Discovery of CNS-Like D3R-Selective Antagonists Using 3D Pharmacophore Guided Virtual Screening


Abstract
:1. Introduction
2. Materials and Methods
2.1. Definition and Whole Workflow of the Study
2.2. Data Collection and Molecular Docking
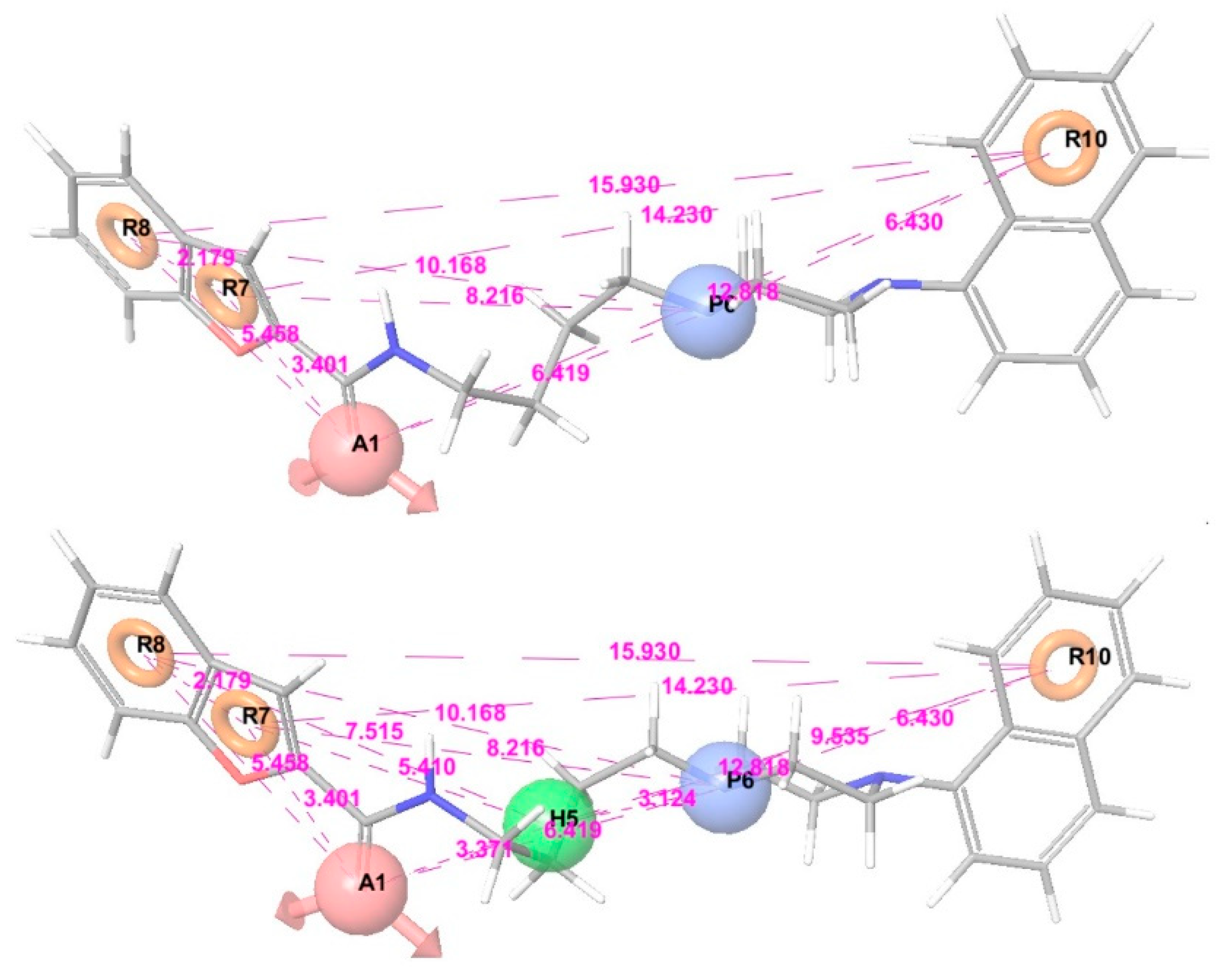
2.3. Identification of Pharmacophore Hypotheses
2.4. Internal and External QSAR Model Validation
2.5. Pharmacophore-Based Virtual Screening
2.6. Hit Selection
2.7. Cell-Based β-Arrestin Assay of D3R and D2R
3. Results and Discussion
3.1. Docking Analysis of D3R Selective Antagonists
3.2. Pharmacophore Modeling and 3D-QSAR Models
3.3. Identification of CNS-Like D3R Antagonist through Virtual Screening
4. The Current Limitation of This Study & Future Possibilities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Contreras, F.; Fouillioux, C.; Bolívar, A.; Simonovis, N.; Hernández-Hernández, R.; Armas-Hernandez, M.J.; Velasco, M. Dopamine, hypertension and obesity. J. Hum. Hypertens. 2002, 16, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Heidbreder, C.A.; Newman, A.H. Current perspectives on selective dopamine D3 receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N. Y. Acad. Sci. 2010, 1187, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Joyce, J.N. Distribution of Dopamine D3 Receptor Expressing Neurons in the Human Forebrain: Comparison with D2 Receptor Expressing Neurons. Neuropsychopharmacology 1999, 20, 60–80. [Google Scholar] [CrossRef]
- Maramai, S.; Gemma, S.; Brogi, S.; Campiani, G.; Butini, S.; Stark, H.; Brindisi, M. Dopamine D3 Receptor Antagonists as Potential Therapeutics for the Treatment of Neurological Diseases. Front. Neurosci. 2016, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.N. Dopamine D3 receptor as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharmacol. Ther. 2001, 90, 231–259. [Google Scholar] [CrossRef]
- Pilla, M.; Perachon, S.; Sautel, F.; Garrido, F.; Mann, A.; Wermuth, C.G.; Schwartz, J.C.; Everitt, B.J.; Sokoloff, P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Koob, G.F.; Caine, S.B. Cocaine addiction therapy—Are we partially there? Nat. Med. 1999, 5, 993–995. [Google Scholar] [CrossRef] [PubMed]
- Levant, B. The D3 Dopamine Receptor: Neurobiology and Potential Clinical Relevance. Pharmacol. Rev. 1997, 49, 231–252. [Google Scholar] [PubMed]
- Newman, A.H.; Grundt, P.; Nader, M.A. Dopamine D3 Receptor Partial Agonists and Antagonists as Potential Drug Abuse Therapeutic Agents. J. Med. Chem. 2005, 48, 3663–3679. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hou, T.; Li, Y. Selectivity and activation of dopamine D3R from molecular dynamics. J. Mol. Model. 2012, 18, 5051–5063. [Google Scholar] [CrossRef] [PubMed]
