
Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
4. Conclusions
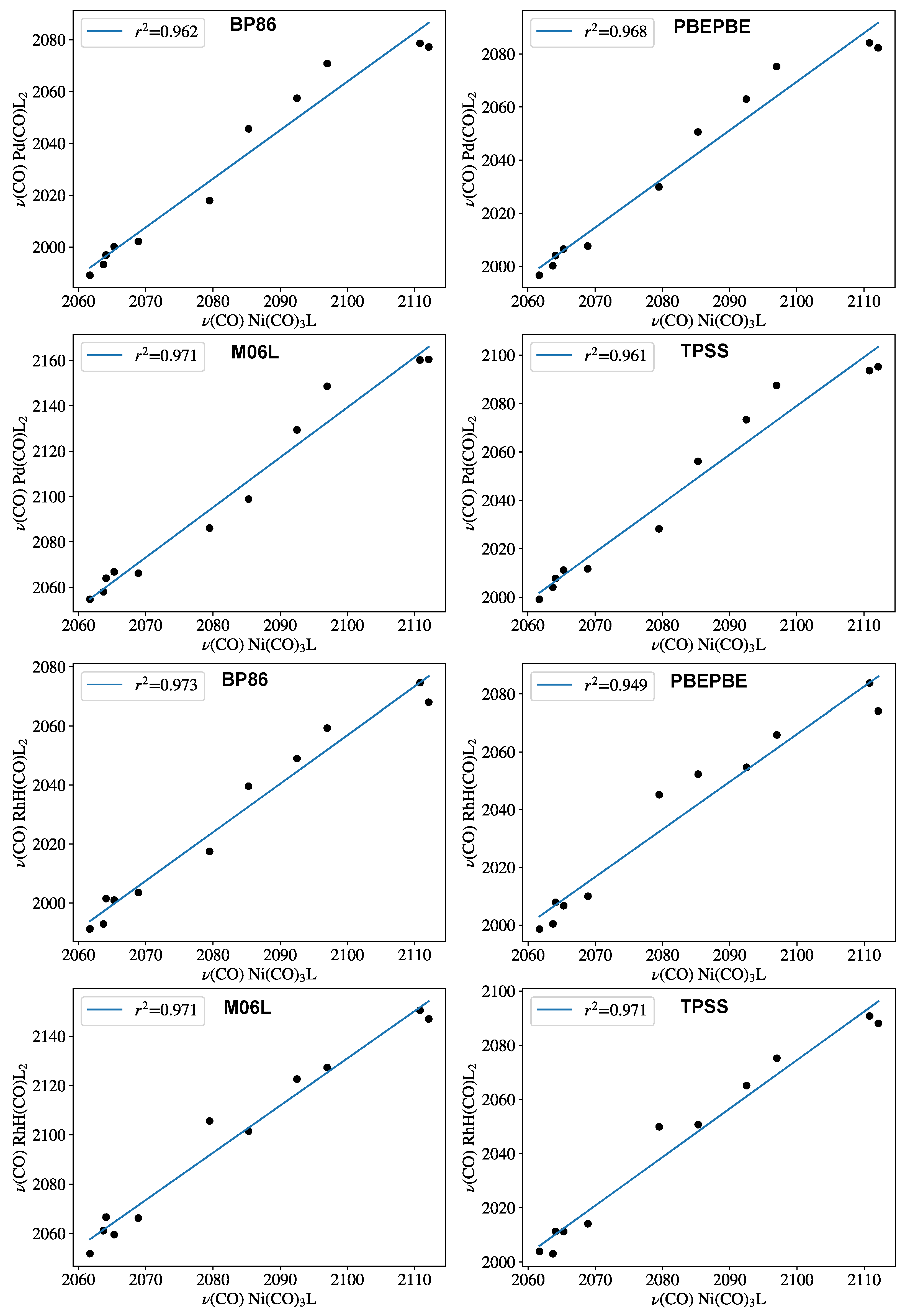
- For both model systems, no major difference in accuracy can be expected for various DFT methods in terms of the linear regression coefficient.
