1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications

 ,
,  and
and
Abstract
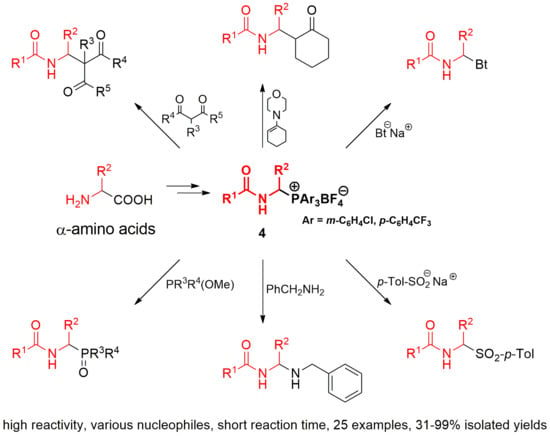
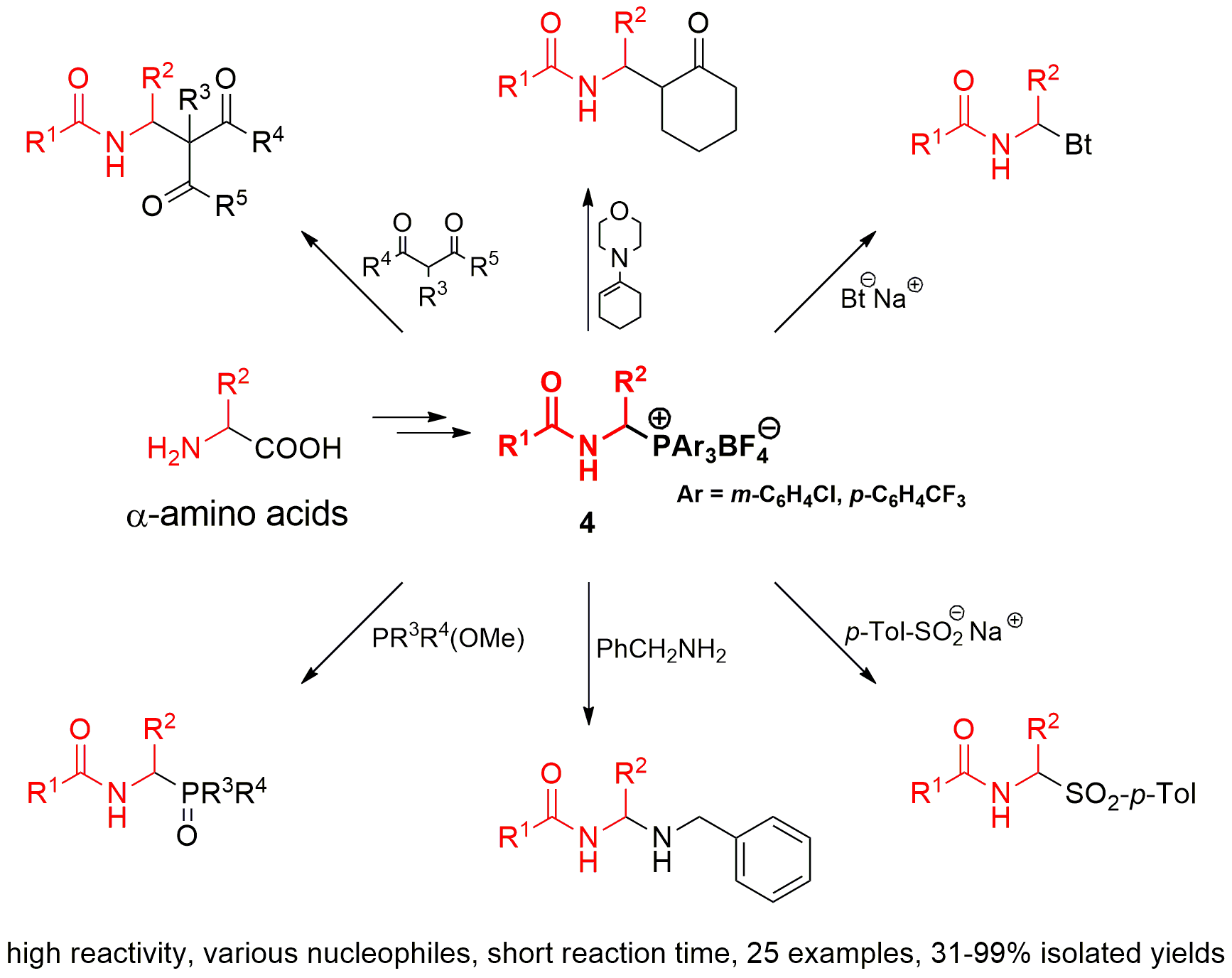
:
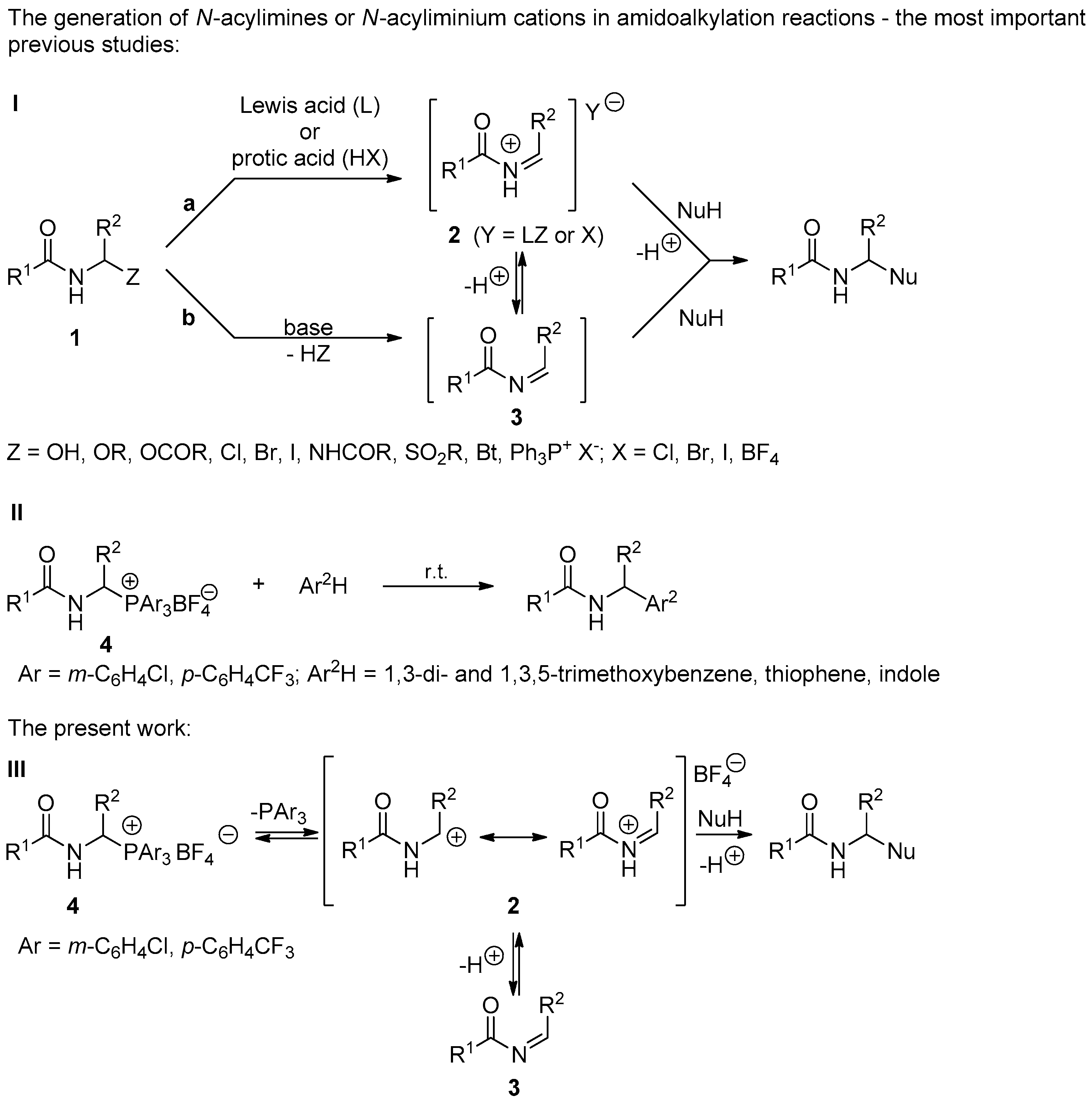
1. Introduction
2. Results and Discussion
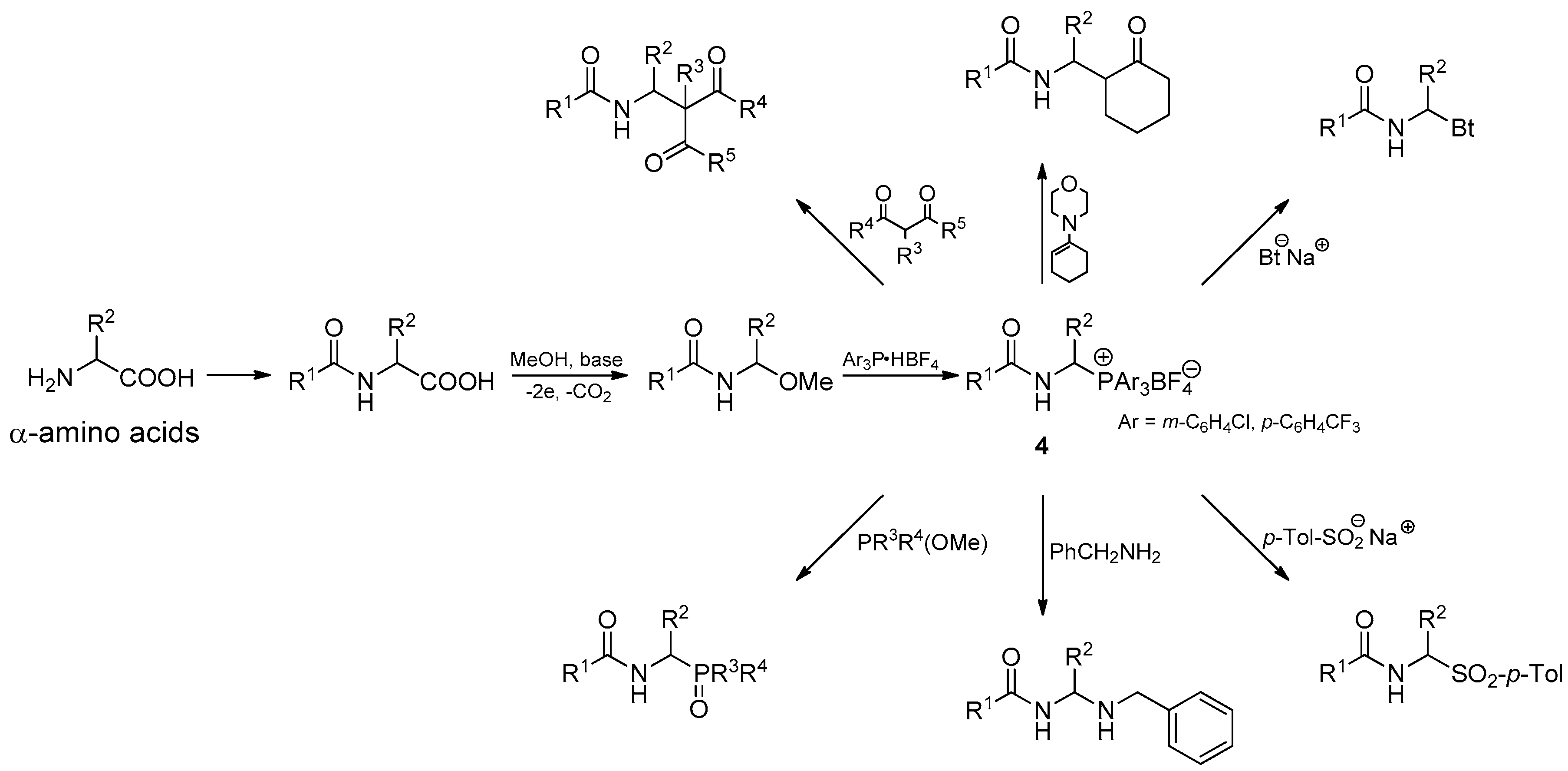
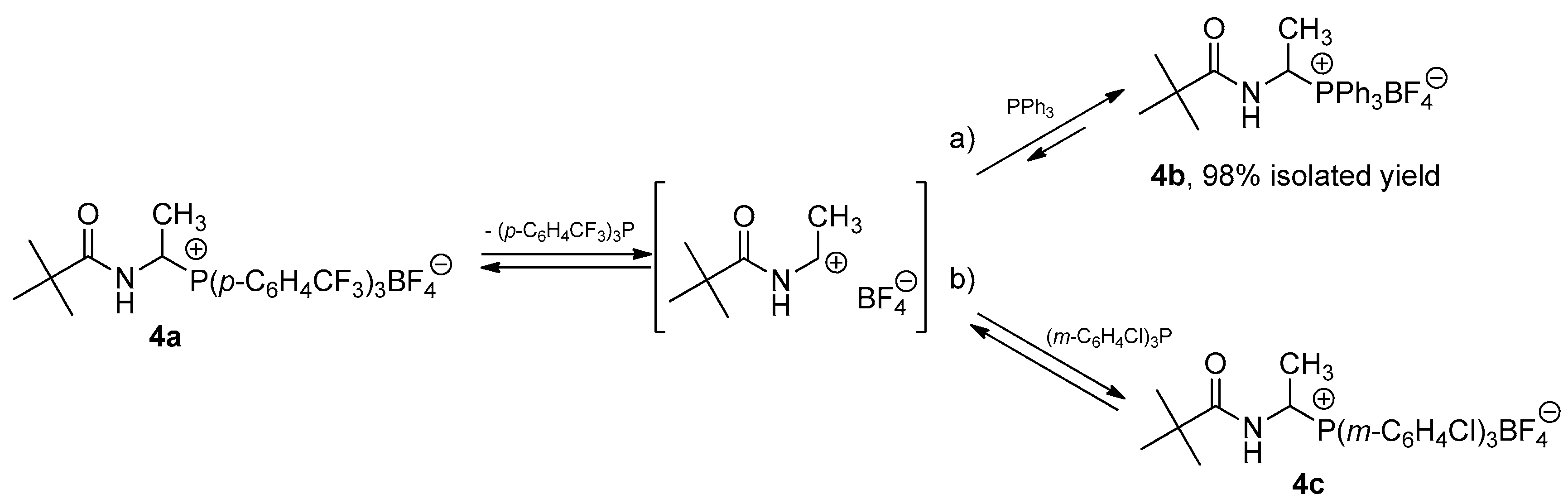
2.1. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength in the Selected Reaction of C-C Bond Formation
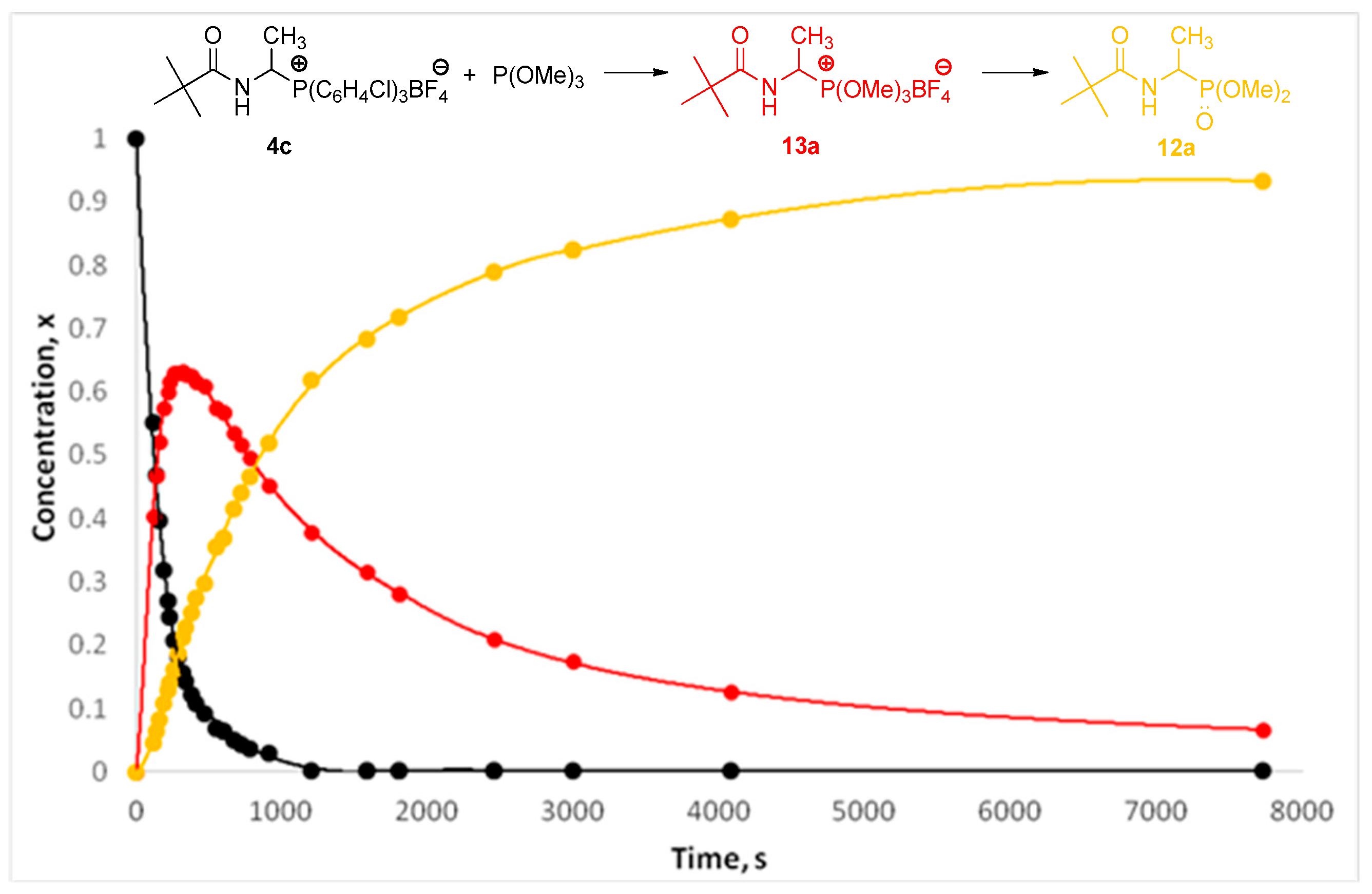
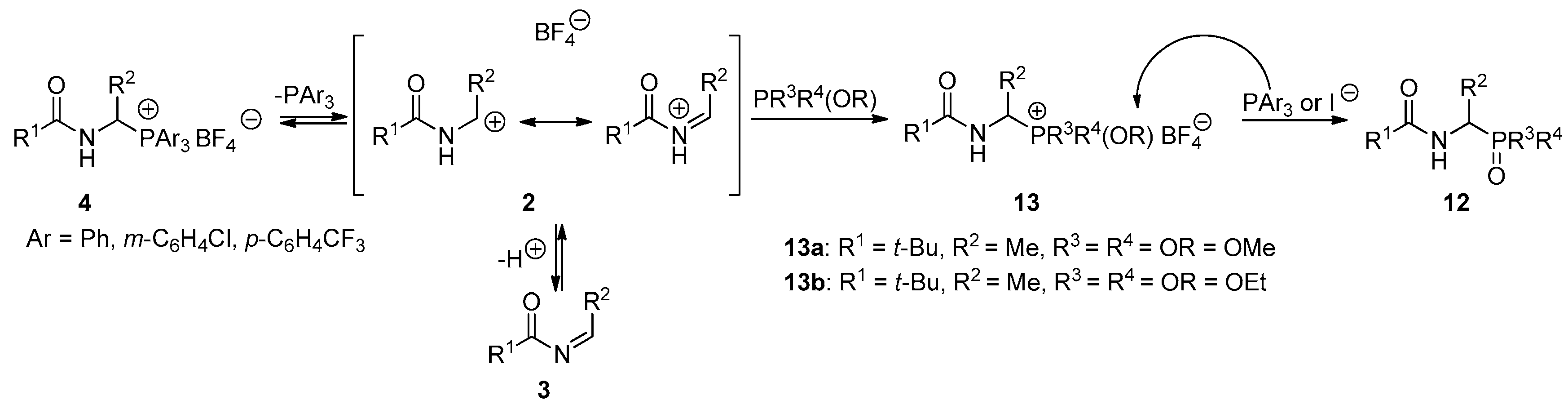
2.2. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength in α-Amidoalkylation of Selected Heteronucleophiles
3. Experimental Section
3.1. General Information
3.2. Syntheses
3.2.1. Substrate Synthesis
3.2.2. Reactivity of 1-(N-acylamino)alkyltriarylphosphonium Salts 4 toward Carbon Nucleophiles
3.2.3. Reactivity of 1-(N-Acylamino)alkyltriarylphosphonium Salts 4 toward Heteronucleophiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mazurkiewicz, R.; Październiok-Holewa, A.; Adamek, J.; Zielińska, K. α-Amidoalkylating agents: Structure, synthesis, reactivity and application. Adv. Heterocycl. Chem. 2014, 111, 43–94. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A.; Krawczyk, M.; Erfurt, K. Catalyst-free Friedel-Crafts reaction of 1-(N-acylamino)alkyltriarylphosphonium salts with electron-rich arenes. Tetrahedron 2018, 74, 2575–2583. [Google Scholar] [CrossRef]