- Griffon, N.; Griffon, N.; Pilon, C.; Sautel, F.; Schwartz, J.C.; Sokoloff, P. Antipsychotics with inverse agonist activity at the dopamine D3 receptor. J. Neural Transm. 1996, 103, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marriott, D.P.; Dougall, I.G.; Meghani, P.; Liu, Y.J.; Flower, D.R. Lead Generation Using Pharmacophore Mapping and Three-Dimensional Database Searching: Application to Muscarinic M3 Receptor Antagonists. J. Med. Chem. 1999, 42, 3210–3216. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.C.; Christoffersen, R.E. Computer-Assisted Drug Design; ACS Publications: Washington, DC, USA, 1979; Chapter 10; pp. 227–241. ISBN 13: 9780841205215. [Google Scholar]
- Boateng, C.A.; Bakare, O.M.; Zhan, J.; Banala, A.K.; Burzynski, C.; Pommier, E.; Slusher, B.S. High Affinity Dopamine D3 Receptor (D3R)-Selective Antagonists Attenuate Heroin Self-Administration in Wild-Type but not D3R. Knockout Mice. J. Med. Chem. 2015, 58, 6195–6213. [Google Scholar] [CrossRef] [PubMed]
- Kurogi, Y.; Guner, O.F. Pharmacophore modeling and three-dimensional database searching for drug design using catalyst. Curr. Med. Chem. 2001, 8, 1035–1055. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Dharmendra, K.Y.; Venkatesan, R.; Afzal, S.; Lee, E.; Yoo, J.; Kim, S.Y.; Kim, M. Identification of novel acetylcholinesterase inhibitors designed by pharmacophore-based virtual screening, molecular docking and Bioassay. Sci. Rep. 2018, in press. [Google Scholar]
- Singh, K.D.; Karthikeyan, M.; Kirubakaran, P.; Nagamani, S. Pharmacophore filtering and 3D-QSAR in the discovery of new JAK2 inhibitors. J. Mol. Graph. Model. 2011, 30, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Chung, J.; Ryu, J.S.; Hah, J.M. Structure tuning of pyrazolylpyrrole derivatives as ERK kinase inhibitors utilizing double tools of 3D-QSAR and side-chain hopping. Bioorg. Med. Chem. Lett. 2011, 21, 4900–4904. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Ryu, J.S.; Hah, J.M. 3D-QSAR studies of 1,2-diaryl-1H-benzimidazole derivatives as JNK3 inhibitors with protective effects in neuronal cells. Bioorg. Med. Chem. Lett. 2013, 23, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Gadhe, C.G.; Lee, E.; Kim, M.H. Finding new scaffolds of JAK3 inhibitors in public database: 3D-QSAR models & shape-based screening. Arch. Pharm. Res. 2015, 38, 2008–2019. [Google Scholar] [PubMed]
- Taha, M.O.; Taha, M.O.; Tarairah, M.; Zalloum, H.; Abu-Sheikha, G. Pharmacophore and QSAR modeling of estrogen receptor β ligands and subsequent validation and in silico search for new hits. J. Mol. Graph. Model. 2010, 28, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Seidel, T.; Seidel, T.; Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov. Today 2011, 7, e221–e228. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lane, J.R.; Chubukov, P.; Liu, W.; Canals, M.; Cherezov, V.; Abagyan, R.; Stevens, R.C.; Katritch, V. Structure-Based Ligand Discovery Targeting Orthosteric and Allosteric Pockets of Dopamine Receptors. Mol. Pharmacol. 2013, 84, 794–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ježko, P.; Žufková, V.; Remko, M. Modelling of absorption, distribution and physicochemical properties of AT1 receptor antagonists/Modelovanie absorpcie, distribúcie a fyzikálnochemických vlastnosti antagonistov AT1 receptorov. Acta Facultatis Pharmaceuticae Universitatis Comenianae. 2015, 62, 20–31. [Google Scholar] [CrossRef]
