
Amide Bond Activation of Biological Molecules

Abstract
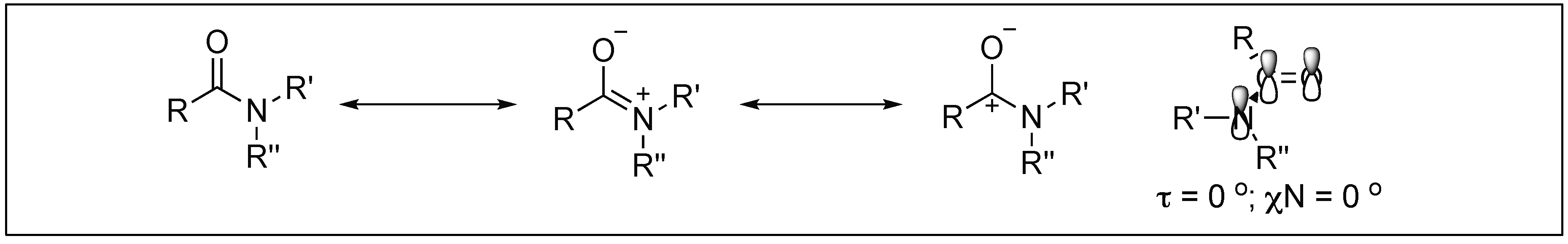
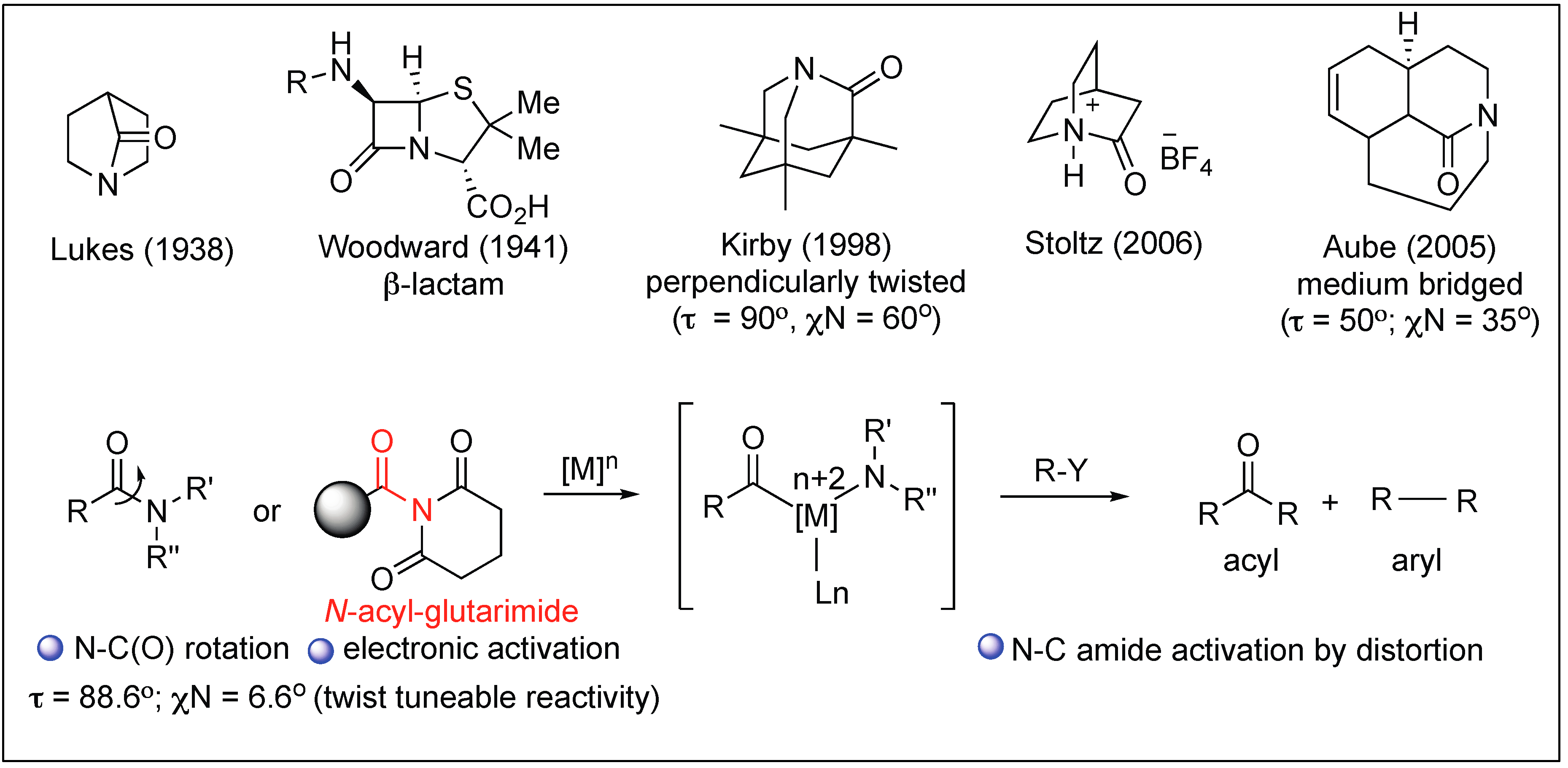
:1. Introduction
2. Biomolecules for the Activation of Amide Bonds—The Enzyme-Directed Hydrolysis of Amides
