
Polymeric Nanocarriers for the Delivery of Antimalarials


Abstract
:
1. Introduction
2. The Life Cycle of Malaria Parasite
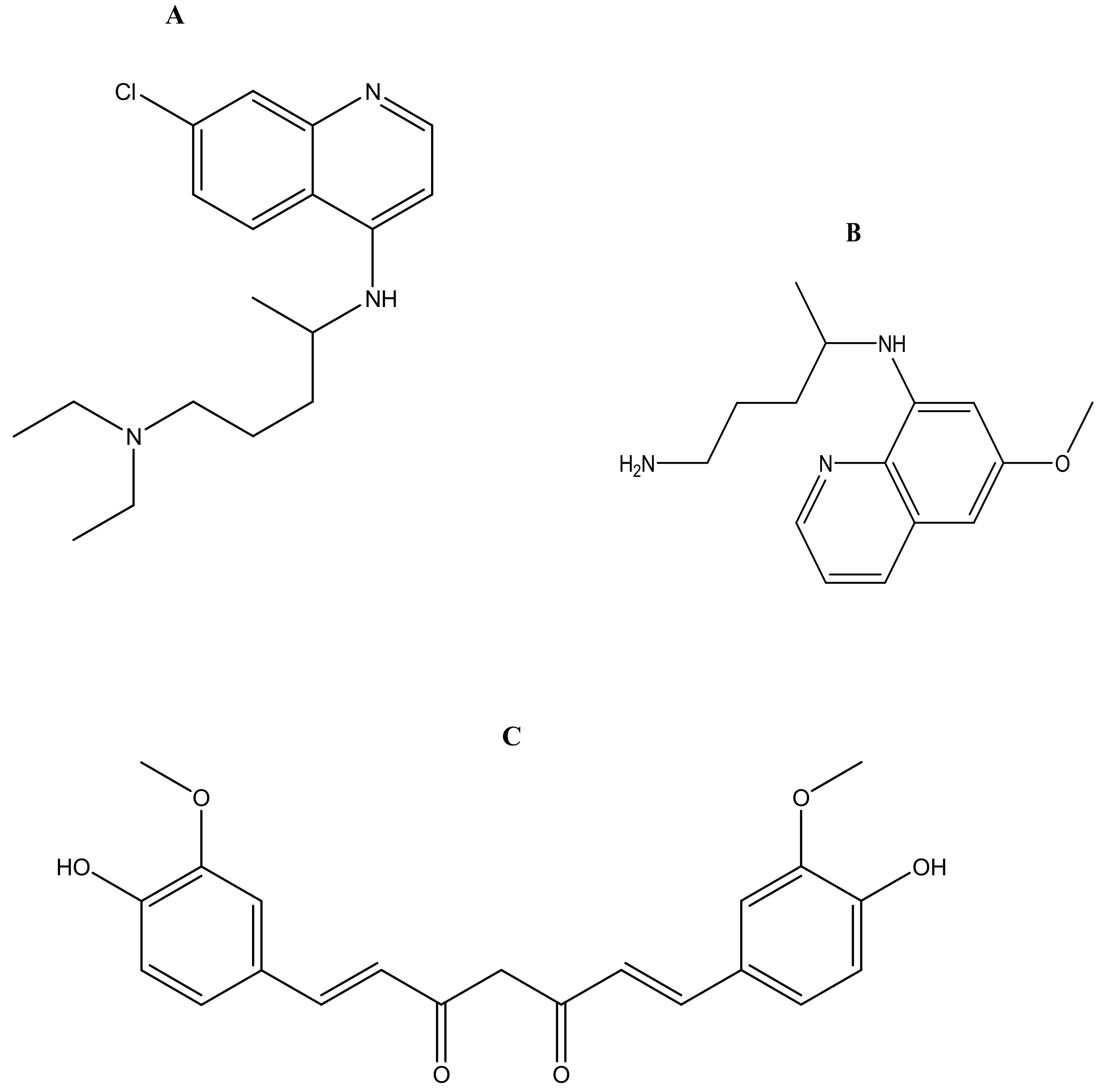
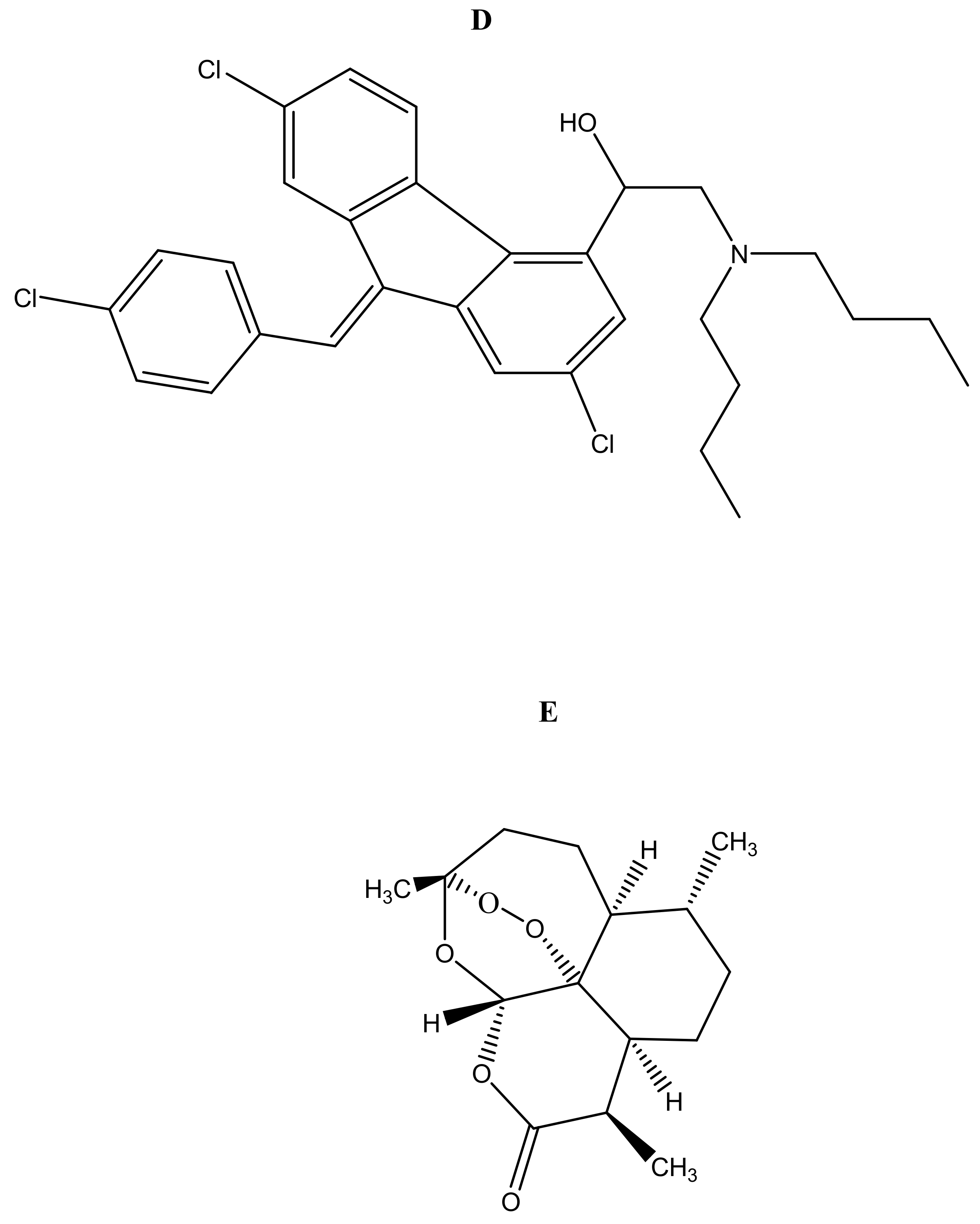
2.1. Antimalarial Resistance