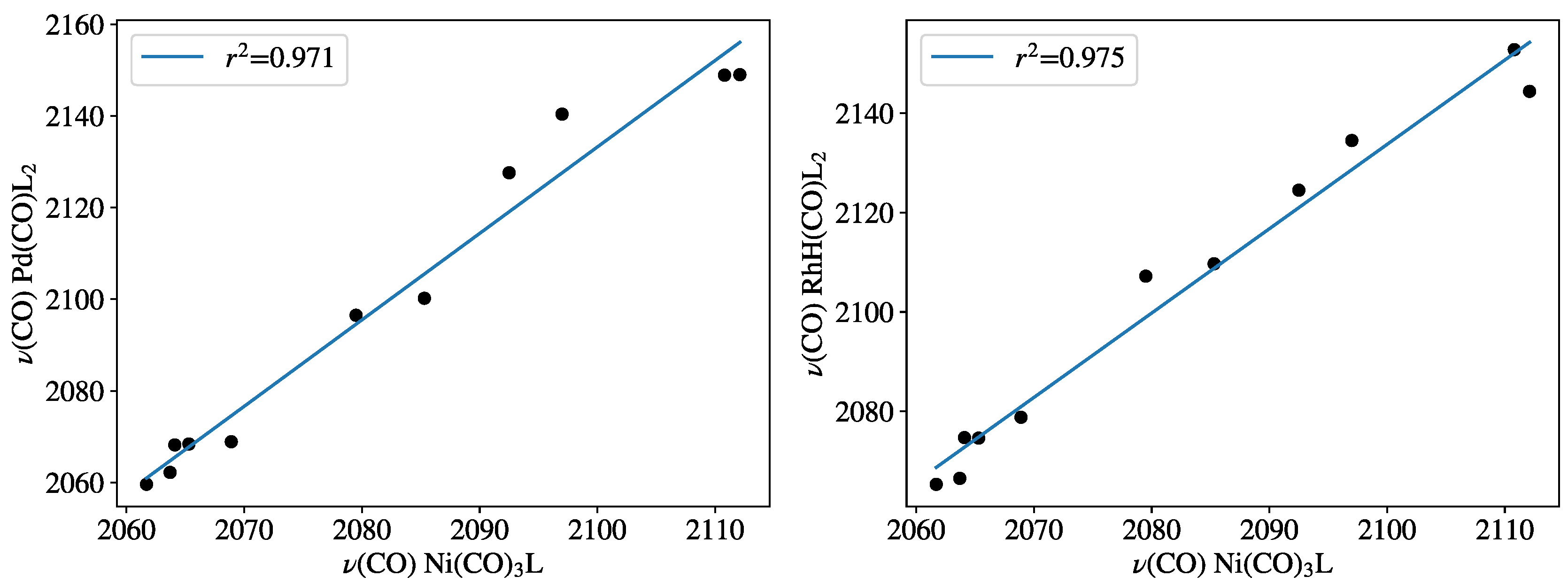
- For monophosphines, both Pd(0)L2(CO) and HRh(I)L2(CO) complexes showed high linearity with the experimental TEP scale.
- For monophosphines, the population of the disynaptic ELF basin V(C,O), associated with the carbonyl ligand, showed good linearity with the experimental TEP scale.
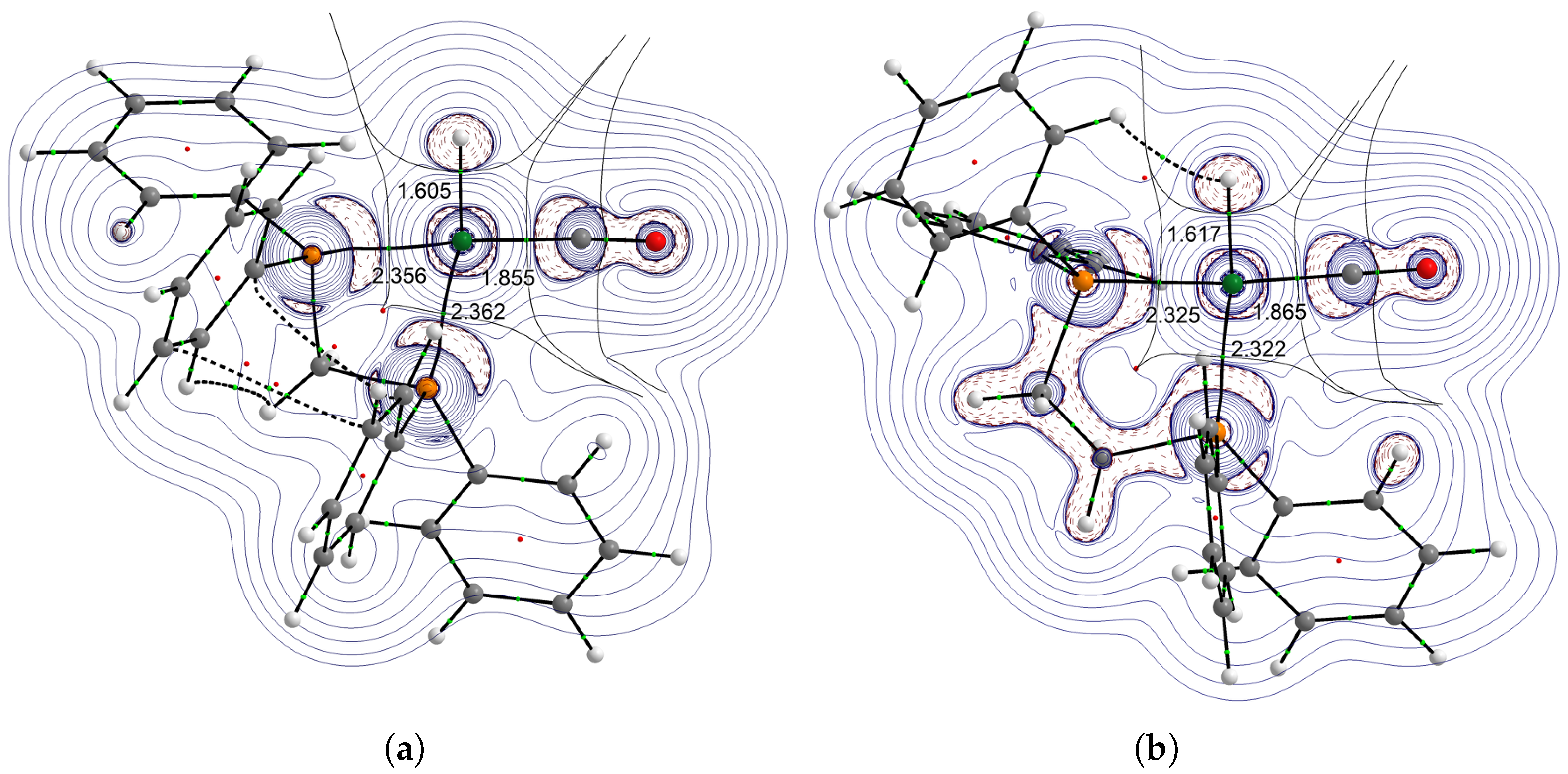
- For diphosphine-containing systems, the bite angle effect showed the opposite trend, with PdL2(CO) complexes revealing a decrease in (CO) by the increase of bite angle.
- In the case of the Pd complexes, the change of substituents on phosphorus caused a change in (CO) being consistent with the donor character of the substituent.
- For diphosphines possessing axial chirality, the nature of the condensed ring does not alter the electronic parameter of the ligand.
- The effect estimated in the HRhL2(CO) complexes is very sensitive to the H–Rh–P angle.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McAuliffe, C.A. Transition Metal Complexes of Phosphorus, Arsenic and Antimony Ligands; Halsted Press: New York, NY, USA, 1973. [Google Scholar]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Hartwig, J.F.; Collman, J.P. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- Brown, T.L.; Lee, K.J. Ligand steric properties. Coord. Chem. Rev. 1993, 128, 89–116. [Google Scholar] [CrossRef]
- Dias, P.B.; de Piedade, M.E.M.; Simões, J.A.M. Bonding and energetics of phosphorus (III) ligands in transition metal complexes. Coord. Chem. Rev. 1994, 135, 737–807. [Google Scholar] [CrossRef]