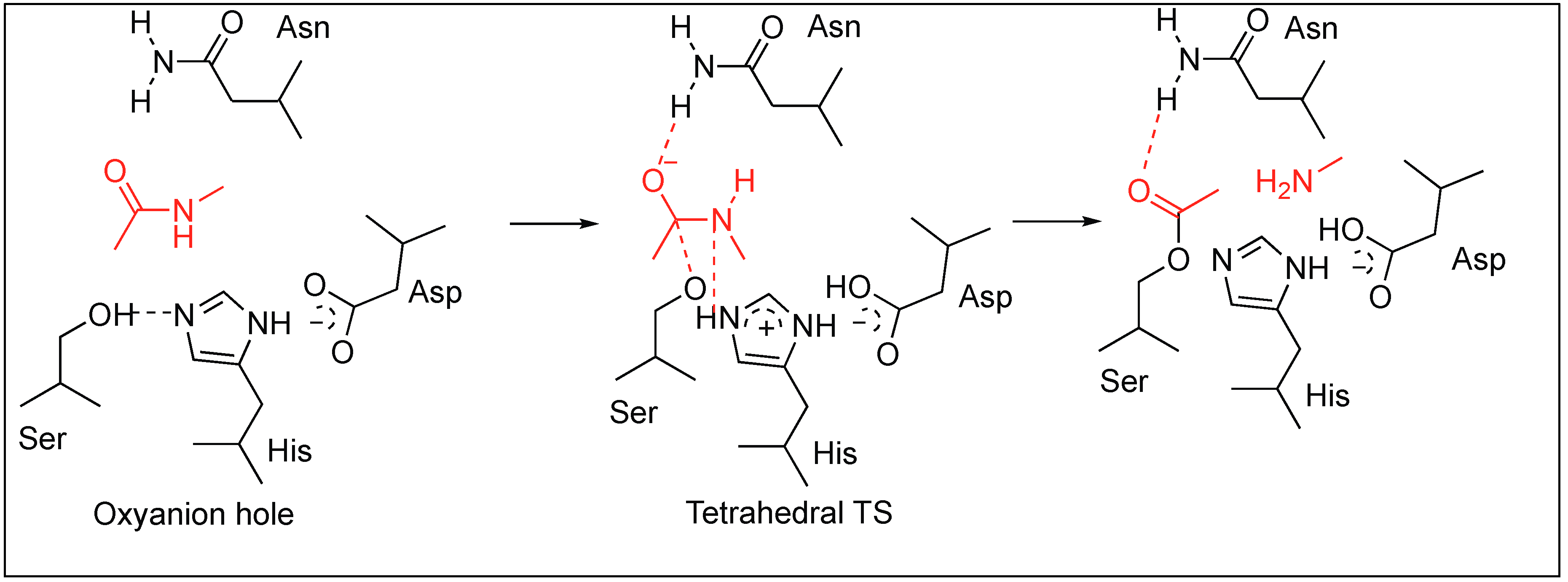
2.1. Serine Proteases
2.2. Cysteine Proteases
2.3. Metalloproteases
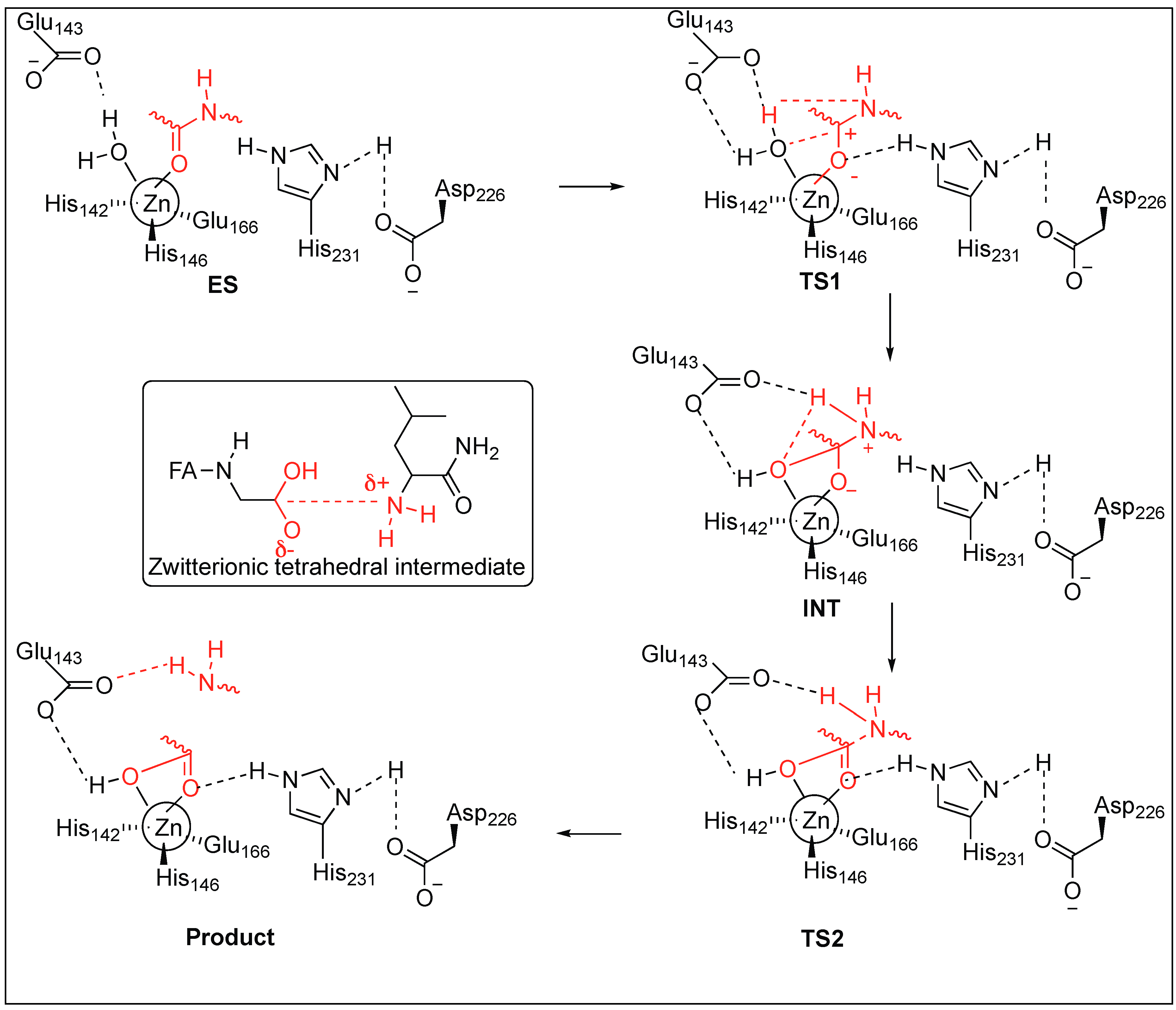
2.3.1. Metalloendopeptidase: Thermolysin
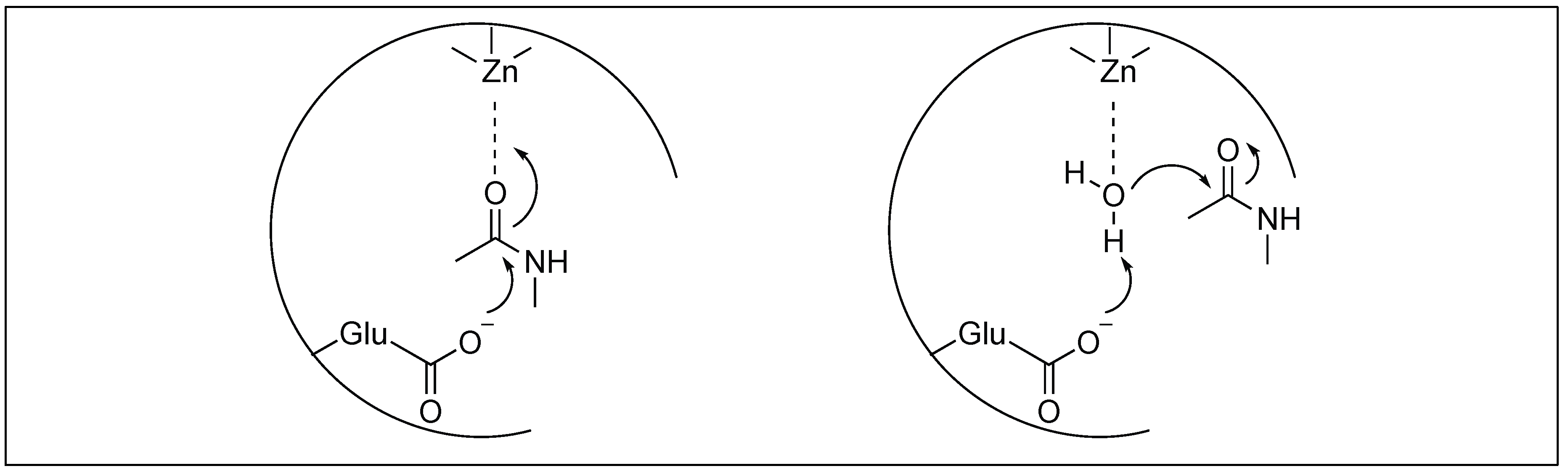