- Adamek, J.; Mazurkiewicz, R.; Węgrzyk, A.; Erfurt, K. 1-Imidoalkylphosphonium salts with modulated Cα-P+ bond strength: Synthesis and application as new active α-imidoalkylating agents. Beilstein J. Org. Chem. 2017, 13, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, K.; Mazurkiewicz, R.; Szymańska, K.; Jarzębski, A.; Magiera, S.; Erfurt, K. Penicillin G acylase-mediated kinetic resolution of racemic 1-(N-acylamino)alkylphosphonic and 1-(N-acylamino)alkylphosphinic acids and their esters. J. Mol. Catal. B Enzym. 2016, 132, 31–40. [Google Scholar] [CrossRef]
- Adamek, J.; Mazurkiewicz, R.; Październiok-Holewa, A.; Grymel, M.; Kuźnik, A.; Zielińska, K. 1-(N-Acylamino)alkyl Sulfones from N-Acyl-α-amino Acids or N-Alkylamides. J. Org. Chem. 2014, 79, 2765–2770. [Google Scholar] [CrossRef] [PubMed]
- Adamek, J.; Mazurkiewicz, R.; Październiok-Holewa, A.; Kuźnik, A.; Grymel, M.; Zielińska, K.; Simka, W. N-[1-(Benzotriazol-1-yl)alkyl] amides from N-acyl-α-amino acids or N-alkylamides. Tetrahedron 2014, 70, 5725–5729. [Google Scholar] [CrossRef]
- Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Comparative Studies on the Amidoalkylating Properties of N-(1-Methoxyalkyl)Amides and 1-(N-Acylamino)Alkyltriphenylphosphonium Salts in the Michaelis–Arbuzov-Like Reaction: A New One-Pot Transformation of N-(1-Methoxyalkyl)Amides into Phosphonic or Phosphinic Analogs of N-Acyl-α-Amino Acids. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 967–980. [Google Scholar] [CrossRef]