- Kamer, P.C.; van Leeuwen, P.W.; Reek, J.N. Wide bite angle diphosphines: Xantphos ligands in transition metal complexes and catalysis. Acc. Chem. Res. 2001, 34, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Kühl, O. Predicting the net donating ability of phosphines do we need sophisticated theoretical methods? Coord. Chem. Rev. 2005, 249, 693–704. [Google Scholar] [CrossRef]
- de Vries, J.G.; Lefort, L. The combinatorial approach to asymmetric hydrogenation: Phosphoramidite libraries, ruthenacycles, and artificial enzymes. Chem. A Eur. J. 2006, 12, 4722–4734. [Google Scholar] [CrossRef] [PubMed]
- Botteghi, C.; Paganelli, S.; Schionato, A.; Marchetti, M. The asymmetric hydroformylation in the synthesis of pharmaceuticals. Chirality 1991, 3, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Kégl, T.; Kollár, L. Chiral Phosphorous Ligands in Asymmetric Catalysis. In Comprehensive Inorganic Chemistry II (Second Edition): From Elements to Applications; Elsevier Ltd.: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Strohmeier, W.; Müller, F.J. Klassifizierung phosphorhaltiger Liganden in Metallcarbonyl-Derivaten nach der π-Acceptorstärke. Chem. Ber. 1967, 100, 2812–2821. [Google Scholar] [CrossRef]
- Tolman, C.A. Electron donor-acceptor properties of phosphorus ligands. Substituent additivity. J. Am. Chem. Soc. 1970, 92, 2953–2956. [Google Scholar] [CrossRef]
- Strohmeier, W.; Müller, F.J. π-Acceptorstärke von Phosphinen als Liganden in Cyclopentadienylmangantricarbonyl und Nickelcarbonyl. Z. Naturforschg. 1967, 22b, 451–452. [Google Scholar] [CrossRef]