- Luco, J.M. Prediction of the Brain-Blood Distribution of a Large Set of Drugs from Structurally Derived Descriptors Using Partial Least-Squares (PLS) Modeling. J. Chem. Inf. Comput. Sci. 1999, 39, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Kelder, J.; Grootenhuis, P.D.; Bayada, D.M.; Delbressine, L.P.; Ploemen, J.P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Ajay; Bemis, G.W.; Murkco, M.A. Designing Libraries with CNS Activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar]
- Shi, L.; Javitch, J.A. The binding site of aminergic G protein–coupled receptors: The transmembrane segments and second extracellular loop. Annu. Rev. pharmacol. 2002, 42, 437–467. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Andrieux, M.; Besançon, R.; Pilon, C.; Martres, M.P.; Giros, B.; Schwartz, J.C. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: Comparison with D2 receptor. Eur. J. Pharmacol. 1992, 225, 331–337. [Google Scholar] [CrossRef]
- Swets, J.A. Measuring the accuracy of diagnostic systems. Science 1988, 240, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Greiner, M.; Pfeiffer, D.; Smith, R.D. Principles and practical application of the receiver-operating characteristic analysis for diagnostic tests. Prev. Vet. Med. 2000, 45, 23–41. [Google Scholar] [CrossRef]
- Venkanna, A.; Kwon, O.W.; Afzal, S.; Jang, C.; Cho, K.H.; Yadav, D.; Kim, K.; Park, H.G.; Chun, K.H.; Kim, S.Y.; et al. Pharmacological use of a novel scaffold, anomeric N,N-diarylamino tetrahydropyran: Molecular similarity search, chemocentric target profiling, and experimental evidence. Sci. Rep. 2017, 7, 12535. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A.; Pennington, L.D. Structure−Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jang, C.; Yadav, D.K.; Kim, M. The Comparison of Automated Clustering Algorithms for Resampling Representative Conformer Ensembles with RMSD Matrix. J. Cheminform. 2017, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Year, E.R.; Cleves, A.E.; Jain, A.N. Chemical Structural Novelty: On-Targets and Off-Targets. J. Med. Chem. 2011, 54, 6771–6785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettles, J.H.; Jenkins, J.L.; Bender, A.; Deng, Z.; Davies, J.W.; Glick, M. Bridging chemical and biological space: “Target fishing” using 2D and 3D molecular descriptors. J. Med. Chem. 2006, 49, 6802–6810. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Sandeep Inakollu, V.S.; Sherman, W. Boosting virtual screening enrichments with data fusion: Coalescing hits from two-dimensional fingerprints, shape, and docking. J. Chem. Inf. Model. 2013, 53, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, V.; Kongsted, J. Virtual screening models for prediction of HIV-1 RT associated RNase H inhibition. PLoS ONE 2013, 8, E73478. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | pKi (D3/D2 Selectivity Ratio) |
|---|---|---|
| 6 |  | 2.73 (397) |
| 14 |  | 3.41 (135) |