- Październiok-Holewa, A.; Adamek, J.; Mazurkiewicz, R.; Zielińska, K. Amidoalkylating Properties of 1-(N-Acylamino)Alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 205–212. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Simka, W.; Gajos, A.; Szymura, K. α-Amidoalkylating Agents from N-Acyl-α-amino Acids: 1-(N-Acylamino)alkyltriphenylphosphonium Salts. J. Org. Chem. 2012, 77, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Październiok-Holewa, A.; Adamek, J.; Zielińska, K.; Piernikarczyk, K.; Mazurkiewicz, R. N-(1-acyloaminoalkyl)amidinium salts derived from DBU or related bases as reactive intermediates in α-amidoalkylation reactions. Arkivoc 2012, 314–329. [Google Scholar] [CrossRef]
- Zaugg, H.E. Recent Synthetic Methods Involving Intermolecular alpha-Amidoalkylation at Carbon. Synthesis 1970, 49–73. [Google Scholar] [CrossRef]
- Zaugh, H.E. α-Amidoalkylation at Carbon: Recent Advances–Part I. Synthesis 1984, 85–110. [Google Scholar] [CrossRef]
- Zaugg, H.E. α-Amidoalkylation at Carbon: Recent Advances–Part II. Synthesis 1984, 181–212. [Google Scholar] [CrossRef]
- Speckamp, W.N.; Hiemstra, H. Intramolecular reactions of N-acyliminium intermediates. Tetrahedron 1985, 41, 4367–4416. [Google Scholar] [CrossRef]
- Hiemstra, H.; Speckamp, W.N. N-Acyliminium Ions as Intermediates in Alkaloid Synthesis. Alkaloids 1988, 32, 271–339. [Google Scholar] [CrossRef]
- Hiemstra, H.; Speckamp, W.N. Comprehensive Organic Synthesis; Throst, B.M., Flemming, I., Eds.; Oxford: Pergamon, Turkey, 1991; Volume 2, pp. 1047–1082. [Google Scholar] [CrossRef]
