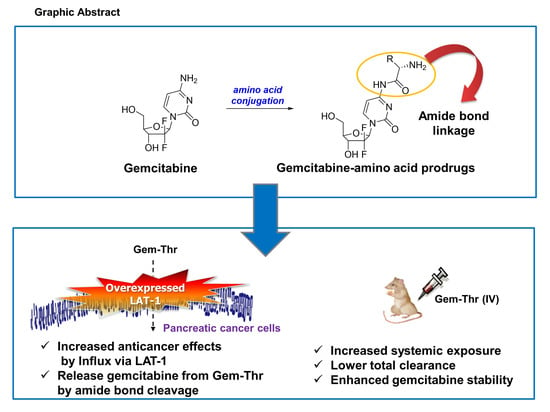
Synthesis of Gemcitabine-Threonine Amide Prodrug Effective on Pancreatic Cancer Cells with Improved Pharmacokinetic Properties

 ,
,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
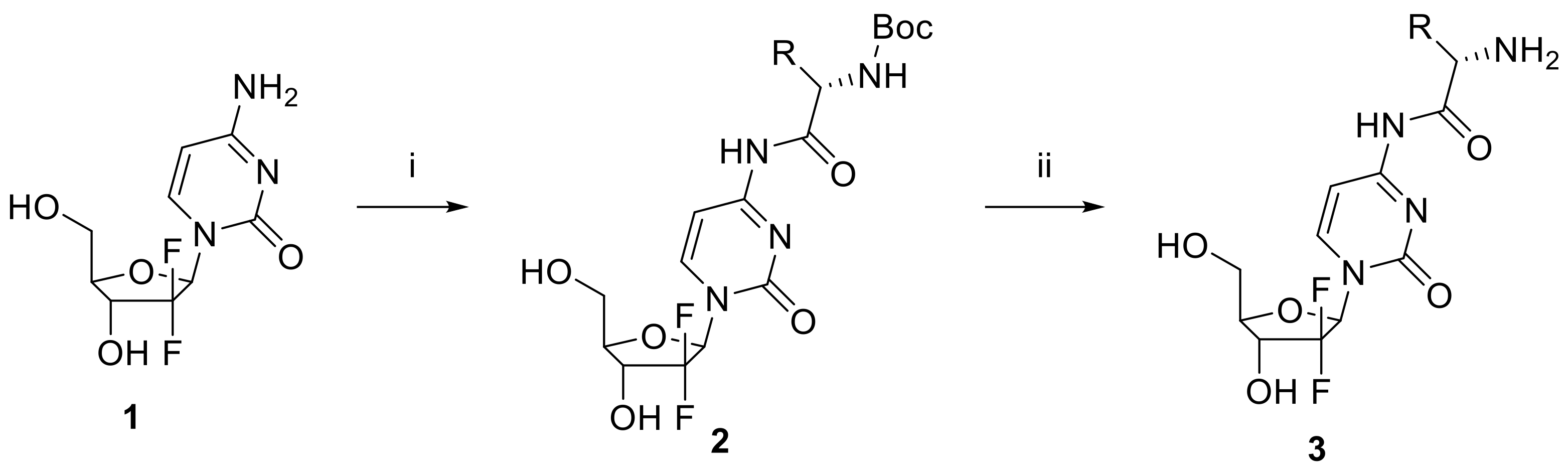
2.1. Synthesis of Gemcitabine Prodrugs with Amino Acids
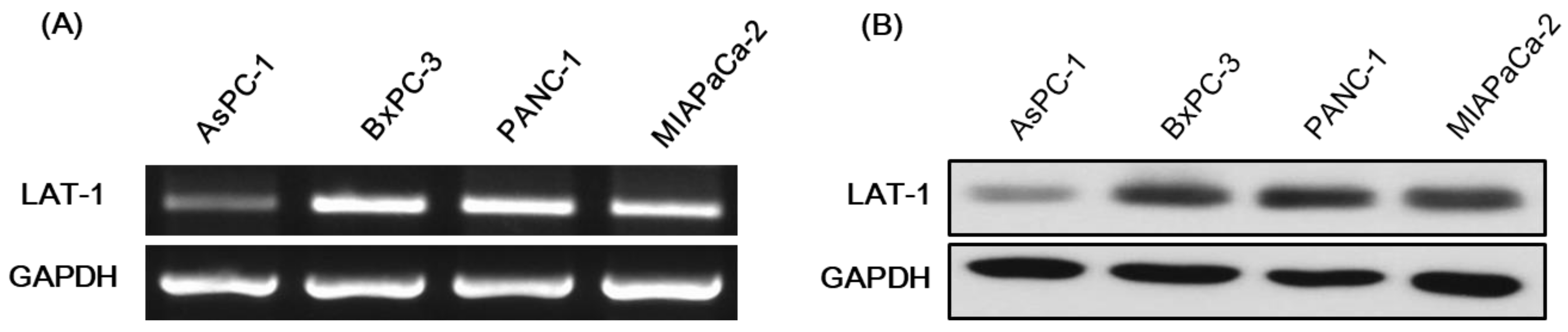
2.2. Expression of LAT-1 in Pancreatic Cancer Cell Lines
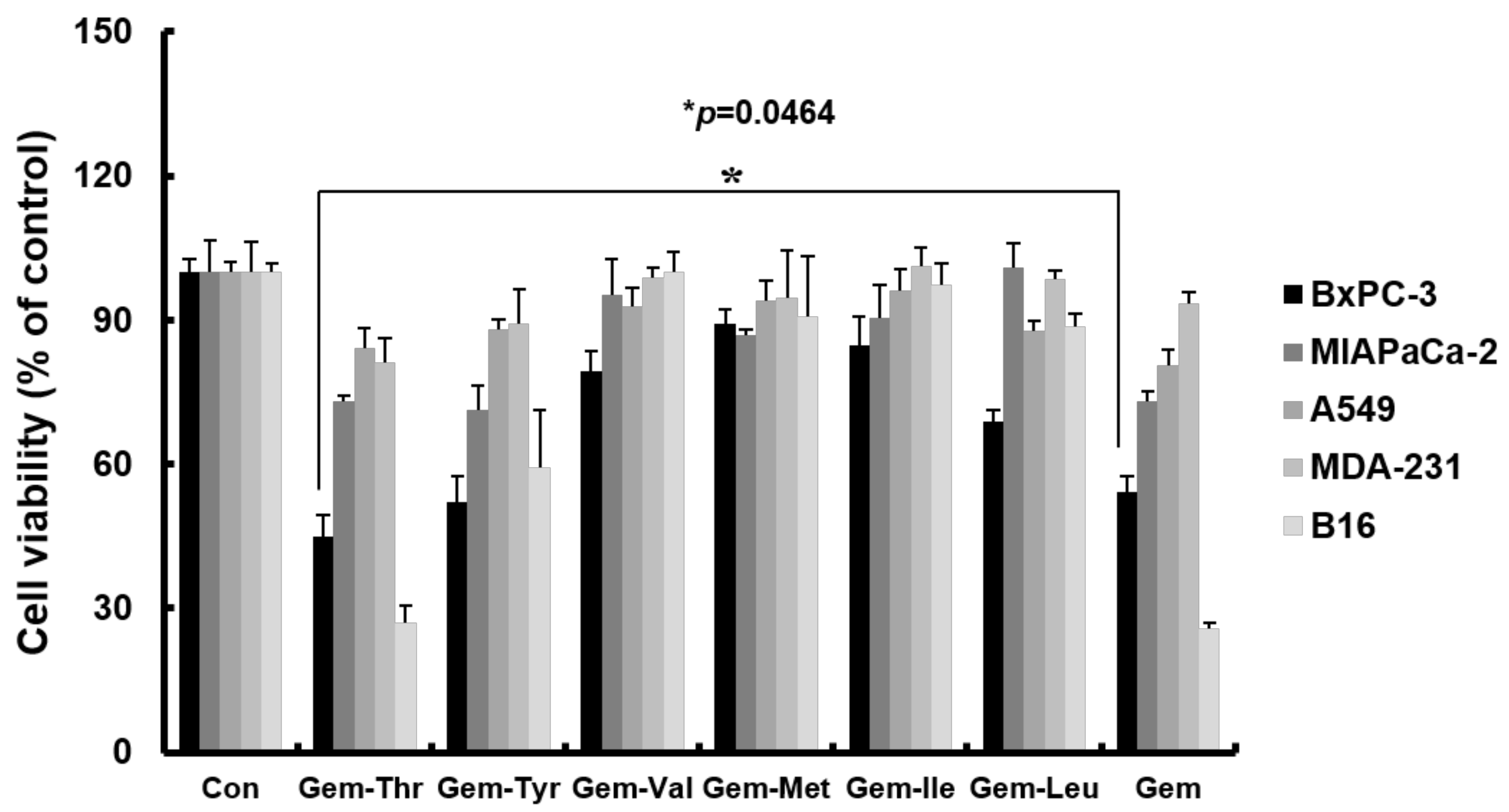
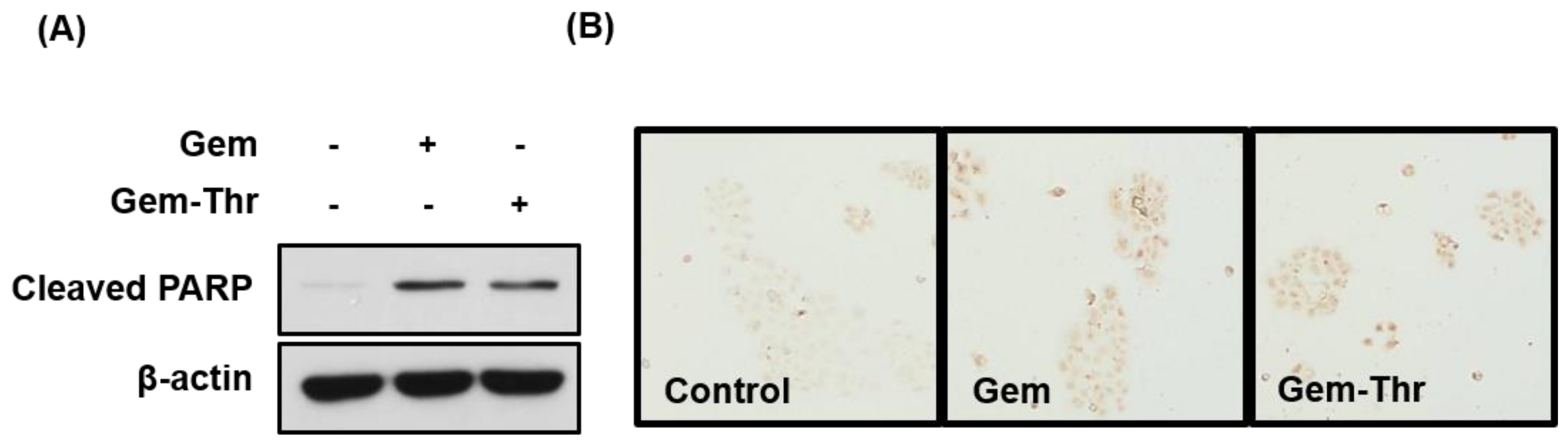
2.3. Anticancer Effects of Prodrugs with Amino Acids in Cancer Cells
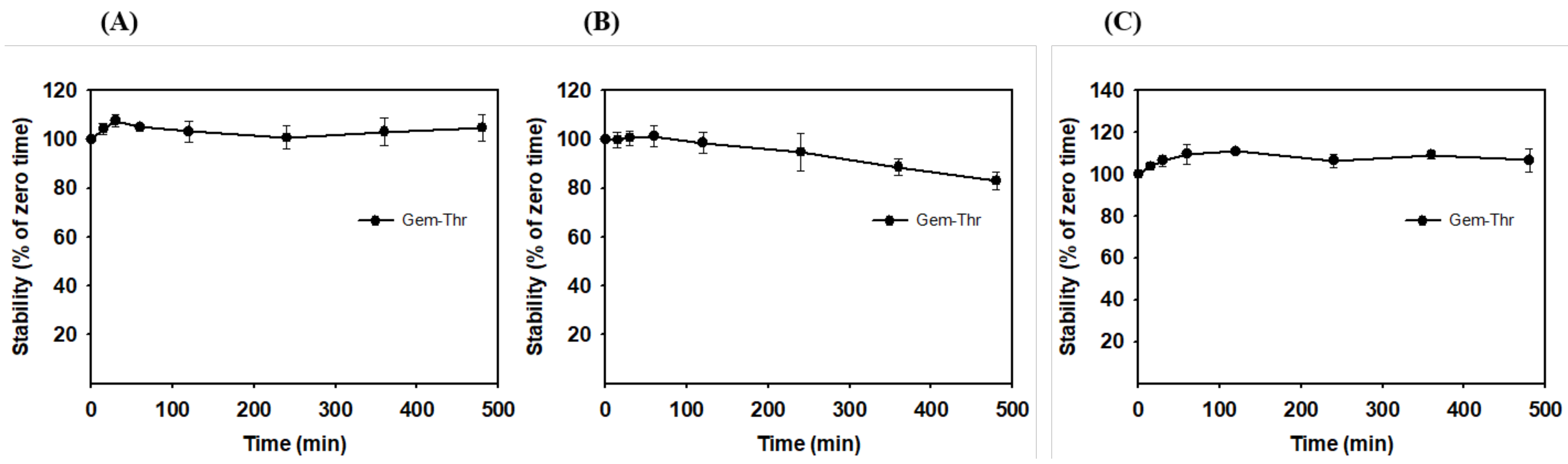
2.4. In Vitro Plasma Stability of Gem-Thr
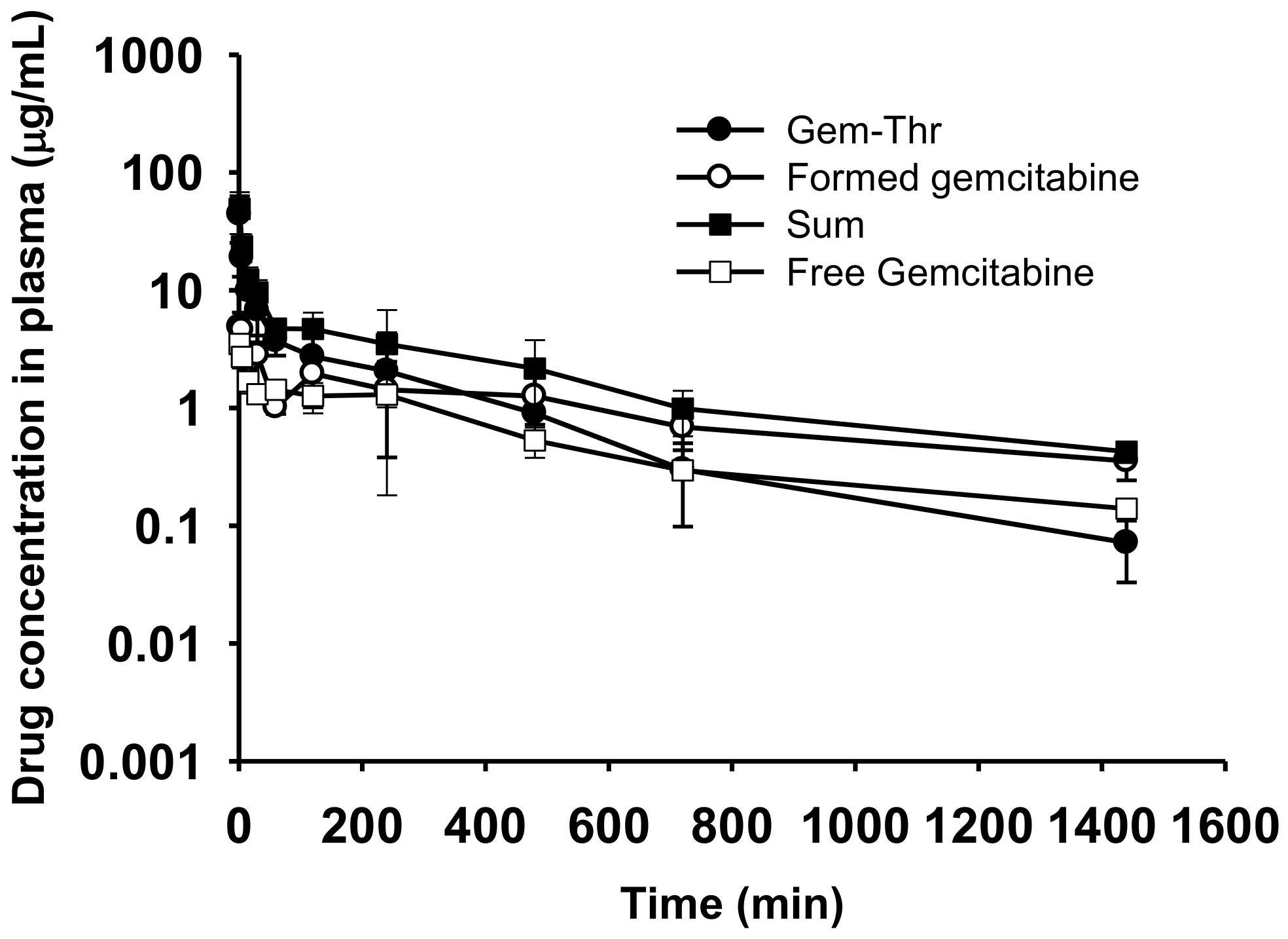
2.5. Comparison of Systemic Pharmacokinetics with Free Gemcitabine
3. Materials and Methods
3.1. Materials
3.2. Synthesis and Characterization of DOX-Val
3.2.1. General Procedure for Preparing Gemcitabine Derivatives
3.2.2. (S)-2-Amino-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)-3-methylbutanamide (Gem-Val)
3.2.3. (2S,3R)-2-Amino-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxylmethyl)tetra-hydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)-3-hydroxybutanamide2-(4-((pyridin-3-ylmethyl)amino)quinazolin-2-yl)phenol (Gem-Thr)
3.2.4. (S)-2-Amino-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)-3-(4-hydroxyphenyl)propanamide (Gem-Tyr)
3.2.5. (S)-2-Amino-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)-4-(methylthio)butanamide (Gem-Met)
3.2.6. (2S,3S)-2-Amino-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)-3-methylpentanamide (Gem-Ile)