- Anton, D.R.; Crabtree, R.H. Metalation-resistant ligands: Some properties of dibenzocyclooctatetraene complexes of molybdenum, rhodium and iridium. Organometallics 1983, 2, 621–627. [Google Scholar] [CrossRef]
- Roodt, A.; Otto, S.; Steyl, G. Structure and solution behaviour of rhodium(I) Vaska-type complexes for correlation of steric and electronic properties of tertiary phosphine ligands. Coord. Chem. Rev. 2003, 245, 121–137. [Google Scholar] [CrossRef]
- Otto, S.; Roodt, A. Quantifying the electronic cis effect of phosphine, arsine and stibine ligands by use of rhodium(I) Vaska-type complexes. Inorg. Chim. Acta 2004, 357, 1–10. [Google Scholar] [CrossRef]
- Suresh, C.H.; Koga, N. Quantifying the Electronic Effect of Substituted Phosphine Ligands via Molecular Electrostatic Potential. Inorg. Chem. 2002, 41, 1573–1578. [Google Scholar] [CrossRef]
- Liu, H.Y.; Eriks, K.; Prock, A.; Giering, W.P. Quantitative analysis of ligand effects (QALE). Systematic study of iron-phosphorus bond lengths and their relationship to steric thresholds. Organometallics 1990, 9, 1758–1766. [Google Scholar] [CrossRef]
- Bartholomew, J.; Fernandez, A.L.; Lorsbach, B.A.; Wilson, M.R.; Prock, A.; Giering, W.P. Comments on Coupling Graphical and Regression Analyses of Ligand Effect Data. Organometallics 1996, 15, 295–301. [Google Scholar] [CrossRef]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2012, 46, 280–288. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intramolecular pnicogen interactions in phosphorus and arsenic analogues of proton sponges. Phys. Chem. Chem. Phys. 2014, 16, 15900–15909. [Google Scholar] [CrossRef]