- Kleinman, E.F. The Bimolecular Aliphatic Mannich and Related Reactions. In Comprehensive Organic Synthesis; Trost, B.M., Ed.; Pergamon Press: Oxford, UK, 1991; Volume 2, pp. 893–951. [Google Scholar]
- Katritzky, A.R.; Lan, X.; Yang, J.Z.; Denisko, O.V. Properties and Synthetic Utility of N-Substituted Benzotriazoles. Chem. Rev. 1998, 98, 409–548. [Google Scholar] [CrossRef] [PubMed]
- Speckamp, W.N.; Moolenaar, M.J. New developments in the chemistry of N-acyliminium ions and related intermediates. Tetrahedron 2000, 56, 3817–3856. [Google Scholar] [CrossRef]
- Mecozzi, T.; Petrini, M.; Profeta, R. Reactivity of chiral exocyclic N-acyliminium ions with aromatic derivatives. Tetrahedron Asymmetry 2003, 14, 1171–1178. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Zhang, H.C.; Cohen, J.H.; Turchi, I.J.; Maryanoff, C.A. Cyclizations of N-acyliminium ions. Chem. Rev. 2004, 104, 1431–1628. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Manju, K.; Singh, S.K.; Meher, N.K. Benzotriazole mediated amino-, amido-, alkoxy- and alkylthioalkylation. Tetrahedron 2005, 61, 2555–2581. [Google Scholar] [CrossRef]
- Petrini, M. α-Amido Sulfones as Stable Precursors of Reactive N-Acylimino Derivatives. Chem. Rev. 2005, 105, 3949–3977. [Google Scholar] [CrossRef] [PubMed]
- Ollevier, T.; Li, Z. The first catalytic Sakurai reaction of N-alkoxycarbonylamino sulfones with allyltrimethylsilane. Org. Biomol. Chem. 2006, 4, 4440–4443. [Google Scholar] [CrossRef] [PubMed]