2.2. Combination Therapy
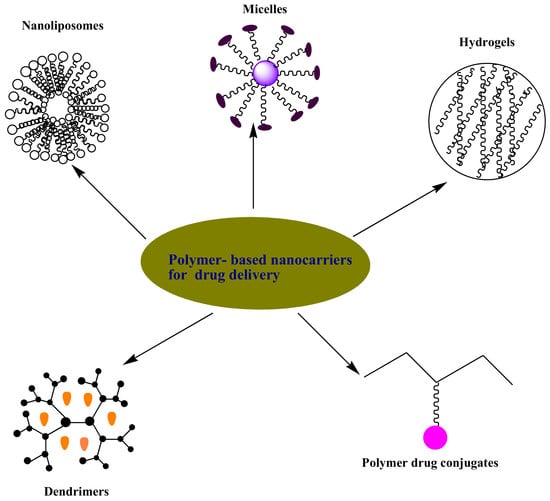
3. Polymer-Based Nanocarriers for Drug Delivery
3.1. Hydrogels
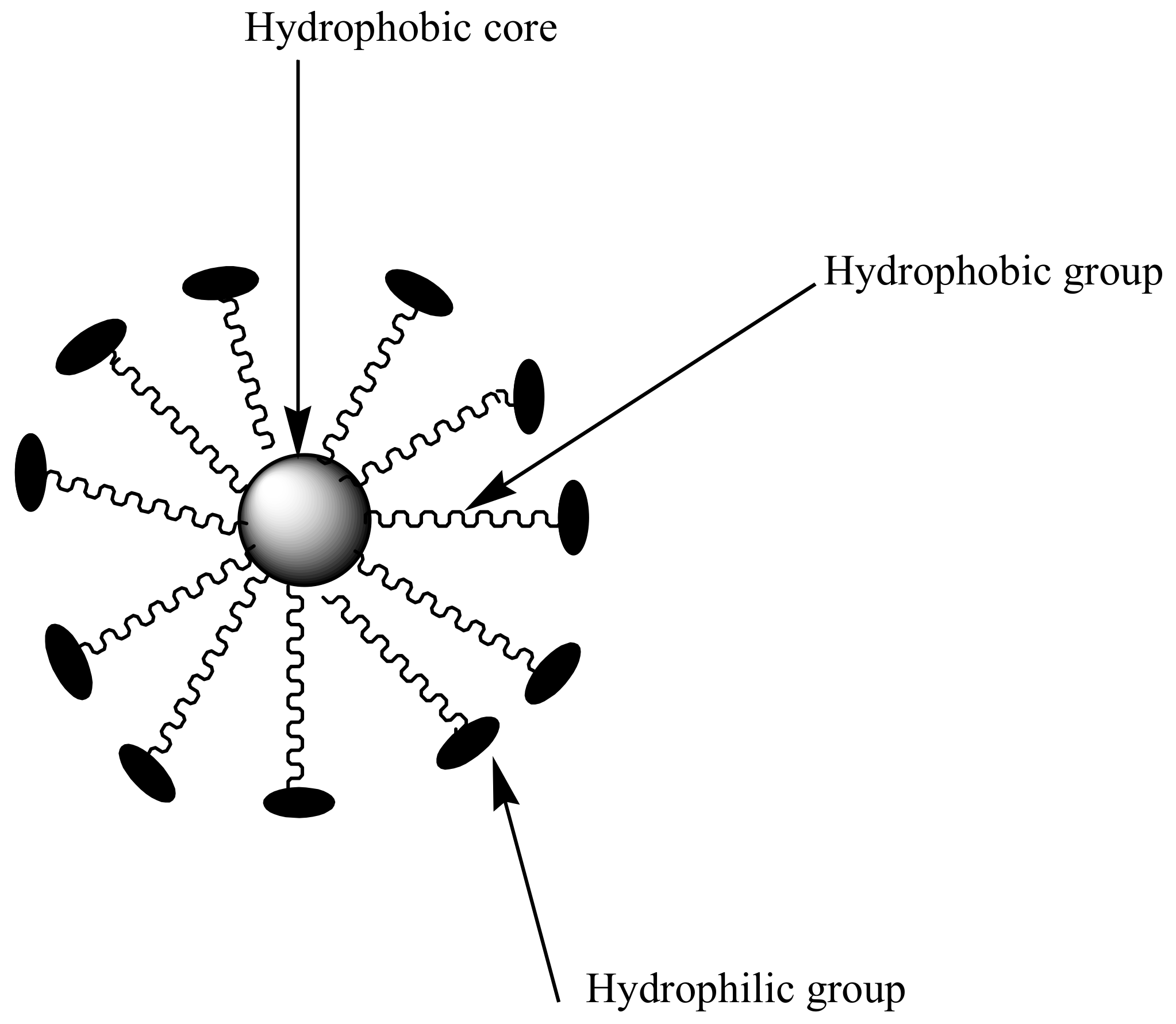
3.2. Micelles
3.3. Nanoliposomes
3.4. Dendrimers
3.5. Polymer-Drug Conjugates
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO Malaria Factsheet updated January 2016. Available online: http://www.who.int/mediacentre/factsheets/fs094/en/. (accessed on 18 February 2016).