- Molloy, A.D.; Sánchez-Sanz, G.; Gilheany, D.G. PP-Rotation, P-Inversion and Metathesis in Diphosphines Studied by DFT Calculations: Comments on Some Literature Conflicts. Inorganics 2016, 4, 36. [Google Scholar] [CrossRef]
- Gusev, D.G. Donor properties of a series of two-electron ligands. Organometallics 2009, 28, 763–770. [Google Scholar] [CrossRef]
- Mitoraj, M.; Michalak, A. Donor-Acceptor Properties of Ligands from the Natural Orbitals for Chemical Valence. Organometallics 2007, 26, 6576–6580. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A. σ-donor and π-acceptor properties of phosphorus ligands: An insight from the natural orbitals for chemical valence. Inorg. Chem. 2010, 49, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Kégl, T.R.; Kollár, L.; Kégl, T. Relationship of QTAIM and NOCV Descriptors with Tolman’s Electronic Parameter. Adv. Chem. 2016, 2016, 4109758. [Google Scholar] [CrossRef]
- Couzijn, E.P.; Lai, Y.Y.; Limacher, A.; Chen, P. Intuitive Quantifiers of Charge Flows in Coordinate Bonding. Organometallics 2017, 36, 3205–3214. [Google Scholar] [CrossRef]
- Perrin, L.; Clot, E.; Eisenstein, O.; Loch, J.; Crabtree, R.H. Computed ligand electronic parameters from quantum chemistry and their relation to Tolman parameters, lever parameters, and Hammett constants. Inorg. Chem. 2001, 40, 5806–5811. [Google Scholar] [CrossRef] [PubMed]
