Monitoring Reaction Paths Using Vibrational Spectroscopies: The Case of the Dehydrogenation of Propane toward Propylene on Pd-Doped Cu(111) Surface


Abstract
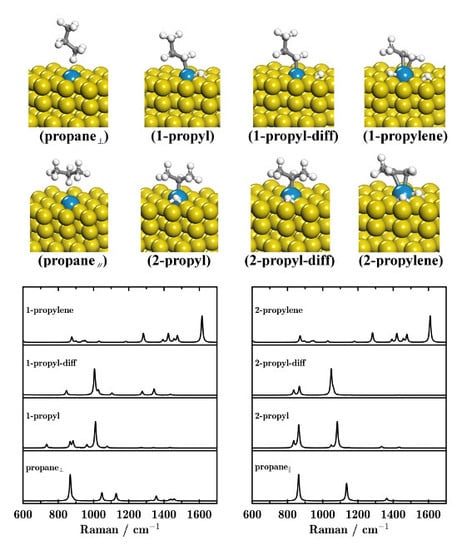
:
1. Introduction
2. Results
3. Computational Methods and Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, R.; Zhang, Y.; Dong, Z.; Jiang, S.; Zhang, C.; Chen, L.; Zhang, L.; Liao, Y.; Aizpurua, J.; Luo, Y.; et al. Chemical Mapping of a Single Molecule by Plasmon-enhanced Raman Scattering. Nature 2013, 498, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Aikens, C.M.; Madison, L.R.; Schatz, G.C. Raman spectroscopy: The effect of field gradient on SERS. Nat. Photonics 2013, 7, 508–510. [Google Scholar] [CrossRef]
- Cong, S.; Yuan, Y.; Chen, Z.; Hou, J.; Yang, M.; Su, Y.; Zhang, Y.; Li, L.; Li, Q.; Geng, F. Noble metal-comparable SERS enhancement from semiconducting metal oxides by making oxygen vacancies. Nat. Commun. 2015, 6, 7800. [Google Scholar] [CrossRef] [PubMed]
- Palonpon, A.F.; Ando, J.; Yamakoshi, H.; Dodo, K.; Sodeoka, M.; Kawata, S.; Fujita, K. Raman and SERS microscopy for molecular imaging of live cells. Nat. Protoc. 2013, 8, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, A.; Chavva, S.R.; Nellore, V.; Priya, B.; May, K.; Matthew, T.; Jones, S.; Vangara, A.; Ray, P.C. Development of a SERS Probe for Selective Detection of Healthy Prostate and Malignant Prostate Cancer Cells Using ZnII. Chem. Asian J. 2017, 12, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Chang, J.; Wang, J.; Zhang, P.; Cao, B.; Geng, Y.; Wang, X.; Pan, K. Fabrication and Formation Mechanism of Ag Nanoplate-Decorated Nanofiber Mats and Their Application in SERS. Chem. Asian J. 2016, 11, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhao, T.; Zhu, P.; Zhu, Y.; Liang, X.; Sun, R.; Wong, C.P. Tailoring Size and Coverage Density of Silver Nanoparticles on Monodispersed Polymer Spheres as Highly Sensitive SERS Substrates. Chem. Asian J. 2016, 11, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Philips, D.S.; Sreejith, S.; He, T.; Menon, N.V.; Anees, P.; Mathew, J.; Sajikumar, S.; Kang, Y.; Stuparu, M.C.; Sun, H.; et al. A Three-Photon Active Organic Fluorophore for Deep Tissue Ratiometric Imaging of Intracellular Divalent Zinc. Chem. Asian J. 2016, 11, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Auer, B.; Skinner, J. IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys. 2008, 128, 224511. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A.C. On the theory of Raman intensities. J. Chem. Phys. 1961, 34, 1476–1484. [Google Scholar] [CrossRef]
- Kurouski, D.; Zaleski, S.; Casadio, F.; Van Duyne, R.P.; Shah, N.C. Tip-enhanced Raman Spectroscopy (TERS) for in situ Identification of Indigo and Iron Gall Ink on Paper. J. Am. Chem. Soc. 2014, 136, 8677–8684. [Google Scholar] [CrossRef] [PubMed]
- Molesky, B.P.; Guo, Z.; Cheshire, T.P.; Moran, A.M. Perspective: Two-dimensional resonance Raman spectroscopy. J. Chem. Phys. 2016, 145, 180901. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.; Lermé, J.; Gehan, H.; Margueritat, J.; Mermet, A. Mechanisms of resonant low frequency Raman scattering from metallic nanoparticle Lamb modes. J. Chem. Phys. 2017, 146, 194201. [Google Scholar] [CrossRef] [PubMed]