2.3.2. Metalloexopeptidase: Carboxypeptidase A
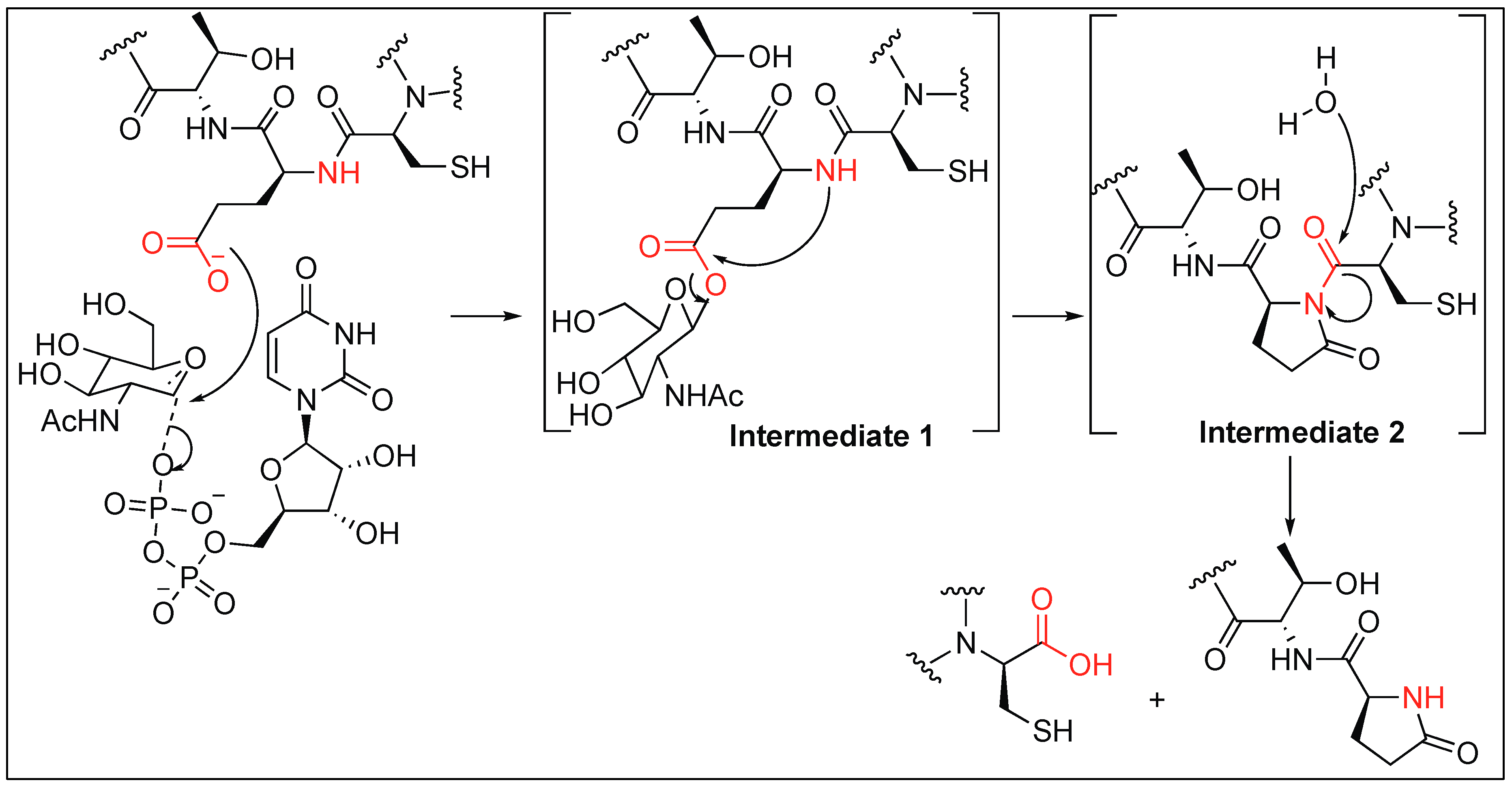
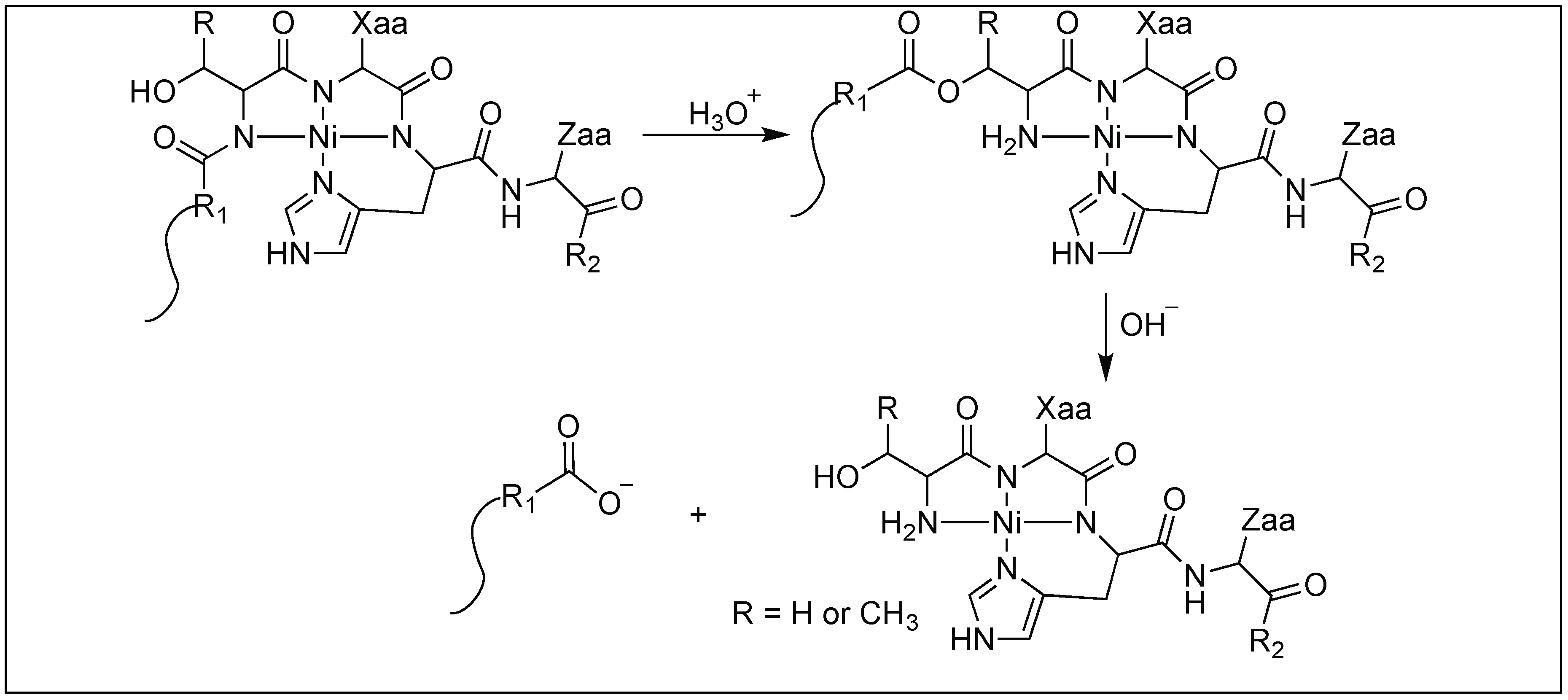
2.3.3. Glutamate Glycosylation for Amide Bond Cleavage
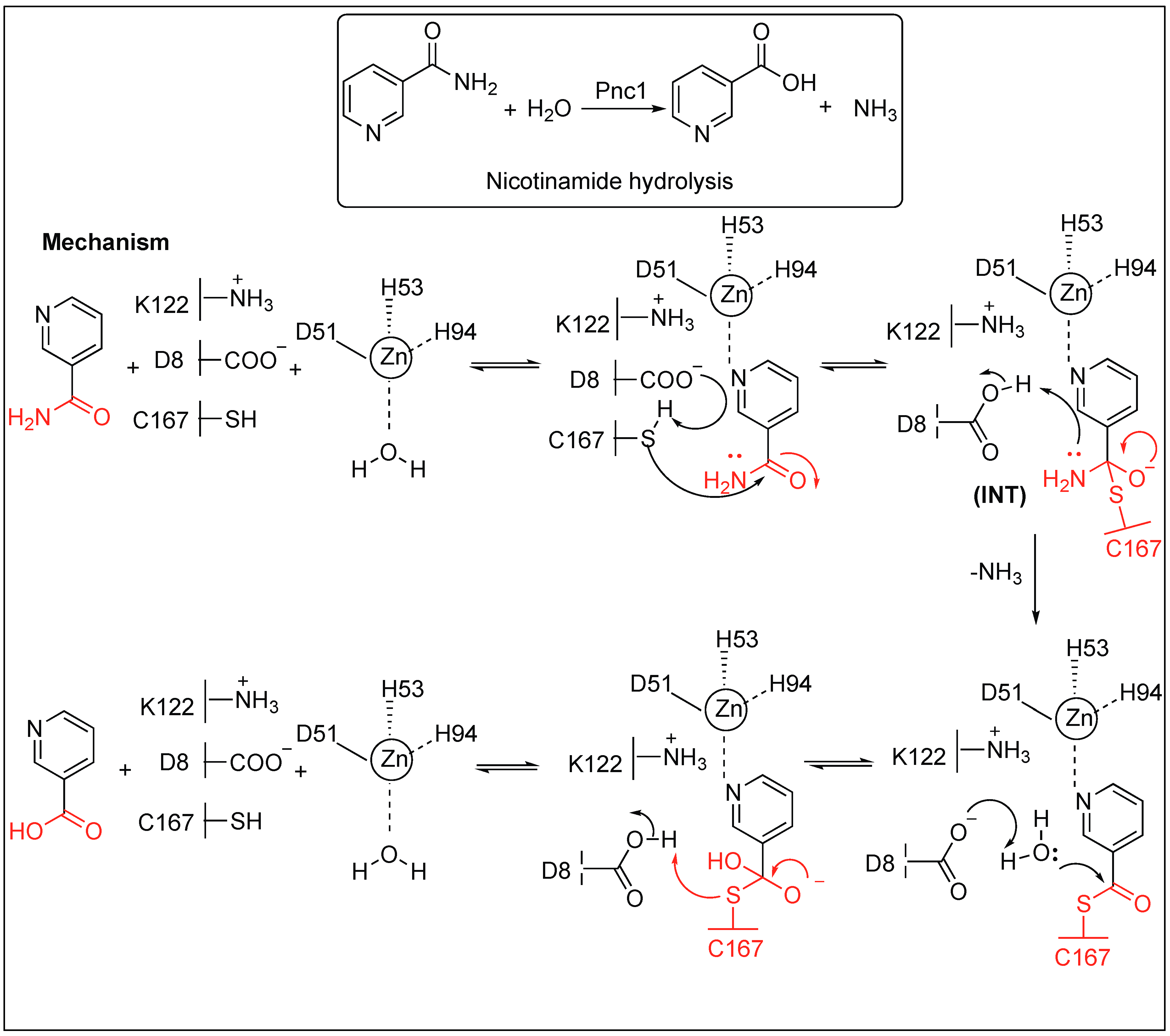