- Valyaev, D.A.; Brousses, R.; Lugan, N.; Fernández, I.; Sierra, M.A. Do ν(CO) Stretching Frequencies in Metal Carbonyl Complexes Unequivocally Correlate with the Intrinsic Electron-Donicity of Ancillary Ligands? Chem. A Eur. J. 2011, 17, 6602–6605. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Scafuri, N.; Bistoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D.; Belpassi, L. When the Tolman Electronic Parameter Fails: A Comparative DFT and Charge Displacement Study of [(L)Ni(CO)3]0/− and [(L)Au(CO)]0/+. Inorg. Chem. 2014, 53, 9907–9916. [Google Scholar] [CrossRef]
- Frank, N. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms Moleculs—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll (Version 15.05.18); TK Gristmill Software: Overland Park, KS, USA, 2015. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Stephens, M.E. Spatial localization of the electronic pair and number distributions in molecules. J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- René, T. Stabilité Structurelle et Morphogénese: Essai D’une Théorie Générale des Modeles; WA Benjamin: New York, NY, USA, 1972. [Google Scholar]
- Putz, M.V. Markovian approach of the electron localization functions. Int. J. Quantum Chem. 2005, 105, 1–11. [Google Scholar] [CrossRef]
- Putz, M. Density functionals of chemical bonding. Int. J. Mol. Sci. 2008, 9, 1050–1095. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
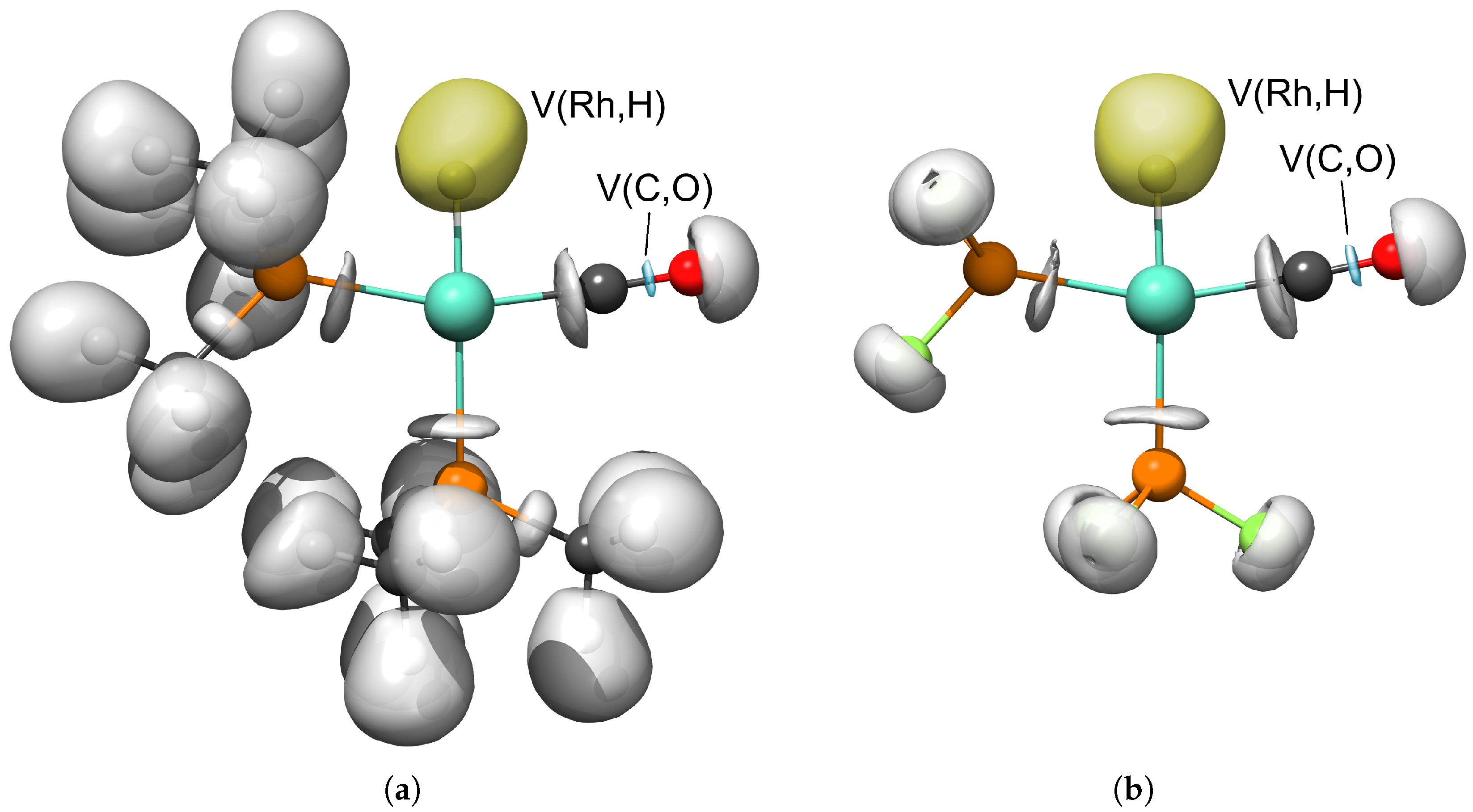{kind=link}
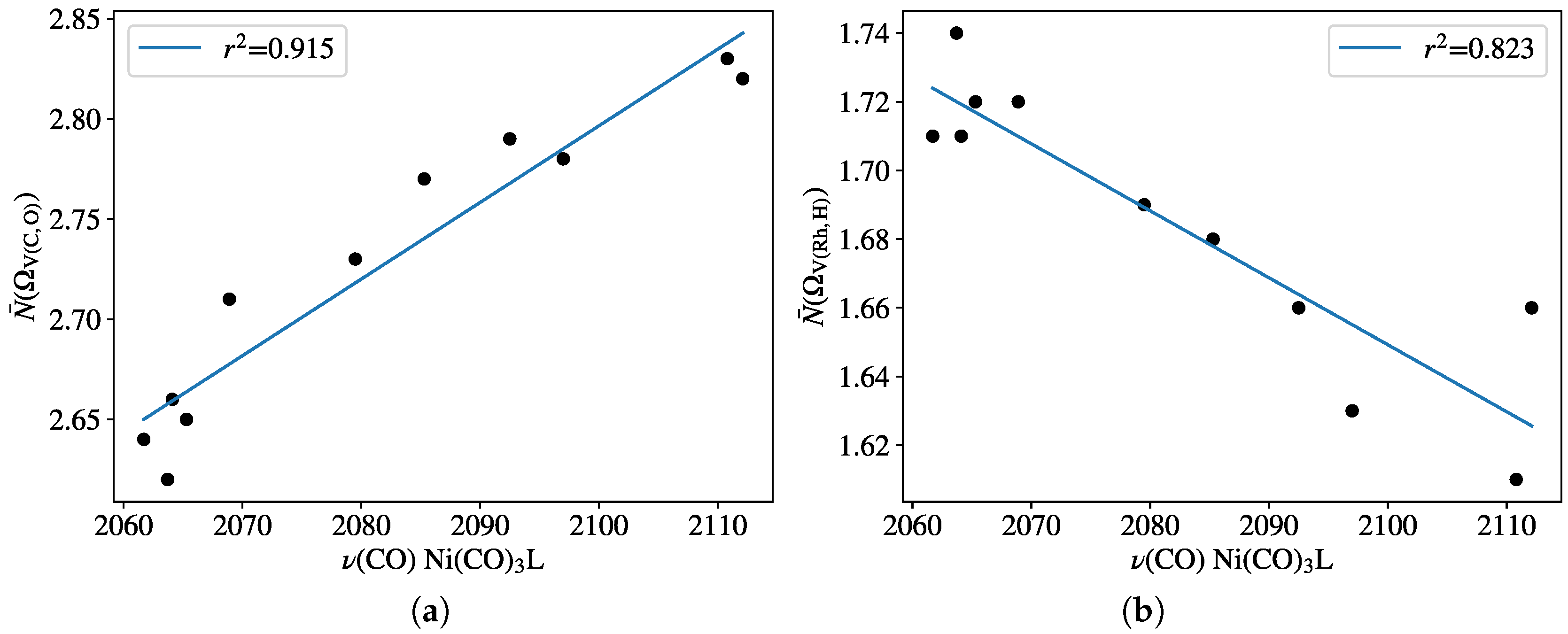{kind=link}
| Ligand | (CO) | (CO) | (CO) |
|---|---|---|---|