- Karhánek, D.; Bučko, T.; Hafner, J. A Density Functional Study of the Adsorption of Methane-thiol on the (111) Surfaces of the Ni-group Metals: I. Molecular and Dissociative Adsorption. J. Phys. Condens. Matter 2010, 22, 265005. [Google Scholar] [CrossRef] [PubMed]
- Karhánek, D.; Bučko, T.; Hafner, J. A Density-functional Study of the Adsorption of Methane-thiol on the (111) Surfaces of the Ni-group Metals: II. Vibrational Spectroscopy. J. Phys. Condens. Matter 2010, 22, 265006. [Google Scholar] [CrossRef] [PubMed]
- Zayak, A.T.; Hu, Y.S.; Choo, H.; Bokor, J.; Cabrini, S.; Schuck, P.J.; Neaton, J.B. Chemical Raman Enhancement of Organic Adsorbates on Metal Surfaces. Phys. Rev. Lett. 2011, 106, 083003. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.B.; Tommasini, M.; Maestri, M. First-principles simulation of Raman Spectra of Adsorbates on Metal Surfaces. ChemPlusChem 2017, 82, 924–932. [Google Scholar] [CrossRef]
- Hu, W.; Duan, S.; Zhang, G.; Ma, Y.; Tian, G.; Luo, Y. Quasi-Analytical Approach for Modeling of Surface-Enhanced Raman Scattering. J. Phys. Chem. C 2015, 119, 28992–28998. [Google Scholar] [CrossRef]
- Hu, W.; Duan, S.; Luo, Y. Theoretical modeling of surface and tip-enhanced Raman spectroscopies. WIREs Comput. Mol. Sci. 2017, 7, e1293. [Google Scholar] [CrossRef]
- Hu, W.; Duan, S.; Zhang, Y.; Ren, H.; Jiang, J.; Luo, Y. Identifying the structure of 4-chlorophenyl isocyanide adsorbed on Au(111) and Pt(111) surfaces by first-principles simulations of Raman spectra. Phys. Chem. Chem. Phys. 2017, 19, 32389–32397. [Google Scholar]
- Sattler, J.J.H.B.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B.M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chem. Rev. 2014, 114, 10613–10653. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.L.; Zhu, Y.A.; Zhou, X.G.; Sui, Z.J.; Chen, D. First-Principles Calculations of Propane Dehydrogenation over PtSn Catalysts. ACS Catal. 2012, 2, 1247–1258. [Google Scholar]
- Lo, J.M.H.; Premji, Z.A.; Ziegler, T.; Clark, P.D. First-Principles Investigation of Selective Oxidation of Propane on Clean and Sulfided V2O5 (010) Surfaces. J. Phys. Chem. C 2013, 117, 11258–11274. [Google Scholar] [CrossRef]
- Yang, M.L.; Zhu, Y.A.; Fan, C.; Sui, Z.J.; Chen, D.; Zhou, X.G. DFT Study of Propane Dehydrogenation on Pt Catalyst: Effects of Step Sites. Phys. Chem. Chem. Phys. 2011, 13, 3257–3267. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Pellin, M.J.; Greeley, J.P.; Marshall, C.L.; Curtiss, L.A.; Ballentine, G.A.; Elam, J.W.; Catillon-Mucherie, S.; Redfern, P.C.; Mehmood, F.; et al. Subnanometre Platinum Clusters as Highly Active and Selective Catalysts for the Oxidative Dehydrogenation of Propane. Nat. Mater. 2009, 8, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, Z.P.; Li, Z.H.; Wang, W.N.; Fan, K.N. Periodic Density Functional Theory Study of Propane Oxidative Dehydrogenation over V2O5 (001) Surface. J. Am. Chem. Soc. 2006, 128, 11114–11123. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Insight into mechanism and selectivity of propane dehydrogenation over the Pd-doped Cu(111) surface. RSC Adv. 2016, 6, 65524–65532. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. J. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B. 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear Optical Properties in the Projector-augmented Wave Methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef]