- Ballini, R.; Palmieri, A.; Petrini, M.; Torregiani, E. Solventless Clay-Promoted Friedel−Crafts Reaction of Indoles with α-Amido Sulfones: Unexpected Synthesis of 3-(1-Arylsulfonylalkyl) Indoles. Org. Lett. 2006, 8, 4093–4096. [Google Scholar] [CrossRef] [PubMed]
- Marianacci, O.; Micheletti, G.; Bernardi, L.; Fini, F.; Fochi, M.; Pettersen, D.; Sgarzani, V.; Ricci, A. Organocatalytic Asymmetric Mannich Reactions with N-Boc and N-Cbz Protected α-Amido Sulfones. Chem. Eur. J. 2007, 13, 8338–8351. [Google Scholar] [CrossRef] [PubMed]
- Yazici, A.; Pyne, S.G. Intermolecular addition reactions of N-acyliminium ions (Part I). Synthesis 2009, 339–368. [Google Scholar] [CrossRef]
- Yazici, A.; Pyne, S.G. Intermolecular addition reactions of N-acyliminium ions (Part II). Synthesis 2009, 513–541. [Google Scholar] [CrossRef]
- Thirupathi, P.; Kim, S.S. InBr3: A Versatile Catalyst for the Different Types of Friedel−Crafts Reactions. J. Org. Chem. 2009, 74, 7755–7761. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Damodar, K.; Bhunia, N. A Simple and Efficient Access to α-Amino Phosphonates from N-Benzyloxycarbonylamino Sulfones Using Indium(III) Chloride. J. Org. Chem. 2009, 74, 5607–5609. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Estibalez, U.; Gómez-SanJuan, A.; García-Calvo, O.; Aranzamendi, E.; Lete, E.; Sotomayor, N. Strategies based on aryllithium and N-acyliminium ion cyclizations for the stereocontrolled synthesis of alkaloids and related systems. Eur. J. Org. Chem. 2011, 3610–3633. [Google Scholar] [CrossRef]