3.2.7. (S)-2-Amino-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)-4-methylpentanamide (Gem-Leu)
3.3. Characterization of Gemcitabine Prodrugs with Amino Acid
3.3.1. Cell Culture
3.3.2. Reverse Transcription-PCR
3.3.3. Western Blot Assays
3.3.4. Cytotoxicity Assay in Pancreatic Cells
3.3.5. Terminal Deoxynucleotidyl Transferase–Mediated Nick End Labeling (TUNEL) Assay
3.4. In Vitro Metabolic Stability of Gem-Thr
3.5. Systemic Pharmacokinetics Study of Gem-Thr in Rats
3.6. Analysis of Gem-Thr and Gemcitabine by LC-MS/MS
3.7. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vitellius, C.; Fizanne, L.; Menager-Tabourel, E.; Nader, J.; Baize, N.; Laly, M.; Lermite, E.; Bertrais, S.; Caroli-Bosc, F.X. The combination of everolimus and zoledronic acid increase the efficacy of gemcitabine in a mouse model of pancreatic adenocarcinoma. Oncotarget 2018, 9, 28069–28082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Suzuki, E.; Mikata, R.; Yasui, S.; Abe, M.; Iino, Y.; Ohyama, H.; Chiba, T.; Tsuyuguchi, T.; Kato, N. Five Cases of Interstitial Pneumonitis Due to Gemcitabine and Nab-Paclitaxel Combination Treatment in Pancreatic Cancer Patients. Pancreas 2018, 47, e42–e43. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Robertson, J.M.; Ye, H.; Margolis, J.; Nadeau, L.; Yan, D. Dose-volume analysis of predictors for gastrointestinal toxicity after concurrent full-dose gemcitabine and radiotherapy for locally advanced pancreatic adenocarcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Bender, D.M.; Bao, J.; Dantzig, A.H.; Diseroad, W.D.; Law, K.L.; Magnus, N.A.; Peterson, J.A.; Perkins, E.J.; Pu, Y.J.; Reutzel-Edens, S.M.; et al. Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J. Med. Chem. 2009, 52, 6958–6961. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.H.; Eiseman, J.L.; Parise, R.A.; Joseph, E.; Covey, J.M.; Egorin, M.J. Modulation of gemcitabine (2′,2′-difluoro-2′-deoxycytidine) pharmacokinetics, metabolism, and bioavailability in mice by 3,4,5,6-tetrahydrouridine. Clin. Cancer Res. 2008, 14, 3529–3535. [Google Scholar] [CrossRef] [PubMed]
- Wickremsinhe, E.; Bao, J.; Smith, R.; Burton, R.; Dow, S.; Perkins, E. Preclinical absorption, distribution, metabolism, and excretion of an oral amide prodrug of gemcitabine designed to deliver prolonged systemic exposure. Pharmaceutics 2013, 5, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, Y.; Wen, X.; Ma, H. Current prodrug strategies, for improving oral absorption of nucleoside analogues. Asian J. Pharm. Sci. 2014, 9, 65–74. [Google Scholar] [CrossRef]
- Tsume, Y.; Incecayir, T.; Song, X.; Hilfinger, J.M.; Amidon, G.L. The development of orally administrable gemcitabine prodrugs with D-enantiomer amino acids: Enhanced membrane permeability and enzymatic stability. Eur. J. Pharm. Biopharm. 2014, 86, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Lorenzi, P.L.; Landowski, C.P.; Vig, B.S.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrugs of the anticancer agent gemcitabine: Synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport. Mol. Pharm. 