2.3.4. Nicotinamidase (Pnc1) for the Hydrolysis of the Amide Bond of Nicotinamide
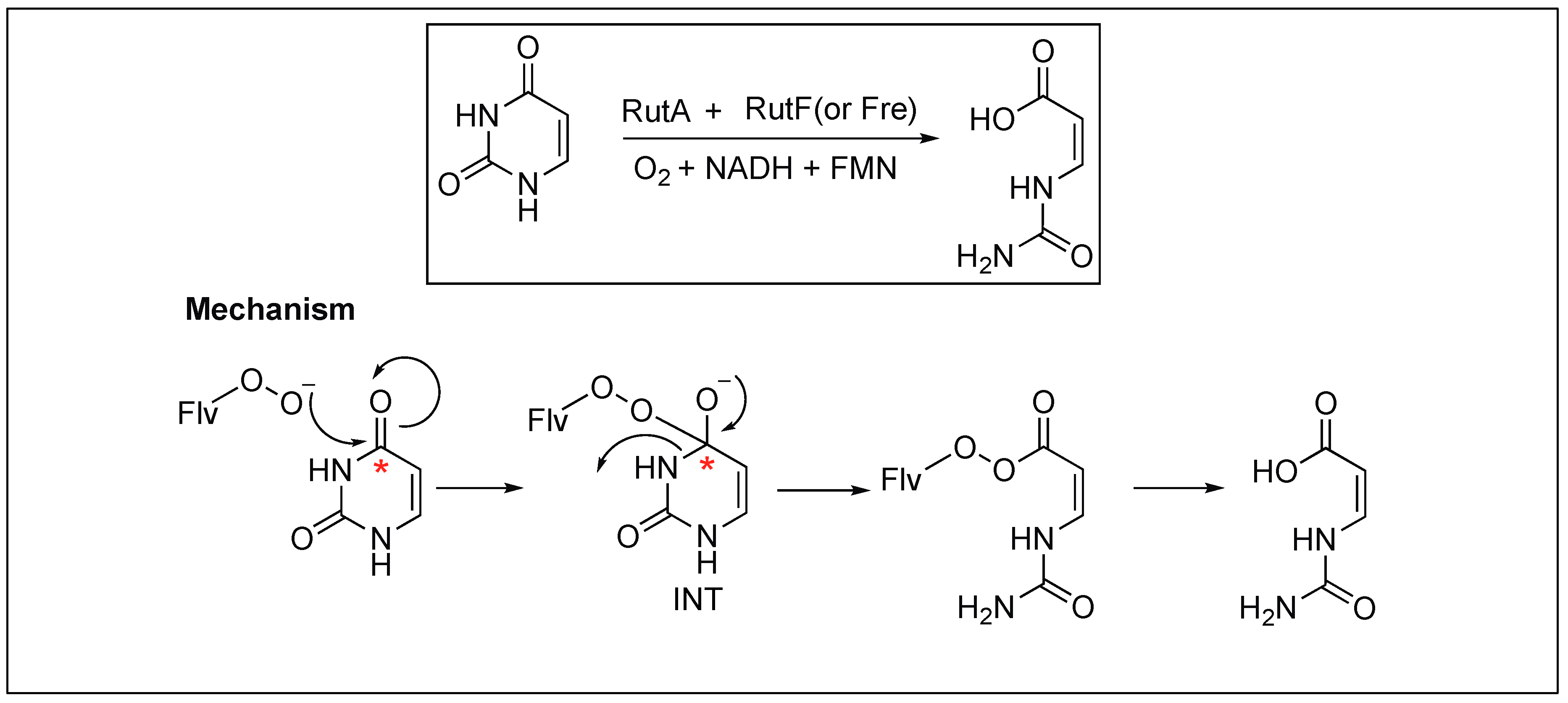
2.3.5. Flavoenzyme-Mediated Hydrolysis of the Amide Bond
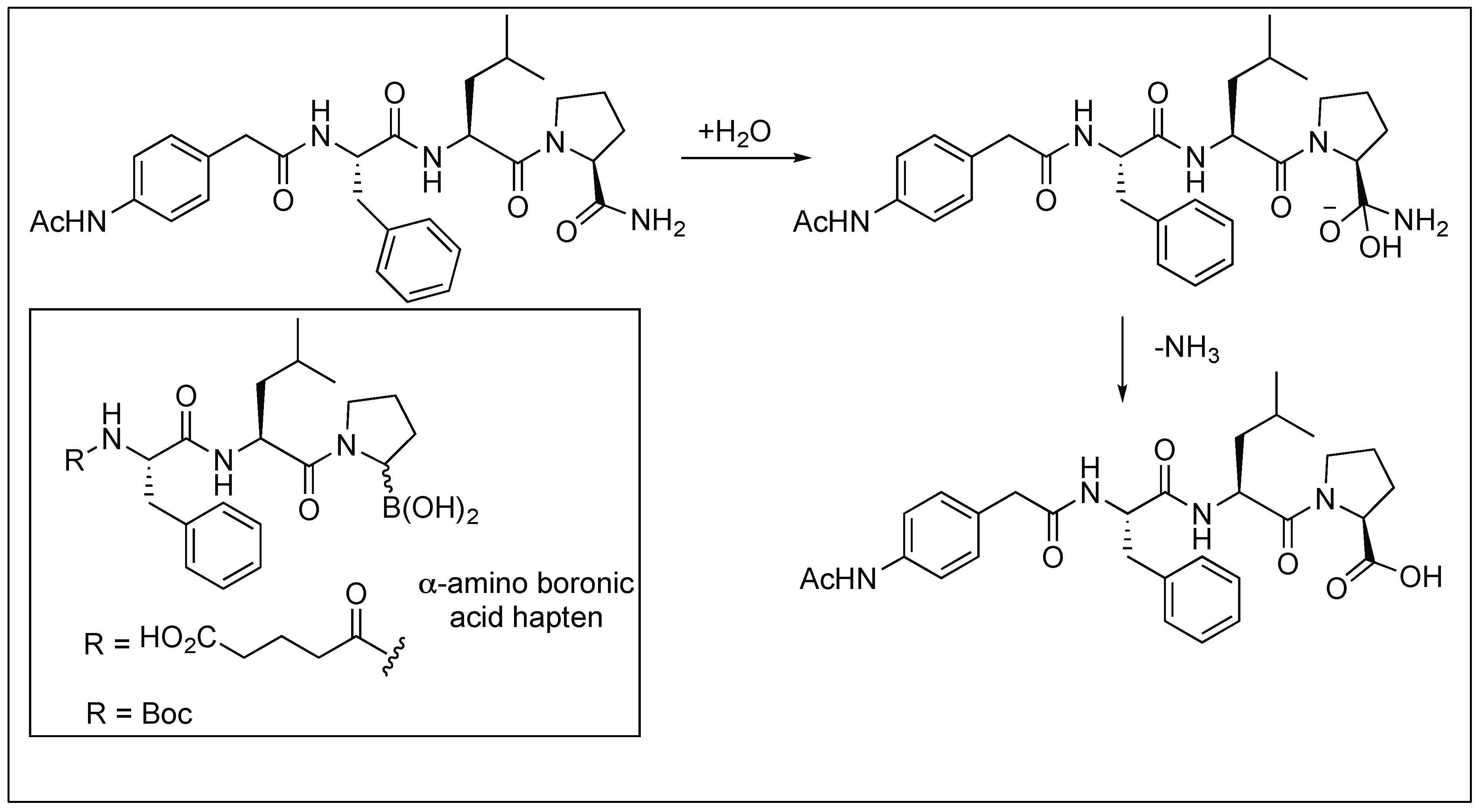
2.3.6. Primary Amide Bond Hydrolysis by Antibodies