| 15 |  | 3.04 (80) |
| 16 |  | 3.93 (109) |
| 17 |  | 3.76 (80) |
| 18 |  | 3.61 (61) |
| 19 |  | 3.89 (65) |
| 20 |  | 3.55 (131) |
| 21 |  | 2.74 (182) |
| 22 |  | 2.46 (19) |
| 23 |  | 1.01 (9.5) |
| 24 |  | 2.60 (27) |
| 25 |  | 1.27 (19) |
| 26 |  | 2.67 (16) |
| 27 |  | 1.35 (15) |
| 28 |  | 1.28 (13) |
| 29 |  | 1.16 (11) |
| 30 |  | 1.42 (27) |
| 31 |  | 1.42 (27) |
| 32 |  | 3.45 (45) |
| 33 |  | 2.44 (71) |
| 34 |  | 2.70 (22) |
| 35 |  | 1.50 (28) |
| 36 |  | 3.24 (69) |
| 37 |  | 2.09 (103) |
| ID | SD | R-squared | F | Stability | RMSE | Q-squared | Pearson-R |
|---|---|---|---|---|---|---|---|
| APRRR215 | 0.436 | 0.8014 | 133.1 | 0.5826 | 0.4819 | 0.7347 | 0.885 |
| HPRRR1639 | 0.38 | 0.8491 | 185.7 | 0.7004 | 0.5094 | 0.7036 | 0.8815 |
| HPRRR1660 | 0.3378 | 0.8807 | 243.7 | 0.6467 | 0.5307 | 0.6783 | 0.8809 |
| HPRRR133 | 0.506 | 0.7324 | 90.3 | 0.8561 | 0.5593 | 0.6426 | 0.8369 |
| ADPRR111 | 0.4345 | 0.8027 | 134.3 | 0.3582 | 0.5645 | 0.6361 | 0.8363 |
| AHPRRR.104 | 0.4177 | 0.8177 | 148 | 0.6505 | 0.4973 | 0.7175 | 0.8591 |
| ADHPRR.82 | 0.4748 | 0.7644 | 107.1 | 0.5688 | 0.5748 | 0.6226 | 0.8293 |
| DHRRR.673 | 0.3455 | 0.8753 | 231.6 | 0.555 | 0.581 | 0.6143 | 0.8167 |
| AHPRRR.551 | 0.4102 | 0.8241 | 154.6 | 0.4573 | 0.5861 | 0.6076 | 0.8147 |
| AHPRRR.554 | 0.4102 | 0.8241 | 154.6 | 0.4573 | 0.5861 | 0.6076 | 0.8147 |
| No | Structure | D3R a(%) | D2R a(%) | Ro5 b | Ro3 c | CNS d | LE e | G-score f | Pred. Actg g |
|---|---|---|---|---|---|---|---|---|---|
| 1 |  | 29.3 | 4 | 0 | 0 | 1 | −0.30 | −7.56 | 3.11, 3.11 |
| 2 |  | 30.8 | 20.6 | 0 | 0 | 1 | −0.31 | −8.02 | 2.68, 2.58 |
| 3 |  | 15.8 | 11.4 | 0 | 0 | 2 | −0.31 | −7.34 | 2.83, 2.87 |
| 4 |  | 13.5 | 17.6 | 0 | 0 | 1 | −0.30 | −7.57 | 2.82, 2.65 |
| 5 |  | 9.1 | −4.4 | 0 | 0 | 2 | −0.33 | −7.87 | 2.98, 2.86 |
| 6 |  | NT h | NT h | 0 | 0 | 1 | −0.32 | −8.32 | 2.54, 2.61 |
| 7 |  | −5.4 | −0.1 | 0 | 0 | 1 | −0.34 | −8.04 | 2.75, 2.84 |
| 8 |  | 11.9 | 3.8 | 0 | 0 | 1 | −0.32 | −8.29 | 2.31, 2.19 |
| 9 |  | 54.1 | 97.2 | 0 | 0 | 1 | −0.30 | −7.87 | 2.55, 2.53 |
| 10 |  | 14.8 | 15.7 | 0 | 0 | 1 | −0.34 | −7.08 | 2.40, 2.40 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Cho, S.J.; Kim, M.-h. Discovery of CNS-Like D3R-Selective Antagonists Using 3D Pharmacophore Guided Virtual Screening. Molecules 2018, 23, 2452. https://doi.org/10.3390/molecules23102452
Lee JH, Cho SJ, Kim M-h. Discovery of CNS-Like D3R-Selective Antagonists Using 3D Pharmacophore Guided Virtual Screening. Molecules. 2018; 23(10):2452. https://doi.org/10.3390/molecules23102452
Chicago/Turabian StyleLee, June Hyeong, Sung Jin Cho, and Mi-hyun Kim. 2018. "Discovery of CNS-Like D3R-Selective Antagonists Using 3D Pharmacophore Guided Virtual Screening" Molecules 23, no. 10: 2452. https://doi.org/10.3390/molecules23102452
APA StyleLee, J. H., Cho, S. J., & Kim, M. -h. (2018). Discovery of CNS-Like D3R-Selective Antagonists Using 3D Pharmacophore Guided Virtual Screening. Molecules, 23(10), 2452. https://doi.org/10.3390/molecules23102452