2005, 2, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Bender, D.M.; Victor, F.; Peterson, J.A.; Boyer, R.D.; Stephenson, G.A.; Azman, A.; McCarthy, J.R. Facile rearrangement of N4-(α-aminoacyl)cytidines to N-(4-cytidinyl)amino acid amides. Tetrahedron Lett. 2008, 49, 2052–2055. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.; Zhao, D.; Ding, D.; Sun, M.; Kou, L.; Luo, C.; Zhang, D.; Yi, X.; Dong, J.; et al. Combination of l-carnitine with lipophilic linkage-donating gemcitabine derivatives as intestinal novel organic cation transporter 2-targeting oral prodrugs. J. Med. Chem. 2017, 60, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.E.; Jin, H.E.; Hong, S.S. Targeting L-type amino acid transporter 1 for anticancer therapy: Clinical impact from diagnostics to therapeutics. Expert Opin. Ther. Targets 2015, 19, 1319–1337. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Sunose, Y.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; Itoh, H.; Nagamori, S.; et al. Prognostic significance of L-type amino acid transporter 1 expression in surgically resected pancreatic cancer. Br. J. Cancer 2012, 107, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, N.; Ichinoe, M.; Mikami, T.; Nakada, N.; Hana, K.; Koizumi, W.; Endou, H.; Okayasu, I. High expression of L-type amino acid transporter 1 (LAT1) predicts poor prognosis in pancreatic ductal adenocarcinomas. J. Clin. Pathol. 