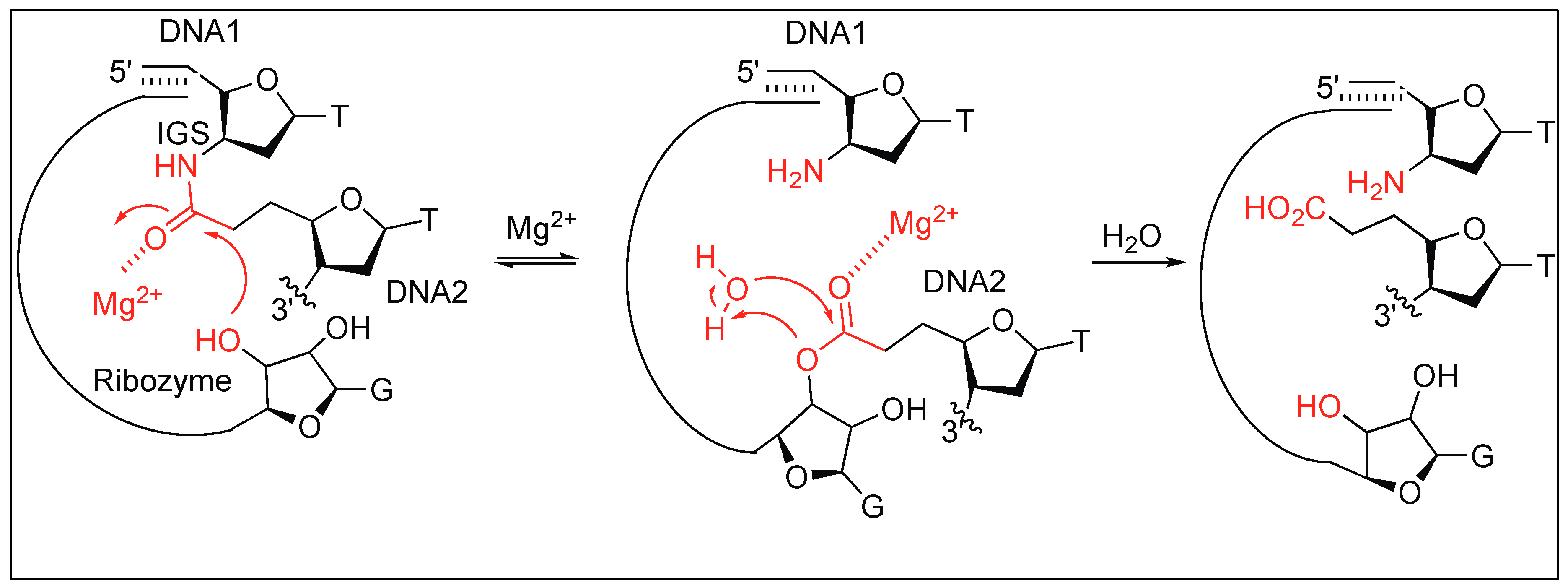
2.3.7. RNA-Assisted Cleavage of Amide Bonds
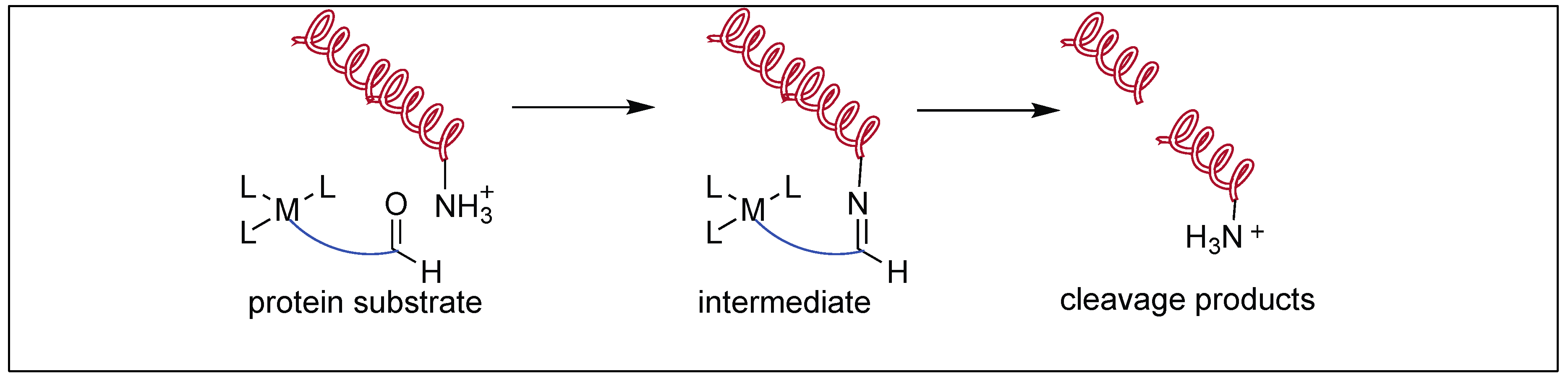
3. Metal Complexes for the Activation of Amide Bonds
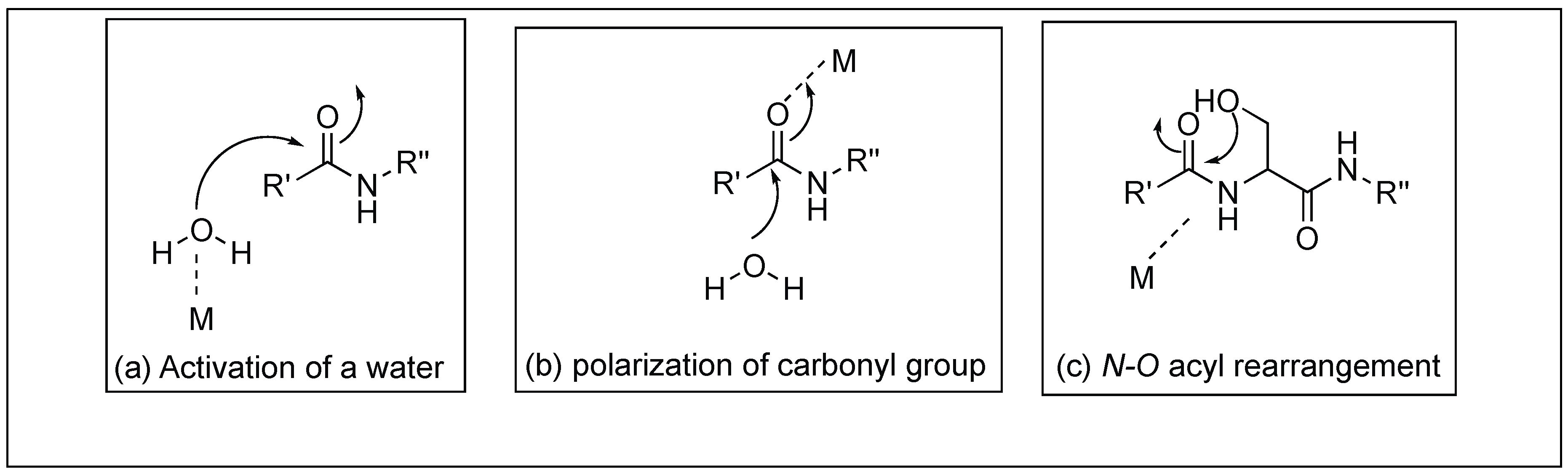
3.1. Lewis Acid Mediated Hydrolysis
