Pharmacokinetic and Metabolism Studies of 12-Riboside-Pseudoginsengenin DQ by UPLC-MS/MS and UPLC-QTOF-MSE


Abstract
:1. Introduction
2. Results
2.1. Pharmacokinetic Study
2.1.1. Method Development
2.1.2. Method Validation
2.1.3. Pharmacokinetic Study
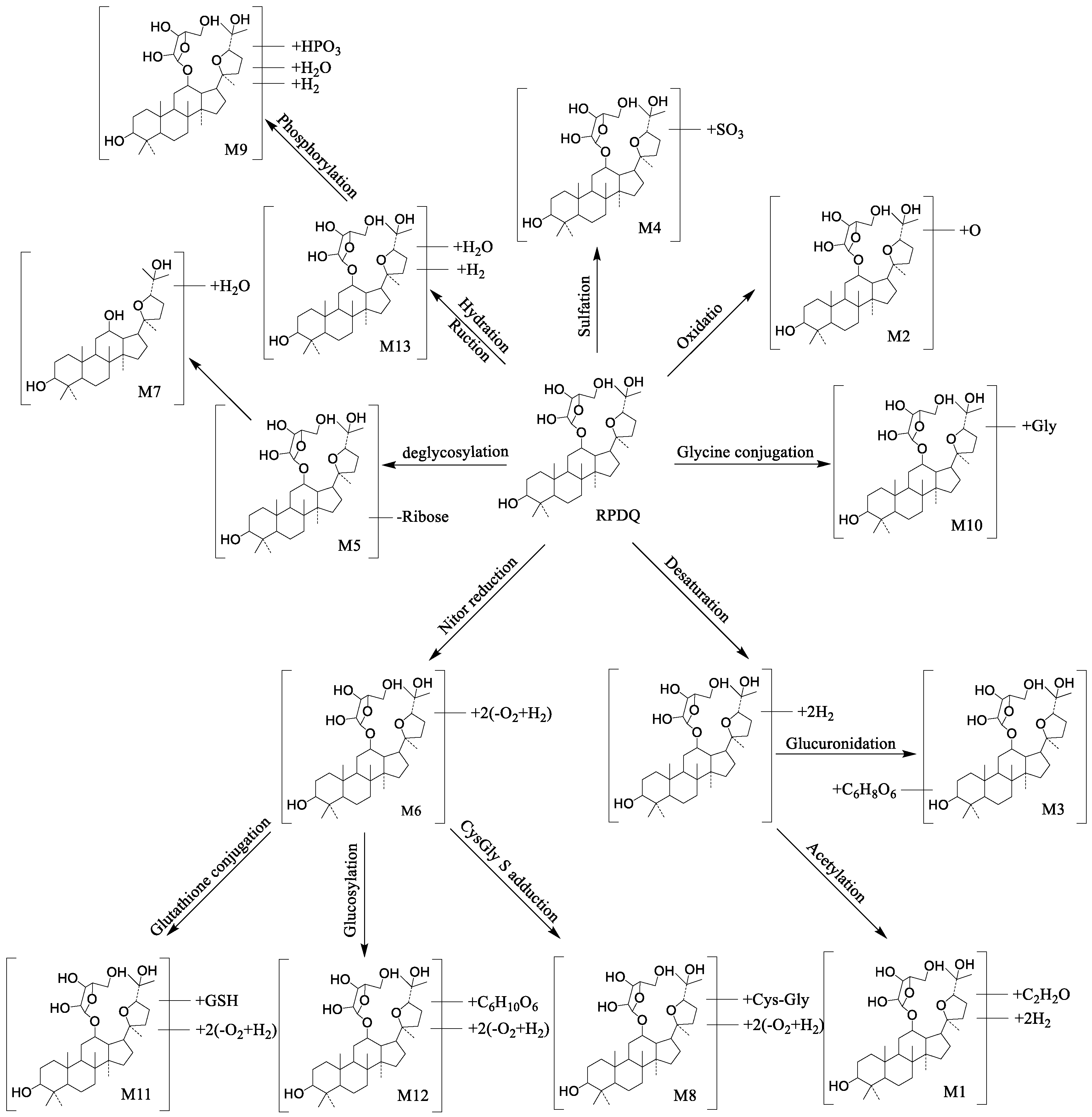
2.1.4. Results of Metabolites Identification of RPDQ
3. Discussion
4. Materials and Methods
4.1. Chemicals and Ragents
4.2. Animals and Drug Administration
4.3. Sample Preparation
4.3.1. Pharmacokinetic Study
4.3.2. Metabolism Study
4.4. Instruments and Experimental Conditions
4.4.1. LC-MS/MS Conditions
4.4.2. UPLC-QTOF/MS Conditions
4.5. Method Validation
4.6. Pharmacokinetic Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Peng, D.; Wang, H.; Qu, C.; Xie, L.; Wicks, S.M.; Xie, J. Ginsenoside Re: Its chemistry, metabolism and pharmacokinetics. Chin. Med. 2012, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K. Pharmacokinetics of ginsenoside Rb1 and its metabolite compound K after oral administration of Korean Red Ginseng extract. J. Ginseng Res. 2013, 37, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wang, R.; Niu, T.; Gao, S.; Yin, T.; You, M.; Jiang, Z.; Hu, M. Inhibition of P-glycoprotein leads to improved oral bioavailability of compound K, an anticancer metabolite of red ginseng extract produced by gut microflora. Drug Metab. Dispos. Biol. Fate Chem. 2012, 40, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lin, Y.; Li, H. The identification of molecular target of (20S) ginsenoside Rh2 for its anti-cancer activity. Sci. Rep. 2017, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shao, Y.; Ma, S.; Wu, X.; Meng, Q. Determination of 20(S)-protopanaxadiol ocotillol type epimers in rat plasma by liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2012, 887–888, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Matsumoto, K.; Yamasaki, K.; Watanabe, H. Majonoside-R2 reverses social isolation stress-induced decrease in pentobarbital sleep in mice: Possible involvement of neuroactive steroids. Life Sci. 1997, 61, 395–402. [Google Scholar] [PubMed]
- Lee, S.Y.; Jeong, J.J.; Le, T.H.V.; Eun, S.H.; Nguyen, M.D.; Park, J.H.; Kim, D.H. Ocotillol, a Majonoside R2 metabolite, ameliorates 2,4,6-trinitrobenzenesulfonic acidinduced colitis in mice by restoring the balance of Th17/Treg cells. J. Agric. Food Chem. 2015, 63, 7024–7031. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.W.; Ma, C.; Zhang, H.Y.; Bi, Y.; Chen, X.; Tian, H.; Xie, X.X.; Meng, Q.G.; Lewis, P.J.; Xu, J.Y. Synthesis and biological evaluation of novel ocotillol-type triterpenoid derivatives as antibacterial agents. Eur. J. Med. Chem. 2013, 68, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.L. Triterpene saponins from Vietnamese ginseng (Panax vietnamensis) and their hepatocytoprotective activity. J. Nat. Prod. 2001, 64, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Li, X.G.; Zhang, L.X.; Meng, X.Y.; Hou, J.R.; Zhang, J. Isolation, identification and content determination of pseudoginsenoside F11 in American ginseng. J. Jilin Agric. Univ. 2006, 6, 645–648. [Google Scholar]
- Toshinobu, M.; Ryoji, K.; Osamu, T.; Zhou, J.; Yang, T.R.; Junzo, S. Saponins of zutziseng, rhizomes of panax japonicus C.A. Meyer var. major (Burk.) C.Y. Wu et K.M. Feng, collected in Yunnan, China. Chem. Pharm. Bull. 1982, 30, 4341–4346. [Google Scholar]
- Zou, K.; Zhu, S.; Chihiro, T.; Cai, S.Q.; Katsuko, K. Dammarane-type riterpene saponins from panax japonicus. J. Nat. Prod. 2002, 65, 346–451. [Google Scholar] [CrossRef] [PubMed]
- Atopkina, L.N.; Shentsova, E.B.; Anisimov, M.M.; Uvarova, N.I. Structural-functional properties of glycosides from Panax ginseng C. A. Mey. and their analogues. II. Haemolytic activity of keto-derivatives of 20(S)-protopanaxadiol and their glycosides. Rastit. Resur. 2000, 36, 89–95. [Google Scholar]
- Bi, Y.; Wang, T.; Meng, Q.; Zhang, J.; Wang, L.; Li, Q.; Zhao, F.; Sun, H. Synthesis and Myocardial Ischemia Protective Effect of Ocotillol-Type Derivatives. Rec. Nat. Prod. 2012, 6, 242–254. [Google Scholar]
- Shen, R.; Cao, X.; Laval, S.; Yu, B. Synthesis of Ocotillol-Type Ginsenosides. J. Org. Chem. 2016, 81, 10279–10294. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Sugimoto, S.; Matsuda, H.; Yoshikawa, M. Medicinal flowers. XVII. New dammarane-type triterpene glycosides from flower buds of American ginseng, Panax quinquefolium L. Chem. Pharm. Bull. 2007, 55, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Sun, J.; Wang, G.; Xie, H.; Zha, W.; Yan, B.; Sun, F.; Hao, H.; Gu, S.; Sheng, L.; et al. Sensitive determination of 20(S)-protopanaxadiol in rat plasma using HPLC–APCI-MS: Application of pharmacokinetic study in rats. J. Pharm. Biomed. Anal. 2008, 48, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Li, Z.; Li, P.; Lin, M.; Han, L.; Wang, F.; Liu, J. Determination of Pseudoginsengenin DQ in rat plasma by UPLC-MS/MS and application of the method in a pharmacokinetic study. J. Chromatogr. B 2013, 933, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Su, C.; Yang, C.; Liu, M.; Tang, L.; Su, W.; Liu, Z. Determination of ginsenosides Rb1, Rb2, and Rb3 in rat plasma by a rapid and sensitive liquid chromatography tandem mass spectrometry method: Application in a pharmacokinetic study. J. Pharm. Biomed. Anal. 2012, 64–65, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Paek, I.B.; Moon, Y.; Kim, J.; Ji, H.Y.; Kim, S.A.; Sohn, D.H.; Kim, J.B.; Lee, S.H. Pharmacokinetics of a ginseng saponin metabolite compound K in rats. Biopharm. Drug Dispos. 