2012, 65, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.Y.; Shim, W.S.; Chang, J.E.; Chong, S.; Kim, D.D.; Chung, S.J.; Shim, C.K. Enhanced intracellular accumulation of a non-nucleoside anti-cancer agent via increased uptake of its valine ester prodrug through amino acid transporters. Xenobiotica 2012, 42, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Maeng, H.J.; Kim, E.S.; Chough, C.; Joung, M.; Lim, J.W.; Shim, C.K.; Shim, W.S. Addition of amino acid moieties to lapatinib increases the anticancer effect via amino acid transporters. Biopharm. Drug Dispos. 2014, 35, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Park, J.H.; Park, S.; Lee, S.Y.; Cho, K.H.; Kim, D.D.; Shim, W.S.; Yoon, I.S.; Cho, H.J.; Maeng, H.J. Enhanced Cellular Uptake and Pharmacokinetic Characteristics of Doxorubicin-Valine Amide Prodrug. Molecules 2016, 21, 1272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, Y.; Qin, Y.; Wang, R.; Li, G.; Sun, C.; Qu, X.; Li, W. Pharmacokinetics and metabolism of SL-01, a prodrug of gemcitabine, in rats. Cancer Chemother. Pharmacol. 2013, 71, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Son, M.K.; Jung, K.H.; Lee, H.S.; Lee, H.; Kim, S.J.; Yan, H.H.; Ryu, Y.L.; Hong, S.S. SB365, Pulsatilla saponin D suppresses proliferation and induces apoptosis of pancreatic cancer cells. Oncol. Rep. 2013, 30, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.S.; Roh, S.H.; Kim, D.K.; Lee, J.G.; Lee, Y.Y.; Hong, S.S.; Kwon, S.W.; Park, J.H. Anti-cancer effect of Betulin on a human lung cancer cell line: A pharmacoproteomic approach using 2 D SDS PAGE coupled with nano-HPLC tandem Mass Spectrometry. Planta