3.1.1. Simple Metal Ions
3.1.2. Oxo Metal Ions
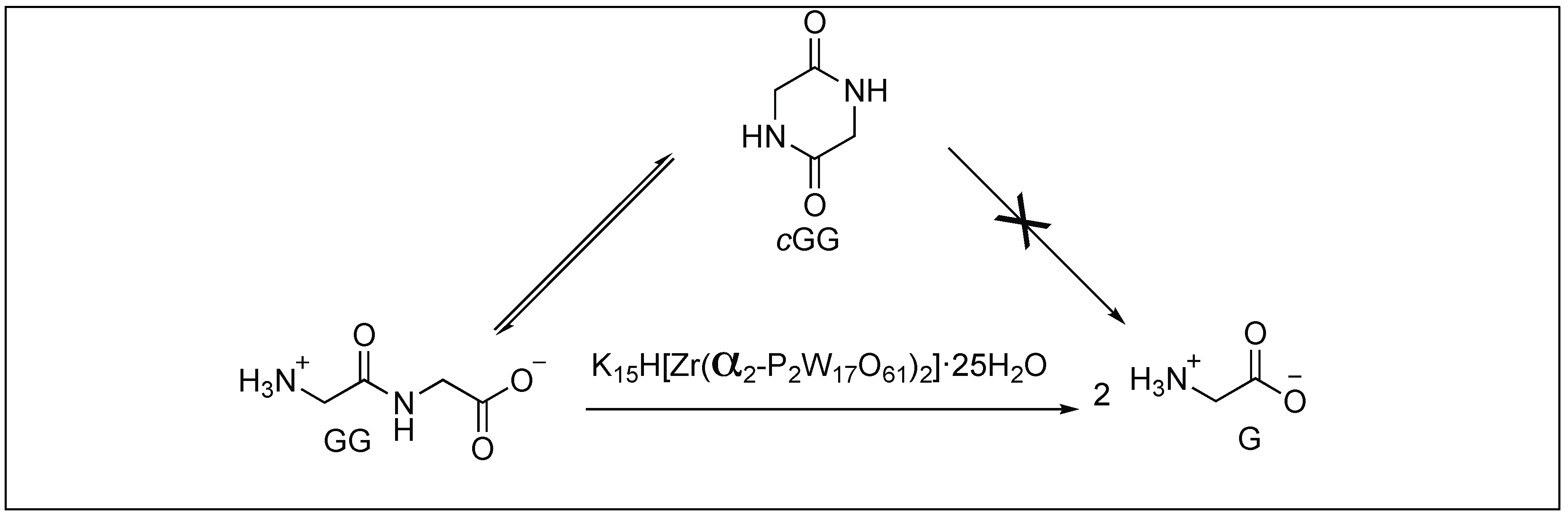
Zirconium Complex Mediated Hydrolysis of Peptide Bonds
Asp-Xaa Selective Hydrolysis of the Peptide Bond by Oxo-Metal Ions
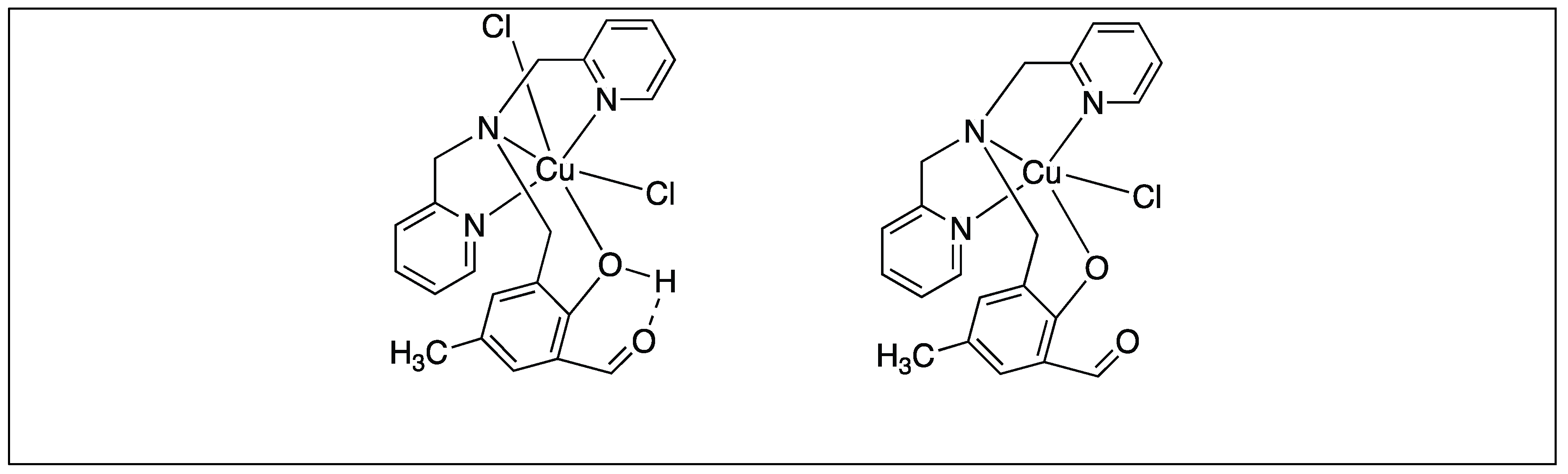