- WHO Factsheet: Malaria Report 2015. Available online: http://www.who.int/malaria/media/world-malaria-report-2015/en/. (accessed on 9 December 2015).
- Baird, J.K. Evidence and implications of mortality associated with acute Plasmodium vivax malaria. Clin. Microbiol. Rev. 2013, 26, 36–57. [Google Scholar] [CrossRef] [PubMed]
- Douglas, N.M.; Anstrey, N.M.; Buffet, P.A.; Poesspoprodjo, J.; Yeo, T.W.; Whiten, N.J. The anaemia of plasmodium vivax malaria. Malar. J. 2012, 10, 11–135. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.R.; Magill, A.J.; Parise, M.E.; Arguin, P.M. Doxycycline for malaria chemoprophylaxis and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am. J. Trop. Med. Hyg. 2011, 4, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.K.; Rogerson, S.J.; Fischer, P.R. The impact of maternal malaria on newborns. Ann.Trop. Pediatr. 2010, 30, 271–82. [Google Scholar] [CrossRef] [PubMed]
- Omari, A.A.; Gamble, C.; Garner, P. Artemether-lumefantrine for uncomplicated malaria: a systematic review. Trop. Med. Int. Health. 2004, 9, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazzinelli, R.T.; Kalantari, P.; Fitzgerald, K.A.; Golenbock, D.T. Innate sensing of Malaria Parasites. Nat. Rev. Immunol. 2014, 14, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Tilley, L.; Dixon, M.W.; Kirk, K. The Plasmodium falciparum-infected red blood cell. Int. J. Biochem. Cell Biol. 2011, 43, 839–42. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, P.J. Artesunate for the Treatment of Severe Falciparum Malaria. N. Engl. J. Med. 2008, 358, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Life Cycle of Malaria Parasite. Available online: http://encarta.msn.com/media_461541582/life cycle of the malaria. (accessed on 3 February 2009).
- Baer, K.; Klotz, C.; Kappe, S.H. Release of hepatic Plasmodium yoelii merozoites into the pulmonary microvasculature. PLoS Pathog. 2007, 3, 171. [Google Scholar] [CrossRef] [PubMed]
- Petersen, I.; Eastman, R.; Lanzer, M. Drug–resistant malaria: Molecular mechanisms and implications for public health. FEBS. Lett. 2011, 585, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Aminake, M.N.; Pradel, G. Antimalarial drugs resistance in Plasmodium falciparum and the current strategies to overcome them. Microbial pathogens and strategies for combating them. Formatex 2013, 2013. 1, 1–774. [Google Scholar]
- Nayyar, G.M.L.; Breman, J.G.; Newton, P.N.; Herrington, J. Poor-quality antimalarial drugs in Southeast Asia and sub-Saharan Africa. Lancet Infect. Dis. 2012, 12, 488–496. [Google Scholar] [CrossRef]
- Krogstad, D.J.; Gluzman, I.Y.; Kyle, D.E.; Oduola, A.M.; Martin, S.K.; Milhous, W.K.; Schlesinger, P.H. Efflux of chloroquine from Plasmodium falciparum: mechanism of chloroquine resistance. Science 1987, 238, 1283–1285. [Google Scholar] [CrossRef] [PubMed]
- Nqoro, X.; Tobeka, N.; Aderibigbe, B.A. Quinoline-based hybrid compounds with antimalarial activity. Molecules. 2017, 22, 2268. [Google Scholar] [CrossRef] [PubMed]
- Saifi, M.A.; Beg, T.; Harrath, A.H.; Altayalan, F.S.H.; Al Quraishy, S. Antimalarial drugs: Mode of action and status of resistance. Afr. J. Pharm. Pharmacol. 2013, 7, 148–156. [Google Scholar] [CrossRef]
- Nosten, F.; Brasseur, P. Combination therapy for malaria: the way forward. Drugs 2002, 62, 1315–1329. [Google Scholar] [CrossRef] [PubMed]
- Kunte, R.; Kunwar, R. WHO Guidelines for the treatment of malaria. Med. J. Armed. Forces India 2016, 67, 376. [Google Scholar] [CrossRef]
- East African Network for Monitoring Antimalarial Treatment (EANMAT). The efficacy of antimalarial monotherapies, sulphadoxine-pyrimethamine and amodiaquine in East Africa: Implications for sub-regional policy. Trop. Med. Int. Health 2003, 8, 860–867. [Google Scholar] [CrossRef]