Med. 2009, 75, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.W.; Lin, J.; Hong, X.Y. Cyclin A2 regulates homologous recombination DNA repair and sensitivity to DNA damaging agents and poly (ADP-ribose) polymerase (PARP) inhibitors in human breast cancer cells. Oncotarget 2017, 24, 90842–90851. [Google Scholar] [CrossRef] [PubMed]
- Hatiboglu, M.A.; Kocyigit, A.; Guler, E.M.; Akdur, K.; Nalli, A.; Karatas, E.; Tuzgen, S. Thymoquinone Induces Apoptosis in B16-F10 Melanoma Cell Through Inhibition of p-STAT3 and Inhibits Tumor Growth in a Murine Intracerebral Melanoma Model. World Neurosurg. 2018, 114, e182–e190. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.M.; Jung, K.H.; Lee, H.; Son, M.K.; Seo, J.H.; Yan, H.H.; Park, B.H.; Hong, S.; Hong, S.S. Synergistic anticancer activity of HS-173, a novel PI3K inhibitor in combination with Sorafenib against pancreatic cancer cells. Cancer Lett. 2013, 331, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Kim, I.B.; Noh, C.K.; Quach, H.P.; Yoon, I.S.; Chow, E.C.Y.; Kim, M.; Jin, H.E.; Cho, K.H.; Chung, S.J.; et al. Effects of 1α,25-dihydroxyvitamin D3, the natural vitamin D receptor ligand, on the pharmacokinetics of cefdinir and cefadroxil, organic anion transporter substrates, in rat. J. Pharm. Sci. 2014, 103, 3793–3805. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic Parameters | Gem-Thr (4 mg/kg) | Free Gemcitabine (4 mg/kg) | ||
|---|---|---|---|---|
| Gem-Thr | Gemcitabine | Sum | ||
| AUC (μg∙min/mL) | 1713.85 ± 1082.40 | 1739.88 ± 282.00 * | 3437.92 ± 1180.56 | 948.38 ± 52.04 |