Various Other Metal Complexes
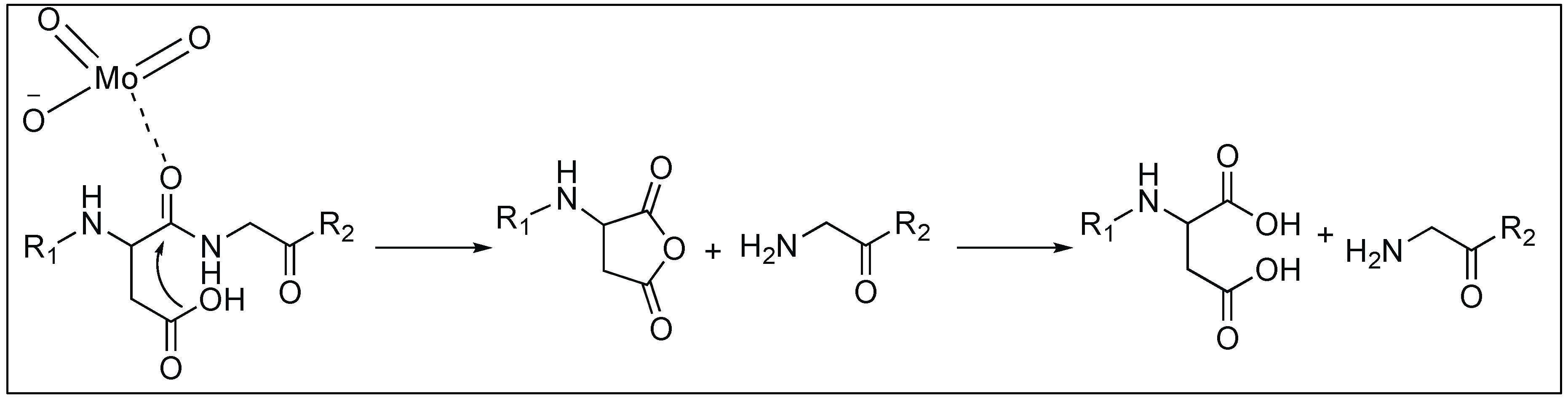
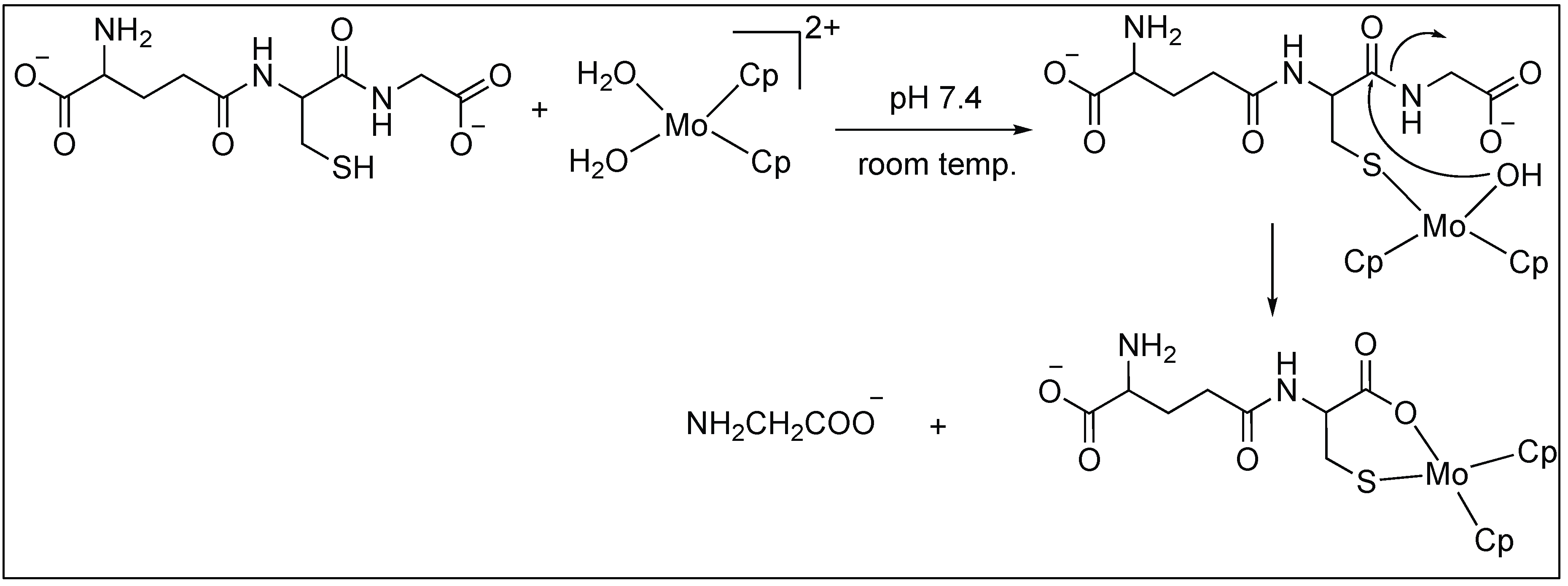
Anchoring at Cys Side Chains: Molybdocene
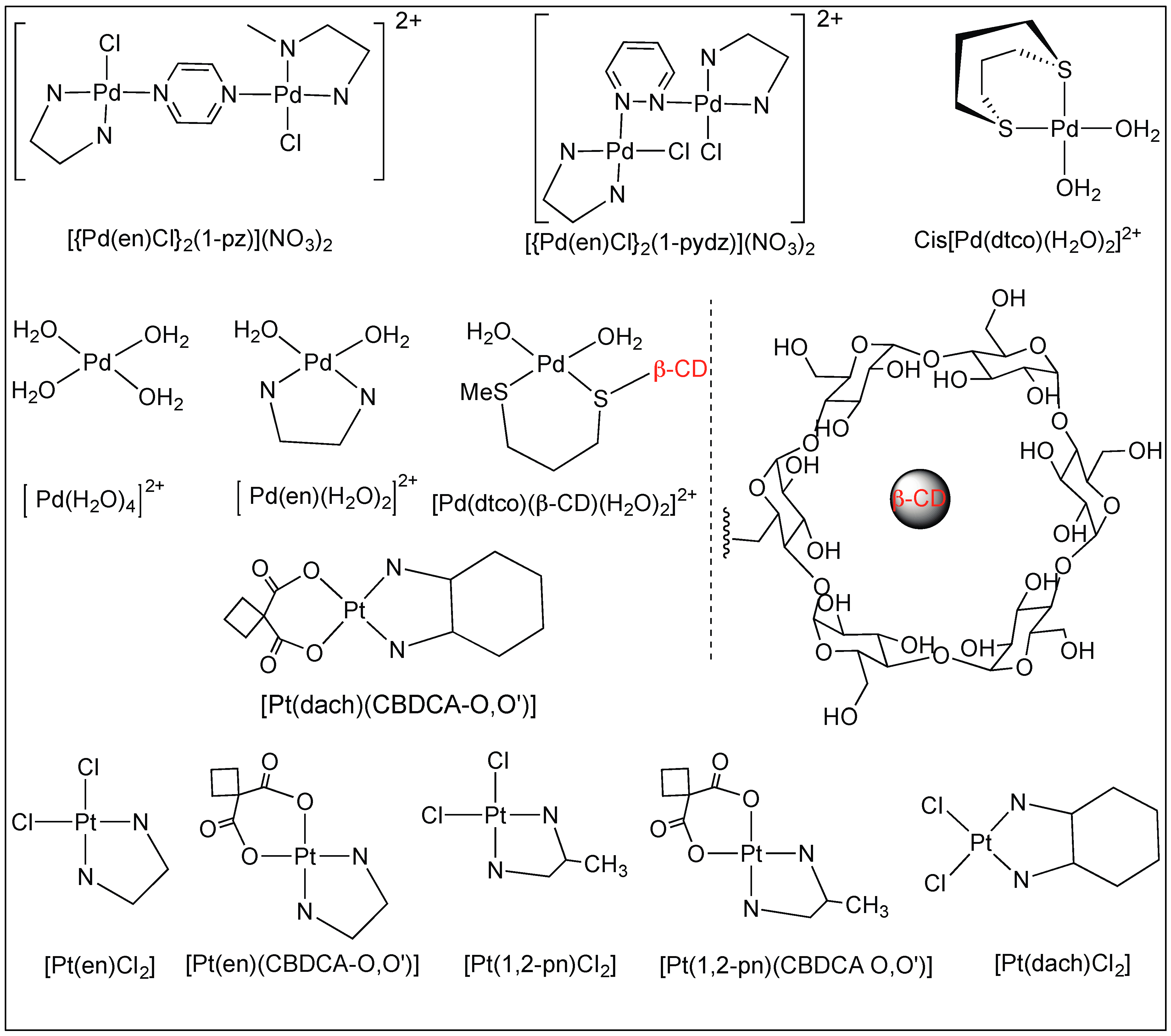
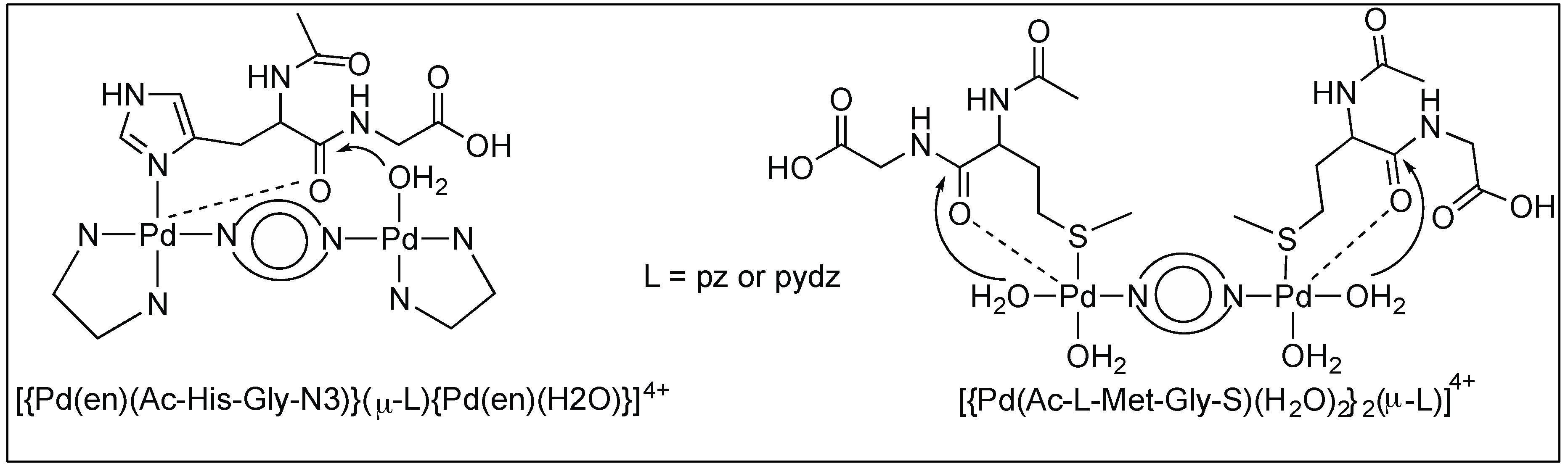
