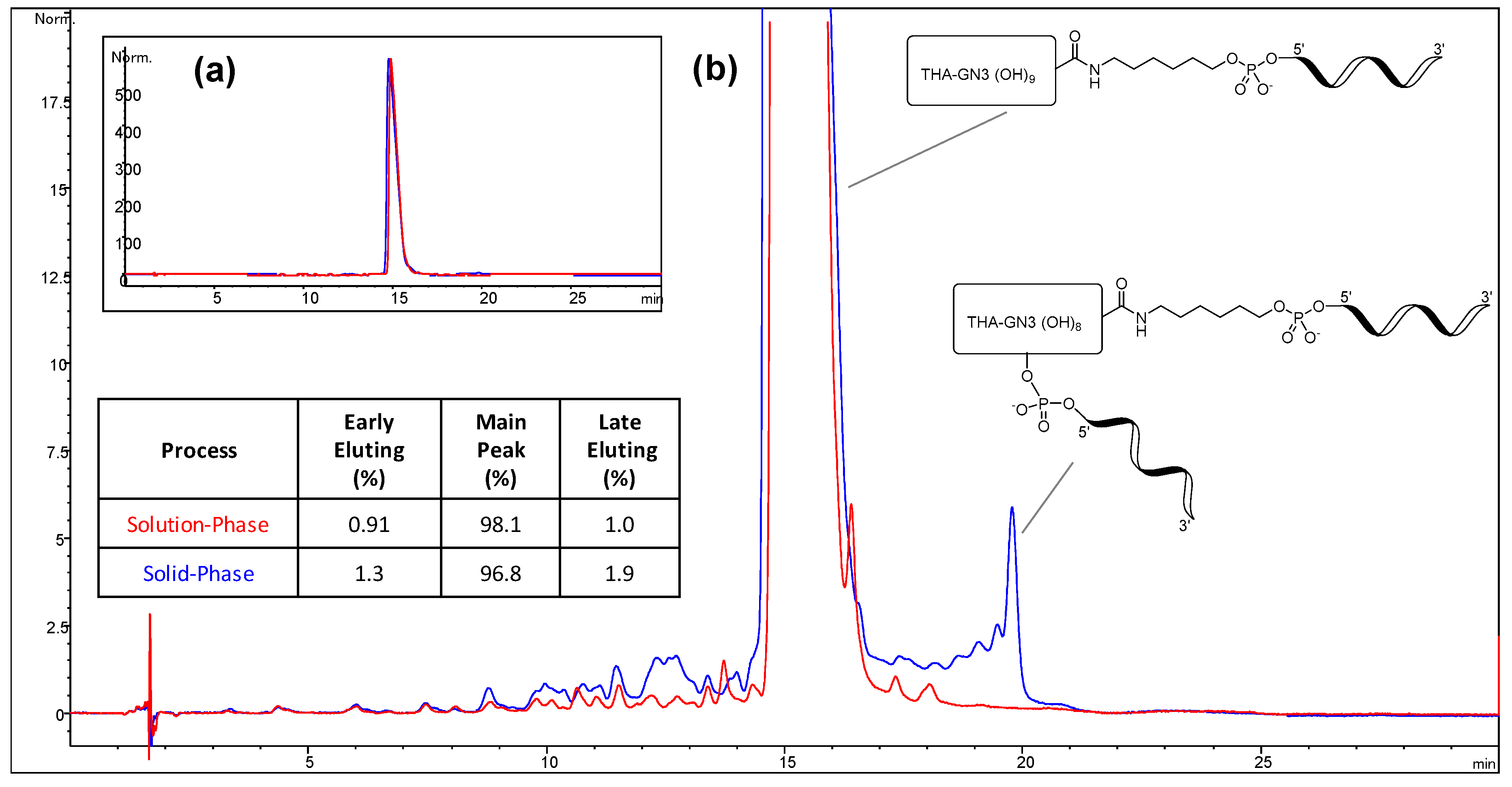
To evaluate the differences between the solution-phase and solid-phase syntheses outlined above, a head-to-head comparison was conducted. Each process was carried out at 1.1 mmol scale using the same 5-10-5 MOE deoxy gapmer oligonucleotide. The two routes were compared with respect to equivalents of GalNAc-THA required, conjugation efficiency, and purity. The results of the study are presented herein.