- Schneider, A.E.; Manolikakes, G. Bi(OTf)3-Catalyzed Multicomponent α-Amidoalkylation Reactions. J. Org. Chem. 2015, 80, 6193–6212. [Google Scholar] [CrossRef] [PubMed]
- Aranzamendi, E.; Arrasate, S.; Sotomayor, N.; González-Díaz, H.; Lete, E. Chiral Brønsted Acid Catalyzed Enantioselective α-Amidoalkylation Reactions: A Joint Experimental and Predictive Study. ChemistryOpen 2016, 5, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Touati, B.; El Bouakher, A.; Azizi, M.S.; Taillier, C.; Othman, R.B.; Trabelsi-Ayadi, M.; Antoniotti, S.; Dunach, E.; Dalla, V. Enolizable Carbonyls and N,O-Acetals: A Rational Approach for Room-Temperature Lewis Superacid-Catalyzed Direct α-Amidoalkylation of Ketones and Aldehydes. Chem. Eur. J. 2016, 22, 6012–6022. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, M.G.; Turova, O.V.; Zlotin, S.G. The progress in the chemistry of N-acyliminium ions and their use in stereoselective organic synthesis. Russ. Chem. Rev. 2017, 86, 1–17. [Google Scholar] [CrossRef]
- Aranzamendi, E.; Sotomayor, N.; González-Díaz, H.; Lete, E. Phenolic Activation in Chiral Brønsted Acid-Catalyzed Intramolecular α-Amidoalkylation Reactions for the Synthesis of Fused Isoquinolines. ACS Omega 2017, 2, 2706–2718. [Google Scholar] [CrossRef]
- Touati, B.; El Bouakher, A.; Azizi, M.S.; Taillier, C.; Othman, R.B.; Trabelsi-Ayadi, M.; Antoniotti, S.; Dunach, E.; Dalla, V. Atom-Economic Catalytic Direct Substitution of N,O-Acetals with Simple Ketones. Eur. J. Org. Chem. 2017, 4445–4460. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 6, 8, 10 and 12 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Phosphonium Salt 4 | 1,3-Dicarbonyl Compound, 5 | Molar Ratio of 4:5:LDA | 6 | Yield, % | ||