| Terminal t1/2 (min) | 236.18 ± 50.94 | 666.83 ± 271.49 | 537.23 ± 227.78 | 532.68 ± 177.90 |
| CL (mL/min/kg) | 2.85 ± 1.33 | 0.60 ± 0.10 * | 1.26 ± 0.39 | 4.23 ± 0.23 |
| Vss (mL/kg) | 662.35 ± 281.40 | 545.57 ± 263.01 * | 770.96 ± 435.31 | 2483.64 ± 867.19 |
| MRT (min) | 237.18 ± 20.87 | 907.18 ± 391.68 * | 577.36 ± 212.90 | 582.06 ± 177.90 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.; Fang, Z.; Jung, H.-Y.; Yoon, J.-H.; Hong, S.-S.; Maeng, H.-J. Synthesis of Gemcitabine-Threonine Amide Prodrug Effective on Pancreatic Cancer Cells with Improved Pharmacokinetic Properties. Molecules 2018, 23, 2608. https://doi.org/10.3390/molecules23102608
Hong S, Fang Z, Jung H-Y, Yoon J-H, Hong S-S, Maeng H-J. Synthesis of Gemcitabine-Threonine Amide Prodrug Effective on Pancreatic Cancer Cells with Improved Pharmacokinetic Properties. Molecules. 2018; 23(10):2608. https://doi.org/10.3390/molecules23102608
Chicago/Turabian StyleHong, Sungwoo, Zhenghuan Fang, Hoi-Yun Jung, Jin-Ha Yoon, Soon-Sun Hong, and Han-Joo Maeng. 2018. "Synthesis of Gemcitabine-Threonine Amide Prodrug Effective on Pancreatic Cancer Cells with Improved Pharmacokinetic Properties" Molecules 23, no. 10: 2608. https://doi.org/10.3390/molecules23102608
APA StyleHong, S., Fang, Z., Jung, H. -Y., Yoon, J. -H., Hong, S. -S., & Maeng, H. -J. (2018). Synthesis of Gemcitabine-Threonine Amide Prodrug Effective on Pancreatic Cancer Cells with Improved Pharmacokinetic Properties. Molecules, 23(10), 2608. https://doi.org/10.3390/molecules23102608