3. Materials and Methods
3.1. Materials
Low water acetonitrile from BDH Chemicals (purchased through VWR, Radnor, PA, USA), deblocking reagent from EMD Millipore (Billerica, MA, USA), oxidation solution from EMD Millipore, xanthane hydride from TCI (Tokyo, Japan), pyridine from Alfa Aesar (Tewksbury, MA, USA), acetic anhydride from Avantor Peformance Chemicals (Center Valley, PA, USA), triethylamine from Burdick and Jackson (Morristown, NJ, USA), and toluene from BDH were all purchased via VWR (Radnor, PA, USA). Molecular sieve packets were purchased from Prime Synthesis (Aston, PA, USA). NittoPhaseHL UnyLinker solid support was purchased from Kinovate Life Sciences (Oceanside, CA, USA).
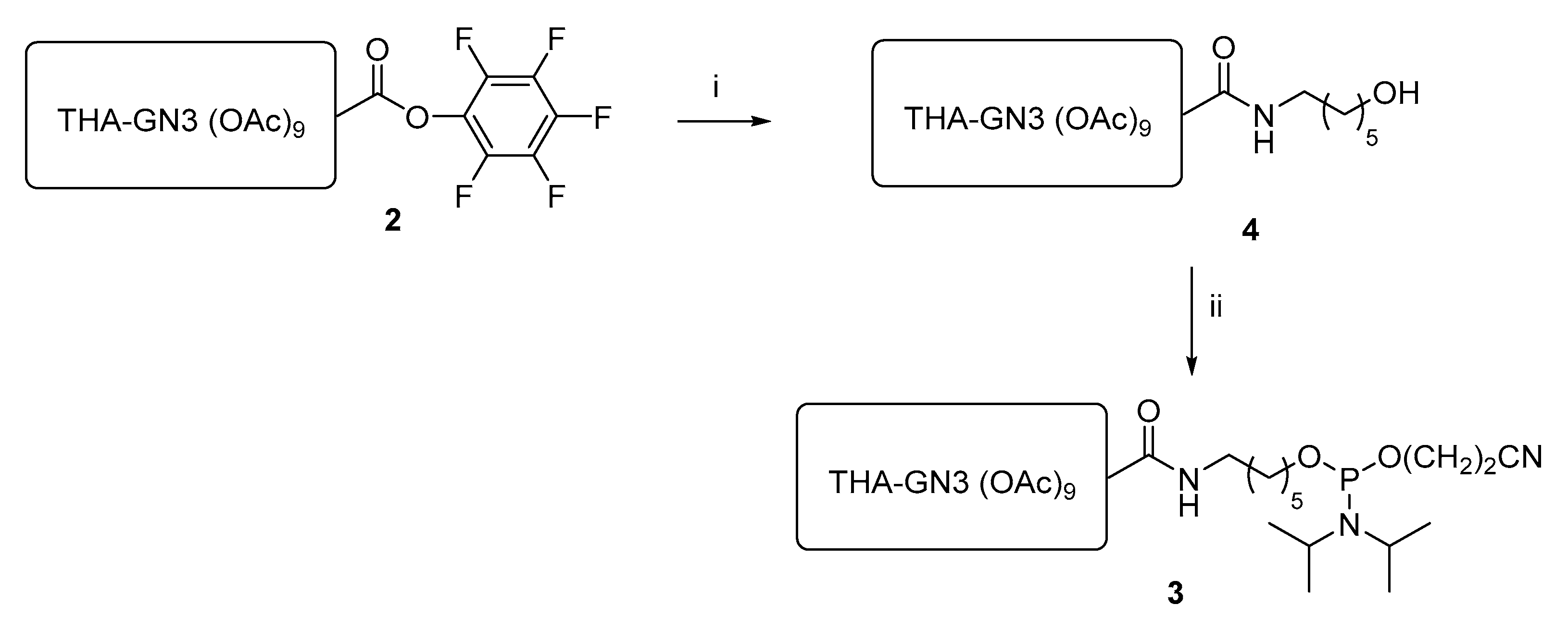
3.2. THA-GalNAc Aminohexyl Alcohol 4
THA-GalNAc PFP ester 2 (150 g, 78.8 mmol) was dissolved in THF (750 mL) with stirring. Triethylamine (27.5 mL, 197 mmol) was added, followed by 6-amino-1-hexanol (9.2 g, 78.8 mmol). The mixture was stirred for 1 h at room temperature and diluted in CH2Cl2 (500 mL). The solution was washed once with 500 mL 5% NaHSO4 solution and twice with 500 mL saturated NaHCO3 solution. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by SiO2 chromatography with a gradient of 5% MeOH in EtOAc up to 20% MeOH in EtOAc. The product fractions were combined and concentrated to a white solid (111.9 g, 77% yield).