| PEt3 (1) | 2061.7 | 2059.6 | 2065.3 |
| PEt2Ph (2) | 2063.7 | 2062.2 | 2066.5 |
| PMe3 (3) | 2064.1 | 2068.2 | 2074.7 |
| PMe2Ph (4) | 2065.3 | 2068.4 | 2074.6 |
| PPh3 (5) | 2068.9 | 2068.9 | 2078.8 |
| P(OMe)3 (6) | 2079.5 | 2096.5 | 2107.2 |
| P(OPh)3 (7) | 2085.3 | 2100.2 | 2109.7 |
| PCl2(OEt) (8) | 2092.5 | 2127.6 | 2124.5 |
| PCl3 (9) | 2097.0 | 2140.4 | 2134.5 |
| PF3 (10) | 2110.8 | 2148.9 | 2152.8 |
| P(CF3)F2 (11) | 2112.1 | 2149.0 | 2144.4 |
| Ligand | (CO) | CEP | θPPdP | (CO) | CEP | θPRhP |
|---|---|---|---|---|---|---|
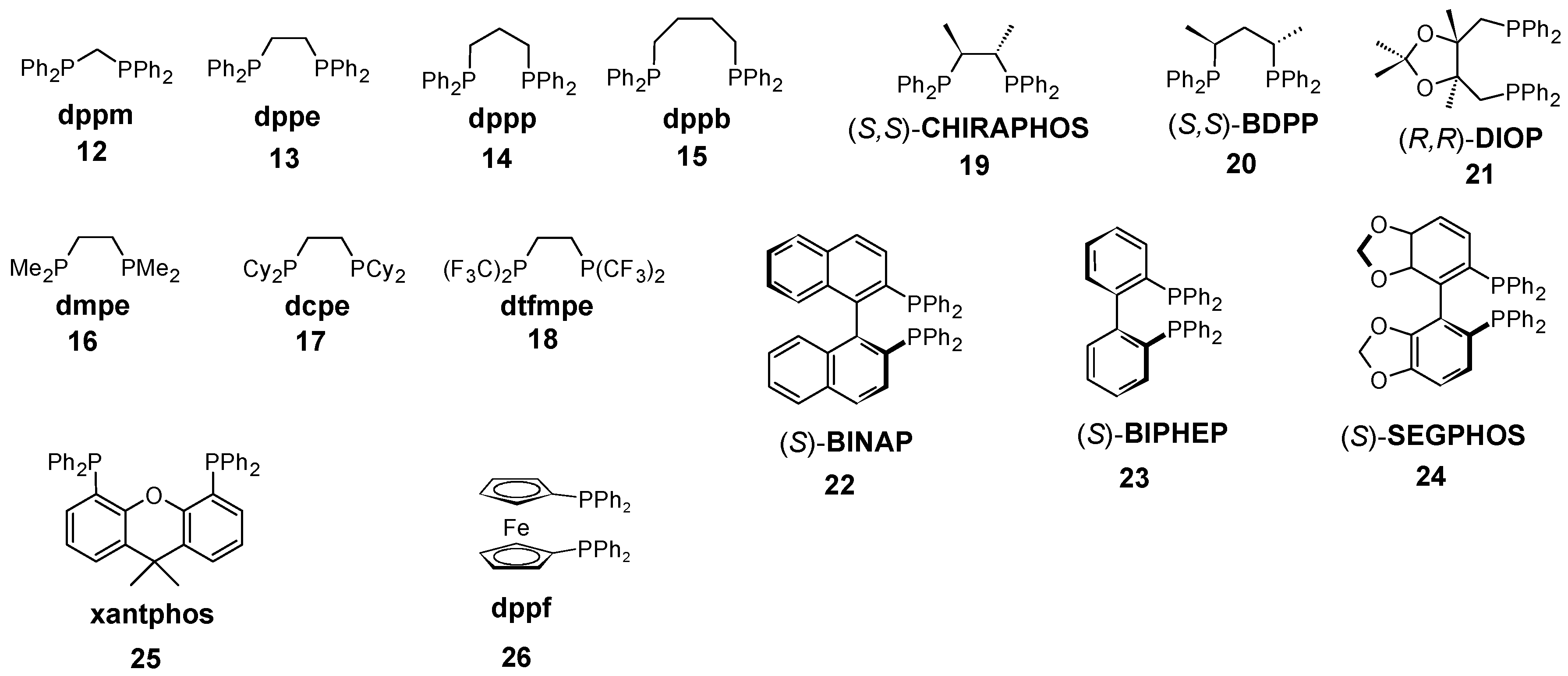
| dppm (12) | 2080.8 | 2072.6 | 69.8 | 2069.8 | 2062.9 | 71.0 |
| dppe (13) | 2076.1 | 2070.1 | 85.1 | 2071.9 | 2064.1 | 84.7 |
| dppp (14) | 2068.4 | 2066.2 | 90.5 | 2074.6 | 2065.7 | 88.7 |
| dppb (15) | 2064.5 | 2064.2 | 96.2 | 2079.0 | 2068.2 | 94.6 |
| dmpe (16) | 2073.5 | 2068.8 | 86.0 | 2075.1 | 2065.9 | 85.5 |
| dcpe (17) | 2058.6 | 2061.1 | 86.2 | 2060.6 | 2057.6 | 85.3 |
| dtfmpe (18) | 2123.1 | 2094.3 | 86.3 | 2125.8 | 2095.1 | 84.9 |
| CHIRAPHOS (19) | 2074.3 | 2069.2 | 85.1 | 2076.3 | 2066.6 | 84.6 |
| BDPP (20) | 2067.7 | 2065.8 | 89.0 | 2071.2 | 2063.7 | 87.7 |
| DIOP (21) | 2069.6 | 2066.8 | 101.5 | 2077.4 | 2067.3 | 95.5 |
| BINAP (22) | 2069.0 | 2066.5 | 93.7 | 2075.9 | 2066.4 | 92.8 |
| BIPHEP (23) | 2069.6 | 2066.8 | 96.4 | 2076.8 | 2066.9 | 93.8 |
| SEGPHOS (24) | 2067.5 | 2065.7 | 94.3 | 2073.9 | 2065.2 | 92.8 |
| xantphos (25) | 2065.0 | 2064.4 | 105.9 | 2070.1 | 2063.1 | 102.8 |
| dppf (26) | 2069.3 | 2066.6 | 103.6 | 2078.6 | 2067.9 | 97.8 |