2010, 27, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, G.; Haitang, X.; Li, H.; Pan, G.; Ian, T. Simultaneous rapid quantification of ginsenoside Rg1 and its secondary glycoside Rh1 and aglycone protopanaxatriol in rat plasma by liquid chromatography-mass spectrometry after solid-phase extraction. J. Pharm. Biomed. Anal. 2005, 38, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Liu, Y.; Hao, H.; Wang, G.; Chen, X.; Ren, H. Determination of 20(S)-ginsenoside Rh1 and its aglycone 20(S)-protopanaxatriol in rat plasma by sensitive LC-APCI-MS method and its application to pharmacokinetic study. Eur. J. Mass Spectrom. 2009, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Hao, H.; Liu, Y.; Zheng, C.; Wang, Q.; Wang, G.; Chen, X. Characterization of pharmacokinetic profiles and metabolic pathways of 20(S)-ginsenoside Rh1 in vivo and in vitro. Planta Med. 2009, 75, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Chen, H.; Li, C.; Liu, W.; Ma, W.; Rui, W. Screening and Identification of the Metabolites of Water Extracts of Raw and Honey-Processed Astragalus, in Rat Urine Based on UHPLC/ESI-Q-TOF-MS and Multivariate Statistical Analysis. J. Am. Soc. Mass Spectrom. 2018, 29, 1919–1935. [Google Scholar]
- Shen, J.; Song, R.; Hodges, T.R.; Heimberger, A.B.; Zhao, H. Identification of metabolites in plasma for predicting survival in glioblastoma. Mol. Carcinog. 2018, 57, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jiang, H.; Jin, M.; Wang, Q.; Sun, Q.; Du, Y.; Gao, L.; Xu, H. UHPLC-Q-TOF-MS/MS based screening and identification of the metabolites in vivo after oral administration of betulin. Fitoterapia 2018, 127, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Su, W.; Zheng, Y.; Liu, H.; Li, P.; Zhang, W.; Liang, Y. UFLC-Q-TOF-MS/MS-Based Screening and Identification of Flavonoids and Derived Metabolites in Human Urine after Oral Administration of Exocarpium Citri Grandis Extract. Molecules 2018, 23, 895. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liao, M.; Cheng, X.; Liang, C.; Diao, X.; Zhang, L. Ultrahigh-performance liquid chromatography coupled with triple quadrupole and time-of-flight mass spectrometry for thescreening and identification of the main flavonoids and their metabolites in rats after oral administration of Cirsium japonicum DC. extract. Rapid Commun. Mass Spectrom. 2018, 30, 1451–1461. [Google Scholar]