1H-NMR (300 MHz, CD3CN) δ 6.99 (t, J = 5 Hz, 3H), 6.85 (d, J = 9 Hz, 3H), 6.71 (m, 1H), 6.50 (m, 1H), 5.35 (d, J = 3 Hz, 3H), 5.26 (dd, J = 5, 11 Hz, 3H), 4.67 (d, J = 9 Hz, 3H), 4.15 (m, 6H), 3.93 (m, 9H), 3.67 (m, 14H), 3.49 (m, 3H), 3.23 (m, 8H), 2.42 (t, J = 6 Hz, 6H), 2.23 (m, 4H), 2.14 (s, 9H), 2.05 (s, 9H), 2.00 (s, 9H), 1.95 (s, 11H), 1.53 (m, 16H), 1.36 (m, 16H); 13C-NMR (75 MHz, CD3CN) δ 173.1, 173.0, 171.5, 170.5, 170.4, 107.9, 101.1, 77.3, 70.5, 70.2, 69.6, 69.3, 67.5, 66.9, 62.3, 61.5, 59.7, 53.5, 51.3, 39.3, 36.7, 36.3, 35.3, 32.5, 31.9, 29.5, 29.2, 29.0, 26.5, 25.5, 25.3, 24.0, 23.3, 22.7, 22.3, 20.7, 14.1.
3.3. THA-GalNAc Phosphoramidite 3
THA-GalNAc aminohexyl alcohol 4 (18.1 g, 9.86 mmol) was dissolved in CH2Cl2 (54 mL) in a round-bottomed flask. The solution was concentrated to dryness by rotary evaporation. The solid was redissolved in CH2Cl2 (54 mL) and the solution concentrated to dryness two more times. The solid was redissolved in CH2Cl2 (54 mL) under positive N2 pressure. The solution was cooled to 0 °C in an ice bath. With stirring, 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphordiamidite (5.94 g, 19.72 mmol) was added dropwise by syringe. The solution stirred for 5 min. Ethylthiotetrazole (ETT, 1.54 g, 11.83 mmol) was added as a solid. The solids slowly dissolved into a homogeneous solution. The reaction mixture stirred for 3 h at 0 °C. After 3 h, the reaction mixture warmed to room temperature and triethylamine (5.0 mL, 35.85 mmol) was added dropwise. The mixture was concentrated to ~⅓ volume by rotary evaporation and loaded onto a SiO2 column (100 g SiO2, ~10 cm diameter) that had been prepared with EtOAc containing 1% NEt3. The column was flushed with 2 L of 1% NEt3 in EtOAc followed by 2 L of 1:3:96 NEt3:acetone:THF. The product eluted during the first 1 L of the latter mobile phase mixture. Product fractions were combined and concentrated to a white solid (15.1 g, 75% yield).
1H-NMR (300 MHz, CD3CN) δ 6.97 (m, 3H), 6.83 (m, 3H), 6.53 (s, 1H), 5.47 (s, 1H), 5.30 (d, J = 3 Hz, 3H), 5.05 (dd, J = 5, 11 Hz, 3H), 4.55 (d, J = 6 Hz, 3H), 4.10 (m, 6H), 3.98 (m, 6H), 3.82 (m, 4H), 3.63 (m, 15H), 3.51 (m, 3H), 3.16 (m, 8H), 2.66 (t, J = 6 Hz, 2H), 2.35 (t, J = 6 Hz, 6H), 2.16 (m, 4H), 2.12 (s, 9H), 2.01 (s, 9H), 1.96 (p, J = 3 Hz, 2H), 1.94 (s, 9H), 1.87 (s, 9H), 1.82 (m, 2H), 1.48 (m, 14H), 1.41 (s, 6H), 1.34 (m, 12H), 1.20 (dd, J = 2, 8 Hz, 12H); 31P-NMR (121.5 MHz, CD3CN) δ 147.05.
3.4. Solid-Phase Synthesis
A total of 2.2 mmol (7.0 g) of NittoPhaseHL UnyLinker solid support loaded at 317 µmol/g was weighed into a plastic weigh boat, slurried in acetonitrile, and transferred to a FineLINE 35 mm column (GE Healthcare, P/N 28946841, Little Chalfont, UK) with the outlet plugged. Additional acetonitrile was added to approximately 10 cm and then the piston was lowered to a height of approximately 7.0 cm. Synthesis was performed using an AKTA OligoPilot 100 Plus synthesizer (GE Healthcare, P/N 18-1136-79) equipped with custom mass flow meters.