- Sternheimer, R. Electronic Polarizabilities of Ions from the Hartree-Fock Wave Functions. Phys. Rev. 1954, 96, 951. [Google Scholar] [CrossRef]
- Kesharwani, M.K.; Brauer, B.; Martin, J.M. Frequency and zero-point vibrational energy scale factors for double-hybrid density functionals (and other selected methods): Can anharmonic force fields be avoided? J. Phys. Chem. A 2014, 119, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, J.; Reiher, M.; Kind, C.; Hess, B.A. Quantum Chemical Calculation of Vibrational Spectra of Large Molecules—Raman and IR Spectra for Buckminsterfullerene. J. Computat. Chem. 2002, 23, 895–910. [Google Scholar] [CrossRef] [PubMed]
- Long, D.A. The Raman effect : A Unified Treatment of the Theory of Raman Scattering by Molecules; Wiley Online Library: Hoboken, NJ, USA, 2002; Volume 1. [Google Scholar]
- Le Ru, E.; Etchegoin, P. Principles of Surface-Enhanced Raman Spectroscopy: And Related Plasmonic Effects; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surfaces | a | b | c | d | e |
|---|---|---|---|---|---|
| Path 1 (kcal/mol) | 37.9 | 32.8 | 31.7 | 29.9 | 28.2 |
| Path 2 (kcal/mol) | 36.7 | 31.6 | 29.6 | 28.6 | 26.3 |
| Mode | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Propane⊥ | 729 | 881 | 867 | 1010 | — | 1128 | 1324 |
| 1-Propyl | 745 | 894 | 888 | 1013 | 1089 | 1114 | 1359 |
| 1-Propyl-diff | 772 | 889 | 891 | 1006 | 1080 | 1088 | 1389 |
| 1-Propylene | — | 876 | 937 | — | 1088 | 1031 | 1400 |
| Propane|| | 710 | — | 863 | — | — | 1136 | 1445 |
| 2-Propyl | — | — | 871 | — | — | 1082 | 1443 |
| 2-Propyl-diff | — | — | 889 | — | — | 1062 | 1449 |
| 2-Propylene | — | 877 | 937 | — | 1087 | 1030 | 1444 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.; Cao, X. Monitoring Reaction Paths Using Vibrational Spectroscopies: The Case of the Dehydrogenation of Propane toward Propylene on Pd-Doped Cu(111) Surface. Molecules 2018, 23, 126. https://doi.org/10.3390/molecules23010126
Hu W, Cao X. Monitoring Reaction Paths Using Vibrational Spectroscopies: The Case of the Dehydrogenation of Propane toward Propylene on Pd-Doped Cu(111) Surface. Molecules. 2018; 23(1):126. https://doi.org/10.3390/molecules23010126
Chicago/Turabian StyleHu, Wei, and Xinrui Cao. 2018. "Monitoring Reaction Paths Using Vibrational Spectroscopies: The Case of the Dehydrogenation of Propane toward Propylene on Pd-Doped Cu(111) Surface" Molecules 23, no. 1: 126. https://doi.org/10.3390/molecules23010126
APA StyleHu, W., & Cao, X. (2018). Monitoring Reaction Paths Using Vibrational Spectroscopies: The Case of the Dehydrogenation of Propane toward Propylene on Pd-Doped Cu(111) Surface. Molecules, 23(1), 126. https://doi.org/10.3390/molecules23010126