- World Health Organization. WHO Information Note on Artemether/Lumefantrine (Coartem); WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Looareesuwan, S.; Vanijanonta, S.; Viravan, C.; Wilairatana, P.; Charoenlarp, P.; Lasserre, R.; Canfield, C.; Kyle, D.E.; Webster, H.K. Randomised trial of mefloquine-tetracycline and quinine-tetracycline for acute uncomplicated falciparum malaria. Acta Trop. 1994, 57, 47–53. [Google Scholar] [CrossRef]
- Arima, H.; Hayashi, Y.; Higashi, T.; Motoyama, K. Recent advances in cyclodextrin delivery techniques. Expert Opin. Drug. Deliv. 2015, 12, 1425. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.; Moreira, R.; Cravo, P.V.L. Malaria Combination Therapies: Advantages and Shortcomings. Mini Rev. Med. Chem. 2008, 8, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P.; Marchisio, M.A.; Duncan, R. Poly (amidoamine)s: Biomedical Applications. Macromol. Rapid Commun. 2002, 23, 332–355. [Google Scholar] [CrossRef]
- Yang, W.W.; Pierstorff, E. Reservoir-Based Polymer Drug Delivery Systems. J. Lab. Autom. 2012, 17, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Kopeček, J. Hydrogel biomaterials: a smart future? Biomaterials 2007, 28, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Kioomars, S.; Heidari, S.; Malaekeh-Nikouei, B.; Shayani, R.M.; Khameneh, B.; Mohajeri, S.A. Ciprofloxacin-imprinted hydrogels for drug sustained release in aqueous media. Pharm. Dev. Technol. 2016, 16, 1–8. [Google Scholar]
- Schuetz, Y.B.; Gurny, R.; Jordan, O. A novel thermoresponsive hydrogel based on chitosan. Eur. J. Pharm. Biopharm. 2008, 68, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, S.; Xiao, L.; Dong, X.; Lu, Q.; Kaplan, D.L. Injectable and pH-responsive silk nanofiber hydrogels for sustained anticancer drug delivery. ACS Appl. Mater Interfaces 2016, 8, 17118–17126. [Google Scholar] [CrossRef] [PubMed]
- Aderibigbe, B.; Sadiku, E.; Jayaramudu, J.; Ray, S.S. Controlled Dual Release Study of Curcumin and a 4-Aminoquinoline Analog from Gum Acacia Containing Hydrogels. J. Appl. Polym. Sci. 2015, 132, 41613. [Google Scholar] [CrossRef]
- Aderibigbe, B.A.; Mhlwatika, Z. Dual release kinetics of antimalarials from soy protein isolate-carbopol-polyacrylamide based hydrogels. Appl. Polym. 2016, 43918, 1–8. [Google Scholar] [CrossRef]
- Dandekar, P.P.; Jain, R.; Patil, S.; Dhumal, R.; Tiwari, D.; Sharma, S.; Vanage, G.; Patravale, V. Curcumin-Loaded Hydrogel Nanoparticles: Application in Anti-Malarial Therapy and Toxicological Evaluation. J. Pharm. Sci. Exp. Pharmacol. 2010, 99, 4992–5010. [Google Scholar] [CrossRef] [PubMed]
- Dreve, S.; Kacso, I.; Popa, A.; Raita, O.; Bende, A.; Borodi, G.; Bratu, I. Chitosan-based nanocarriers for antimalarials. AIP Conf. Proc. 2012, 1425, 17–21. [Google Scholar]
- Liu, J.; Pang, Y.; Zhang, S.; Cleveland, C.; Yin, X.; Booth, L.; Lin, J.; Lee, Y.A.; Mazdiyasni, H.; Saxton, S.; Kirtane, A.R. Triggerable tough hydrogels for gastric resident dosage forms. Nat. Commun. 2017, 8, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musabayane, C.T.; Munjeri, O.; Matavire, T.P. Transdermal delivery of chloroquine by amidated pectin hydrogel matrix patch in the rat. Ren. Fail. 2003, 25, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Mavondo, G.A.; Tagumirwa, M.C. Asiatic acid-pectin hydrogel matrix patch transdermal delivery system influences parasitaemia suppression and inflammation reduction in P. berghei murine malaria infected Sprague–Dawley rats. Asian Pac. J. Trop. Biomed. 2016, 9, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, K. Polymer Micelles for Drug Delivery. Chapter 19. Available online: http://kinam. com/Articles/PMicelles%20Papers/Kim,%20SW%2010%20Pol%20Micelle%20BookCh.pdf (accessed on 23 December 2016).