Dichloroacetic acid (10% by volume) in toluene was used for deblocking of the 4,4′-dimethoxytrityl (DMTr) groups from the 5′-hydroxyl group of the nucleotide. 4,5-Dicyanoimidazole (1.0 M) in the presence of
N-methylimidazole (0.10 M) was used as the activator during the coupling step. During the coupling step, 1.45 equivalents of 0.20 M phosphoramidite solution (2′-deoxy and 2′-
O-methoxyethyl nucleosides) and a flow ratio of 1:1 (
v/
v) of phosphoramidite solution to activator solution was used. Phosphoramidite and activator solutions were prepared using low-water acetonitrile (water content <30 ppm) and were dried further by the addition of molecular sieve packets (Prime Synthesis, P/N SP-MT10). Phosphorothioate linkages were introduced by sulfurization of phosphite triesters with 0.20 M solution of xanthane hydride in pyridine. Phosphate diester linkages were incorporated via oxidation of phosphite triesters using a solution of iodine in pyridine/water (90/10,
v/
v). Unreacted hydroxyl groups were acetylated using
N-methylimidazole/pyridine/acetonitrile (20/30/50,
v/
v/
v) and acetic anhydride/acetonitrile (20/80,
v/
v) delivered in a 1:1 (
v/
v) flow ratio. At the end of synthesis, the support-bound oligonucleotide was treated with a solution of triethylamine/acetonitrile (1:1,
v/
v) to remove acrylonitrile formed by deprotection of the cyanoethyl group from the phosphorothioate triester. Reagent delivery volumes and contact times are detailed in
Table 4. Subsequently, the support-bound oligonucleotide was incubated with 160 mL of concentrated aqueous ammonium hydroxide at 55 °C for approximately 15 h to complete the cleavage from the support, elimination of UnyLinker molecules to liberate the 3′-hydroxy group of the oligonucleotide, and deprotection of nucleobase-protecting groups. After allowing the crude mixture to cool to room temperature it was filtered (0.45 µm aPES) and the native support was rinsed with 160 mL of purified water.
3.5. Work-Up and Isolation of Solid-Phase GalNAc-ASO Conjugate
Precipitation of purified oligonucleotide was performed by adding 1.0 part purified oligonucleotide solution to 8.0 or 9.0 parts ethanol by volume in a glass bottle. The precipitation mixture was shaken vigorously by hand and allowed to settle overnight at room temperature. Supernatant was decanted to waste and precipitated oligonucleotide was reconstituted in water. Replicate runs (n = 6) demonstrated that measured recovery was 98–100% by IP-HPLC-UV.
3.6. Solution-Phase GalNAc-THA PFP Ester Conjugation
The following procedure is based on a previously reported protocol [
8]. Aminohexyl-ASO (1.8 g, 0.23 mmol) was dissolved in 0.06 M sodium tetraborate buffer at pH 9.3 (18 mL) and stirred at room temperature. A solution of THA-GalNAc PFP ester
2 (1.137 g, 0.60 mmol, 2.6 eq. vs. aminohexyl-ASO, 1.8 eq. vs. 1.1 mmol solid-phase synthesis scale) dissolved in acetonitrile (6.4 mL) was added to the aminohexyl-ASO solution over about one minute. The mixture was stirred at room temperature for 3.25 h, at which point in-process analysis indicated the reaction had progressed to >99%. Concentrated aqueous ammonia was added (10 mL), and the reaction mixture stirred for 24 h at room temperature. The crude solution was purified by SAX chromatography using SOURCE 30Q media (GE Healthcare) and a gradient of Buffer A (20 mM NaOH in water) and Buffer B (2 M NaCl, 20 mM NaOH in water). Product fractions were combined and desalted by ultrafiltration (Sartorius 5 kD Hydrosart membrane). The conjugation efficiency was >99%, and the overall yield of the process was estimated to be 58%. HPLC purity of the final product was 97.7%.
3.7. IP-HPLC-UV-MS Analysis
A sample of crude solution was weighed into a centrifuge, vacuum centrifuged to dryness at room temperature, and reconstituted in approximately 1 mL of water. Reconstituted solution was transferred to a 10 mL volumetric flask and brought to volume with water. Purified and isolated samples were prepared by dilution in water. Prepared samples were analyzed on an Agilent 1200 series HPLC (Santa Clara, CA, USA) coupled to an Agilent 6130 Quadrapole mass spectrometer using an XBridge C18 3.5 µm 2.1×150 mm column (Waters P/N 186003023, Milford, MA, USA) using the gradient shown in
Table 5. UV detection was set to 260 nm with a 4-nm bandwidth and a reference wavelength of 400 nm with an 80 nm bandwidth. Column temperature was set to 50.0 °C.

 and
and 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}