|---|---|---|---|---|---|---|---|
| R1 | R2 | Ar | |||||
| 1 | t-Bu | Me | m-C6H4Cl | diethyl malonate | 1:2:1 | 6a | 40 |
| 2 | t-Bu | Me | m-C6H4Cl | diethyl malonate | 1:8:1 | 6a | 65 |
| 3 | t-Bu | Me | p-C6H4CF3 | diethyl malonate | 1:8:1 | 6a | 67 |
| 4 | t-Bu | Me | m-C6H4Cl | ethyl acetoacetate | 1:8:1 | 6b | 62 a |
| 5 | Bn | i-Bu | m-C6H4Cl | ethyl acetoacetate | 1:8:1 | 6c | 83 b |
| 6 | Bn | i-Bu | m-C6H4Cl | dimethyl malonate | 1:8:1 | 6d | 63 |
| 7 | Bn | i-Bu | m-C6H4Cl | diethyl malonate | 1:8:1 | 6e | 52 |
| 8 | BnO | Bn | m-C6H4Cl | ethyl acetoacetate | 1:8:1 | 6f | 30 c |

| Entry | Phosphonium Salt 4 | 8 | Yield, % | ||
|---|---|---|---|---|---|
| R1 | R2 | Ar | |||
| 1 | t-Bu | Me | m-C6H4Cl | 8a | 63 a |
| 2 | Bn | i-Bu | m-C6H4Cl | 8b | 62 b |
| 3 | BnO | i-Bu | m-C6H4Cl | 8c | 40 c |
| 4 | BnO | Bn | m-C6H4Cl | 8d | 33 d |
| 5 | BnO | CH2Ot-Bu | p-C6H4CF3 | 8e | 31 c |

| Entry | Phosphonium Salt 4 | Nu−Na+/NuH | Temp., °C | Time, Min. | Molar Ratio of 4:9 | 10 | Yield, % | ||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Ar | |||||||
| 1 | t-Bu | Me | p-C6H4CF3 | Bt−Na+ | 20 | 15 | 1:1 | 10a | 99 |
| 2 | BnO | Bn | m-C6H4Cl | Bt−Na+ | 20 | 15 | 1:1 | 10b | 70 |
| 3 | t-Bu | Me | p-C6H4CF3 | TolSO2−Na+ | 20 | 15 | 1:1 | 10c | 88 |
| 4 | t-Bu | Me | m-C6H4Cl | Bn-NH2 | 20 | 5 | 1:4 | 10d | 91 |
| 5 | Bn | i-Bu | p-C6H4CF3 | Bn-NH2 | 20 | 5 | 1:4 | 10e | 55 |