- Li, Y.; Peng, Y.; Wang, M.; Zhou, G.; Zhang, Y.; Li, X. Rapid screening and identification of the differences between metabolites of Cistanche deserticola and C. tubulosa water extract in rats by UPLC-Q-TOF-MS combined pattern recognition analysis. J. Pharm. Biomed. Anal. 2016, 131, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qi, L.; Li, P.; Wen, X.; Zhu, Y.; Liu, E.; Gong, Z.; Yang, X.; Ren, M.; Li, Y.; et al. Identification of metabolites of Danggui Buxue Tang in rat urine by liquid chromatography coupled with electrospray ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 23, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, J.; Xie, H. Study on absorption mechanism of ginsenoside Rg 1 using rat everted gut sac. Chin. J. Clin. Pharm. Ther. 2004, 9, 510–513. [Google Scholar]
- Gui, J.; Yang, W.; Li, Y. Simultaneous enantiomer determination of 20 (R)- and 20 (S)-ginsenoside-Rg2 in rat plasma after intravenous administration using HPLC method. J. Chromatogr. B 2007, 850, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Qian, T.; Wong, R.N.S.; Jiang, Z. Liquid chromatography–electrospray ionization mass spectrometry for metabolism and pharmacokinetic studies of ginsenoside Rg3. Anal. Chim. Acta 2003, 492, 283–293. [Google Scholar] [CrossRef]
- Santagata, S.; Di, C.E.; Carducci, C. Development of a new UPLC-ESI-MS/MS method for the determination of biopterin and neopterin in dried blood spot. Clin. Chim. Acta 2017, 466, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sang, M.; Liu, E.; Banahene, P.O.; Zhang, Y.; Wang, T. Rapid profiling and pharmacokinetic studies of major compounds in crude extract from Polygonum multiflorum by UHPLC-Q-TOF-MS and UPLC-MS/MS. J. Pharm. Biomed. Anal. 2017, 140, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Qiu, Y.; Chen, J.; Mu, S.; Sun, L. Simultaneous determination of nineteen major active compounds in Qiangshen tablet by UPLC-ESI-MS/MS. J. Pharm. Biomed. Anal. 2016, 128, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, Z.; Li, J.; Ito, Y.; Sun, W. A new quantitation method of protodioscin by HPLC-ESI-MS/MS in rat plasma and its application to the pharmacokinetic study. Steroids 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xue, J.; Li, B.; Xue, J.; Li, B.; Lin, X.; Wang, Z. Simultaneous quantification of triterpenoid saponins in rat plasma by UHPLC–MS/MS and its application to a pharmacokinetic study after oral total saponin of Aralia elata leaves. J. Sep. Sci. 2016, 39, 4360–4368. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, N.; Wang, Z.; Qi, Z. Rapid characterization of chemical constituents of Platycodon grandiflorum and its adulterant Adenophora stricta by UPLC-QTOF-MS/MS. J. Mass Spectrom. 2017, 52, 643–656. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 12-riboside-pseudoginsengenin DQ, 20R-panaxadiol and pseudoginsengenin DQ are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (ng/mL) | Inter-Day Measured Concentration (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | Intra-Day Measured Concentration (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) |