- Danafar, H.; Rostamizadeh, K.; Davaran, S.; Hamidi, M. Drug-conjugated PLA–PEG– PLA copolymers: A novel approach for controlled delivery of hydrophilic drugs by micelle formation. Pharm. Dev. Technol. 2017, 22, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.F.; Cristea, M.; Winnik, F.M. Polymeric micelles for oral drug delivery: Why and how? Int. Res. J. Pure Appl. Chem. 2004, 76, 321–1335. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, J.Z.; Hu, J.; Guo, Q.; Yang, D. Preparation of amphiphilic copolymers for covalent loading of paclitaxel for drug delivery system. J. Polym. Sci. A Polym. Chem. 2014, 52, 366–74. [Google Scholar] [CrossRef]
- Cabral, H.; Kataoka, K. Progress of drug-loaded polymeric micelles into clinical studies. J. Control. Release 2014, 190, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danafar, H. MPEG–PCL copolymeric nanomicelles in drug delivery systems. Cogent Med. 2016, 3, 1142411. [Google Scholar] [CrossRef]
- Bae, Y.; Nishiyama, N.; Fukushima, S.; Koyama, H.; Yasuhiro, M.; Kataoka, K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug. Chem. 2005, 16, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Manjili, H.R.K.; Malvandi, H.; Mousavi, M.; Danafar, H. Preparation and Physicochemical Characterization of Biodegradable mPEG-PCL Core-Shell Micelles for Delivery of Artemisinin. J. Pharm. Sci. Exp. Pharmacol. 2016, 22, 234–243. [Google Scholar] [Green Version]
- Ramazani, A.; Keramati, M.; Malvandi, H.; Danafar, H.; Kheiri Manjili, H. Preparation and in vivo evaluation of anti-plasmodial properties of artemisinin-loaded PCL-PEG-PCL nanoparticles. Pharm. Dev. Technol 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, D.; Bhadra, S.; Jain, N.K. Pegylated Lysine Based Copolymeric Dendritic Micelles For Solubilization And Delivery Of Artemether. J. Pharm. Pharm. Sci. 2005, 8, 467–482. [Google Scholar] [PubMed]
- Manjili, H.K.; Malvandi, H.; Mousavi, M.S.; Attari, E.; Danafar, H. In vitro and in vivo delivery of artemisinin loaded PCL–PEG–PCL micelles and its pharmacokinetic study. Artif. Cells Nanomed. Biotechnol. 2018, 46, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Aderibigbe, B.A. Design of Drug Delivery Systems Containing Artemisinin and Its Derivatives. Molecules 2017, 22, 323. [Google Scholar] [CrossRef] [PubMed]
- Çag˘das, M.; Sezer, A.D.; Bucak, S. Liposomes as Potential Drug Carrier Systems for Drug Delivery. In Nanotechnology and Nanomaterials. Application of Nanotechnology in Drug Delivery; Sezer, A.D., Ed.; Intech: Rijeka, Croatia, 2014; pp. 1–50. [Google Scholar]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Mufamadi, M.S.; Pillay, V.; Choonara, Y.E.; Du Toit, L.C.; Modi, G.; Naidoo, D.; Ndesendo, V.M.K. A Review on Composite Liposomal Technologies for Specialized Drug Delivery. J. Drug. Deliv. Sci Technol. 2011, 2011, 19. [Google Scholar] [CrossRef] [PubMed]
- Chimanuka, B.; Gabriëls, M.; Detaevernier, M.R.; Plaizier-Vercammen, J.A. Preparation of β-artemether liposomes, their HPLC-UV evaluation and relevance for clearing recrudescent parasitaemia in Plasmodium chabaudi malaria-infected mice. J. Pharm. Biomed. Anal. 2002, 28, 13–22. [Google Scholar] [CrossRef]