| Ligand | (RhH) | r | (Rh,H) | (RhH) | |
|---|---|---|---|---|---|
| PEt3 (1) | 1920.8 | 1.618 | 100.4 | 0.858 | 0.131 |
| PEt2Ph (2) | 1931.6 | 1.615 | 95.6 | 0.873 | 0.132 |
| PMe3 (3) | 1927.3 | 1.618 | 100.9 | 0.860 | 0.131 |
| PMe2Ph (4) | 1948.3 | 1.611 | 98.4 | 0.876 | 0.133 |
| PPh3 (5) | 1964.7 | 1.606 | 102.6 | 0.849 | 0.135 |
| P(OMe)3 (6) | 1949.0 | 1.618 | 100.7 | 0.844 | 0.132 |
| P(OPh)3 (7) | 1984.5 | 1.607 | 106.9 | 0.831 | 0.135 |
| PCl2(OEt) (8) | 2014.5 | 1.598 | 96.4 | 0.866 | 0.139 |
| PCl3 (9) | 1993.0 | 1.602 | 101.2 | 0.837 | 0.135 |
| PF3 (10) | 1970.6 | 1.614 | 99.9 | 0.841 | 0.134 |
| P(CF3)F2 (11) | 1967.5 | 1.617 | 100.2 | 0.829 | 0.133 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kégl, T.R.; Pálinkás, N.; Kollár, L.; Kégl, T. Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters. Molecules 2018, 23, 3176. https://doi.org/10.3390/molecules23123176
Kégl TR, Pálinkás N, Kollár L, Kégl T. Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters. Molecules. 2018; 23(12):3176. https://doi.org/10.3390/molecules23123176
Chicago/Turabian StyleKégl, Tímea R., Noémi Pálinkás, László Kollár, and Tamás Kégl. 2018. "Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters" Molecules 23, no. 12: 3176. https://doi.org/10.3390/molecules23123176
APA StyleKégl, T. R., Pálinkás, N., Kollár, L., & Kégl, T. (2018). Computational Characterization of Bidentate P-Donor Ligands: Direct Comparison to Tolman’s Electronic Parameters. Molecules, 23(12), 3176. https://doi.org/10.3390/molecules23123176