|---|---|---|---|---|---|---|
| 15 | 15.52 ± 0.84 | 5.41 | 3.47 | 15.43 ± 0.55 | 3.51 | 2.87 |
| 200 | 182.41 ± 6.87 | 3.48 | −8.82 | 188.79 ± 3.56 | 1.79 | −5.61 |
| 800 | 775.02 ± 17.75 | 2.23 | −3.12 | 788.99 ± 12.04 | 1.51 | −1.3 |
| Concentration | Mean | Accuracy | ||
|---|---|---|---|---|
| (ng /mL) | (ng /mL) | (%) | ||
| Short-term stability (25 °C, 4 h) | RPDQ | 15 | 14.85 ± 0.72 | −1.00 |
| 200 | 188.12 ± 14.00 | −5.94 | ||
| 800 | 773.99 ± 26.01 | −3.25 | ||
| IS | 100 | 92.34 ± 3.12 | −7.66 | |
| Long-term stability (10°C, 16 h) | RPDQ | 15 | 15.33 ± 1.61 | 2.2 |
| 200 | 186.96 ± 13.41 | −6.52 | ||
| 800 | 816.23 ± 15.82 | 2.03 | ||
| IS | 100 | 91.34 ± 2.31 | −8.66 | |
| Freezing stability (−20 °C, 14 d) | RPDQ | 15 | 15.41 ± 1.47 | 2.74 |
| 200 | 179.07 ± 14.73 | −10.47 | ||
| 800 | 786.32 ± 19.88 | −1.71 | ||
| IS | 100 | 94.74 ± 4.25 | −5.26 | |
| Freeze–thaw stability (freeze–thaw cycles) | RPDQ | 15 | 14.96 ± 0.58 | −0.03 |
| 200 | 188.7 ± 16.57 | −5.65 | ||
| 800 | 785.21 ± 19.19 | −1.85 | ||
| IS | 100 | 94.11 ± 3.47 | −5.89 | |
| Concentration after Dilution (ng/mL) | Mean (ng/mL) | Accuracy (%) | RSD (%) |
|---|---|---|---|
| 15 | 14.82 ± 0.61 | −1.20 | 4.11 |
| 200 | 187.79 ±12.19 | −6.11 | 6.49 |
| 800 | 784.17 ± 21.17 | −1.98 | 2.70 |
| Dose (mg/kg) | t1/2 (h) | Tmax (h) | AUC(0–60) (ug/L/h) | AUC(0–∞) (ug/L/h) | F (%) | Vz (L/kg) | CLz (L/h/kg) |
|---|---|---|---|---|---|---|---|
| 6 | 12.59 ± 1.28 | 7.0 ± 1.55 | 9212.53 ± 608.41 | 9568.25 ± 545.64 | 5.55% | 11.45 ± 1.55 | 0.63 ± 0.036 |
| 12 | 12.83 ± 0.56 | 7.5 ± 1.63 | 17,108.67 ± 987.76 | 17,685.11 ± 985.93 | 5.15% | 12.53 ± 0.59 | 0.68 ± 0.022 |
| 24 | 13.74 ± 1.11 | 7.0 ± 1.55 | 40,358.37 ± 3441.44 | 42,553.53 ± 3207.59 | 6.08% | 11.28 ± 1.69 | 0.57 ± 0.044 |
| 0.3 (i.v) | 2.94 ± 0.51 | 0.25 | 8298.18 ± 258.5. | 8325.73 ± 270.09 | -- | 11.52 ± 0.02 | 0.04 ± 0.001 |
| No. | RT (min) | Formula | Measured Mass (m/z) | Calculate Mass (m/z) | Error (ppm) | Fragment Ions | Metabolic Pathway | Source |
|---|---|---|---|---|---|---|---|---|
| RPDQ | 16.65 | C35H60O8 | 609.4346 | 609.8540 | 1.3 | 549.3856, 477.3938, 459.3833, 441.3727, 381.3152, 283.2420, 143.1067 | parent | P,B, F,U |
| M1 | 13.09 | C37H58O9 | 647.4144 | 647.8590 | −1.4 | 475.3782, 457.3675, 439.3569, 421.3464, 357.2788, 355.2632, 143.1067 | Desaturation Acetylation | F |
| M2 | 13.10 | C35H60O9 | 625.4330 | 625.8534 | 3.3 | 493.3866, 457.3649, 475.3781, 439.3565, 143.1024 | Oxidation | F,U |