- Shakeel, K.; Raisuddin, S.; Ali, S.; Imam, S.S.; Rahman, M.A.; Jain, G.K.; Ahmad, F.J. Development and in vitro/in vivo evaluation of artemether and lumefantrine co-loaded nanoliposomes for parenteral delivery. J. Liposome Res. 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.; Moles, E.; Urbán, P.; Prohens, R.; Busquets, M.A.; Sevrin, C.; Grandfils, C.; Fernàndez-Busquets, X. Application of heparin as a dual agent with antimalarial and liposome targeting activities toward Plasmodium-infected red blood cells. Nanomedicine 2014, 10, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Urbán, P.; Estelrich, J.; Adeva, A.; Cortés, A.; Fernàndez-Busquets, X. Study of the efficacy of antimalarial drugs delivered inside targeted immunoliposomal nanovectors. Nanoscale Res. Lett. 2011, 6, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, V.; Rohra, S.; Raza, M.; Hasan, G.M.; Dutt, S.; Ghosh, P.C. Stearylamine Liposomal Delivery of Monensin in combination with free Artemisinin eliminates blood stages of P. falciparum in culture and P. berghei infection in Murine Malaria. Antimicrob Agents. Ch. 2016, 60, 1304–1318. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kanamoto, T.; Nakashima, H.; Yoshida, T. Synthesis of a new amphiphilic glycodendrimer with antiviral functionality. Carbohydr. Polym. 2012, 90, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Nanjwade, B.K.; Bechra, H.M.; Derkar, G.K.; Manvi, F.V.; Nanjwade, V.K. Dendrimers: Emerging polymers for drug-delivery systems. Eur. J. Pharm. Sci. 2009, 38, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Gangadharan, D.; Dhandhala, N.; Dixit, D.; Thakur, R.S.; Popat, K.M.; Anand, P.S. Investigation of solid supported dendrimers for water disinfection. J. Appl. Polym. Sci. 2012, 124, 138491. [Google Scholar] [CrossRef]
- Bhadra, D.; Yadav, A.K.; Bhadra, S.; Jain, H.K. Glycodendrimeric nanoparticulate carriers of primaquine phosphate for liver targeting. Int. J. Pharm. 2005, 295, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Gupta, U.; Jain, N.K. Glycoconjugated peptide dendrimers-based nanoparticulate system for the delivery of chloroquine phosphate. Biomaterials 2007, 28, 3349–3359. [Google Scholar] [CrossRef] [PubMed]
- Movellan, J.; Urbán, P.; Moles, E.; Fuente, J.M.; Sierra, T.; Serrano, J.L.; Fernàndez-Busquets, X. Amphiphilic dendritic derivatives as nanocarriers for the targeted delivery of antimalarial drugs. Biomaterials 2014, 13, 7940–7950. [Google Scholar] [CrossRef] [PubMed]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly (ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.S.; Aher, N.; Patil, R.; Khandare, J. Poly (ethylene glycol)-Prodrug Conjugates: Concept, Design, and Applications. J. Drug. Deliv. 2012, 2012, 17. [Google Scholar] [CrossRef] [PubMed]
- Ringsdorf, H. Structure and properties of pharmacologically active polymers. J. Polym. Sci.: Polym Symposia 1975, 5, 135–153. [Google Scholar] [CrossRef]
- Agrawal, N.; Rohini, A.J.; Mukerjee, A. Polymeric Prodrugs: Recent Achievements and General Strategies. J. Antivir. Antiretrovir 2013, 15, 1–12. [Google Scholar]
- Van, S.; Das, S.K.; Wang, X.; Feng, Z.; Jin, Y.; Hou, Z.; Chen, F.; Pham, A.; Jiang, N.; Howell, S.B.; Yu, L. Synthesis, characterization, and biological evaluation of poly (L-γ-glutamyl-glutamine)-paclitaxel nanoconjugate. Int. J. Nanomedicine 2010, 5, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. Polymer–drug conjugation, recent achievements and general strategies. Prog. Polym. Sci. 2007, 32, 933–961. [Google Scholar] [CrossRef]