| Entry | Phosphonium Salt 4 | P-Nu 11 | Temp., °C | Time, h | Molar Ratio of 4:11 | 12 | Yield, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Ar | R | R3 | R4 | ||||||
| 1 | t-Bu | Me | m-C6H4Cl | Me | OMe | OMe | 20 | 3 | 1:1.5 | 12a | 47 |
| 2 | t-Bu | Me | m-C6H4Cl | Me | OMe | OMe | 20 | 3 | 1:1.5 a | 12a | 85 |
| 3 | Bn | i-Bu | m-C6H4Cl | Me | OMe | OMe | 20 | 3 | 1:1.5 a | 12b | 77 |
| 4 | BnO | CH2Ot-Bu | p-C6H4CF3 | Me | OMe | OMe | 20 | 3 | 1:1.5 a | 12c | 61 |
| 5 | t-Bu | Me | m-C6H4Cl | Me | Ph | OMe | 60 | 2 | 1:1.5 a | 12d | 69 |
| 6 | BnO | i-Bu | p-C6H4CF3 | Me | Ph | Ph | 20 | 3 | 1:1.5 a | 12e | 83 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamek, J.; Węgrzyk, A.; Kończewicz, J.; Walczak, K.; Erfurt, K. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules 2018, 23, 2453. https://doi.org/10.3390/molecules23102453
Adamek J, Węgrzyk A, Kończewicz J, Walczak K, Erfurt K. 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules. 2018; 23(10):2453. https://doi.org/10.3390/molecules23102453
Chicago/Turabian StyleAdamek, Jakub, Anna Węgrzyk, Justyna Kończewicz, Krzysztof Walczak, and Karol Erfurt. 2018. "1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications" Molecules 23, no. 10: 2453. https://doi.org/10.3390/molecules23102453
APA StyleAdamek, J., Węgrzyk, A., Kończewicz, J., Walczak, K., & Erfurt, K. (2018). 1-(N-Acylamino)alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules, 23(10), 2453. https://doi.org/10.3390/molecules23102453