| M3 | 13.34 | C41H64O14 | 781.4389 | 781.9464 | 2.7 | 723.4678, 573.4149, 545.3472, 315.2318 | Desaturation Glucuronidation | U |
| M4 | 13.49 | C35H60O11S | 689.3936 | 689.9172 | 1.1 | 675.3792, 670.3783, 539.3401, 521.3295, 503.3189, 441.3708, 143.1067 | Sulfation | F |
| M5 | 21.39 | C30H52O4 | 477.3932 | 477.7394 | 1.4 | 459.3832, 441.3727, 423.3621, 381.3152, 283.2420, 143.1067 | Deglycosylation | P,B, F,U |
| M6 | 25.20 | C35H64O4 | 549.4867 | 549.8882 | −1.8 | 541.3839, 521.3837, 477.3575, 416.4376, 283.2457 | Nitor reduction | F,U |
| M7 | 18.06 | C30H50O3 | 459.3815 | 459.7241 | 4.7 | 457.3675, 441.3727, 439.3570, 421.3465, 45.3515, 381.3117, 143.1067 | Desaturation Hydration | P,B, F,U |
| M8 | 25.65 | C40H72N2O7S | 725.5117 | 726.0818 | −2.2 | 715.4351, 557.4126, 465.3574, 382.3957, 340.3538, 143.1254 | CysGly S adduction | P,F |
| M9 | 27.34 | C35H65O12P | 709.4263 | 709.8651 | 1.9 | 505.3441, 495.4044, 481.3888, 463.3781 | Phosphorylation | P,F,U |
| M10 | 27.35 | C37H63NO9 | 666.4598 | 666.9054 | 3.2 | 551.39423, 534.4177, 516.3819, 495.3707, 143.1052 | Glycine conjugation | P,B |
| M11 | 27.65 | C45H79N3O9S | 838.5641 | 839.1964 | 3.8 | 718.4459, 549.4627, 520.3758, 513.35745, 270.3118, 143.0924 | Glutathione conjugation | P,B, F,U |
| M12 | 27.91 | C41H72O10 | 725.5152 | 725.0123 | 3.1 | 557.3817, 549.4758, 443.3841, 143.1014 | Glucosylation | P,B, F,U |
| M13 | 28.18 | C35H64O9 | 629.4687 | 629.8852 | 1.0 | 594.4491, 569.4427, 461.3625, 303.3046, 143.2017 | Reduction Hydration | B |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Lin, H.; Zhu, H.; Yang, N.; Zhou, B.; Wang, C.; Li, P.; Liu, J. Pharmacokinetic and Metabolism Studies of 12-Riboside-Pseudoginsengenin DQ by UPLC-MS/MS and UPLC-QTOF-MSE. Molecules 2018, 23, 2499. https://doi.org/10.3390/molecules23102499
Wang Z, Lin H, Zhu H, Yang N, Zhou B, Wang C, Li P, Liu J. Pharmacokinetic and Metabolism Studies of 12-Riboside-Pseudoginsengenin DQ by UPLC-MS/MS and UPLC-QTOF-MSE. Molecules. 2018; 23(10):2499. https://doi.org/10.3390/molecules23102499
Chicago/Turabian StyleWang, Zhenzhou, Hongqiang Lin, Hailin Zhu, Na Yang, Baisong Zhou, Cuizhu Wang, Pingya Li, and Jinping Liu. 2018. "Pharmacokinetic and Metabolism Studies of 12-Riboside-Pseudoginsengenin DQ by UPLC-MS/MS and UPLC-QTOF-MSE" Molecules 23, no. 10: 2499. https://doi.org/10.3390/molecules23102499
APA StyleWang, Z., Lin, H., Zhu, H., Yang, N., Zhou, B., Wang, C., Li, P., & Liu, J. (2018). Pharmacokinetic and Metabolism Studies of 12-Riboside-Pseudoginsengenin DQ by UPLC-MS/MS and UPLC-QTOF-MSE. Molecules, 23(10), 2499. https://doi.org/10.3390/molecules23102499