- Mukaya, H.E.; Van Zyl, R.L.; Van Vuuren, N.J.; Mbianda, X.Y. Synthesis and characterization of water-soluble polyaspartamides containing platinum (II) complex and bisphosphonate as potential antimalarial drug. Polym. Bull. 2016, 74, 3161–3178. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, R.K.; Sharma, R.; Murthy, R.S.; Bhardwaj, T.R. Design, synthesis and evaluation of antimalarial potential of polyphosphazene linked combination therapy of primaquine and dihydroartemisinin. Eur. J. Pharm. Sci. 2014, 66C, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Aderibigbe, B.A.; Neuse, E.W. Macromolecular conjugates of 4-and 8-aminoquinoline compounds. J. Inorg. Organomet. Polym. Mater. 2012, 22, 429–438. [Google Scholar] [CrossRef]
- Aderibigbe, B.A.; Neuse, E.W.; Sadiku, E.R.; Shina, R.S; Smith, P.J. Synthesis, characterization, and antiplasmodial activity of polymer-incorporated aminoquinolines. J. Biomed. Mater. Res. A 2014, 102, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Rajic, Z.; Kos, G.; Zorc, B.; Singh, P.P.; Singh, S. Macromolecular prodrugs. XII. Primaquine conjugates: Synthesis and preliminary antimalarial evaluation. Acta Pharm. 2009, 59, 107–115. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimalarials | Classification | Mode of Action | Polymer Carriers | References |
|---|---|---|---|---|
| Primaquine | Hypnozoiticidal and gametocytocidal | Primaquine interferes with the electron transport in the parasite during respiration process | Nanoliposomes Dendrimers Polymer drug conjugates | 57 63, 65 74,77 |
| Chloroquine | Blood schizontocides | Chloroquine accumulate in the acidic food vacuoles of intraerythrocytic trophozoites and thereby prevent haemoglobin degradation | Nanoliposomes Hydrogels Dendrimers | 58 38, 39 54, 65 |
| Artemisinin Dihydroartemisinin | Gametocytocidal | Involves the heme-mediated decomposition of the peroxide bridge to produce carbon-centred free radicals | Micelles Polymer-drug conjugates | 47, 49 74 |
| Curcumin | Blood schizontocides | Curcumin inhibits the activity of enzymes and lipid peroxides | Hydrogels | 33, 34, 35 |
| Artemether Beta-Artemether | Gametocytodal | It acts against erythrocytic stages of P. falciparum and inhibits nucleic acid and protein synthesis. | Micelles Nanoliposomes | 50 55 |
| Lumefantrine | Blood schizontocides | Lumefantrine is believed inhibits nucleic and formation of β-hematin by forming a complex with hemin | Nanoliposomes Hydrogels | 56 37 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mhlwatika, Z.; Aderibigbe, B.A. Polymeric Nanocarriers for the Delivery of Antimalarials. Molecules 2018, 23, 2527. https://doi.org/10.3390/molecules23102527
Mhlwatika Z, Aderibigbe BA. Polymeric Nanocarriers for the Delivery of Antimalarials. Molecules. 2018; 23(10):2527. https://doi.org/10.3390/molecules23102527
Chicago/Turabian StyleMhlwatika, Zandile, and Blessing Atim Aderibigbe. 2018. "Polymeric Nanocarriers for the Delivery of Antimalarials" Molecules 23, no. 10: 2527. https://doi.org/10.3390/molecules23102527
APA StyleMhlwatika, Z., & Aderibigbe, B. A. (2018). Polymeric Nanocarriers for the Delivery of Antimalarials. Molecules, 23(10), 2527. https://doi.org/10.3390/molecules23102527