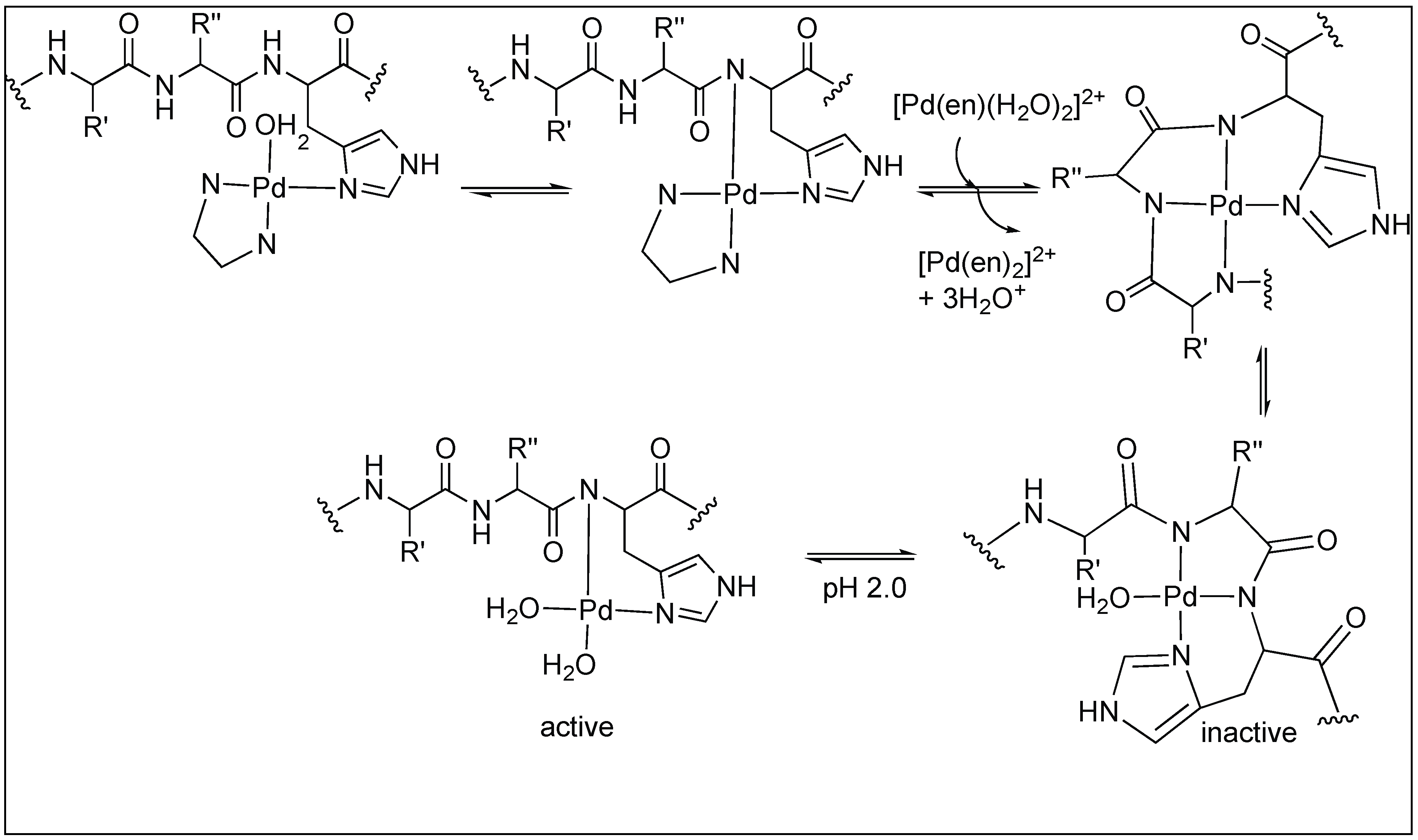
3.1.3. Anchoring at Met, His, and Cys Side Chains: Palladium(II) Complexes for the Cleavage of Amide Bonds
Pyrazine and Pyridazine Palladium(II)-Aqua Dimers
Platinum Complexes for the Cleavage of the Amide Bond
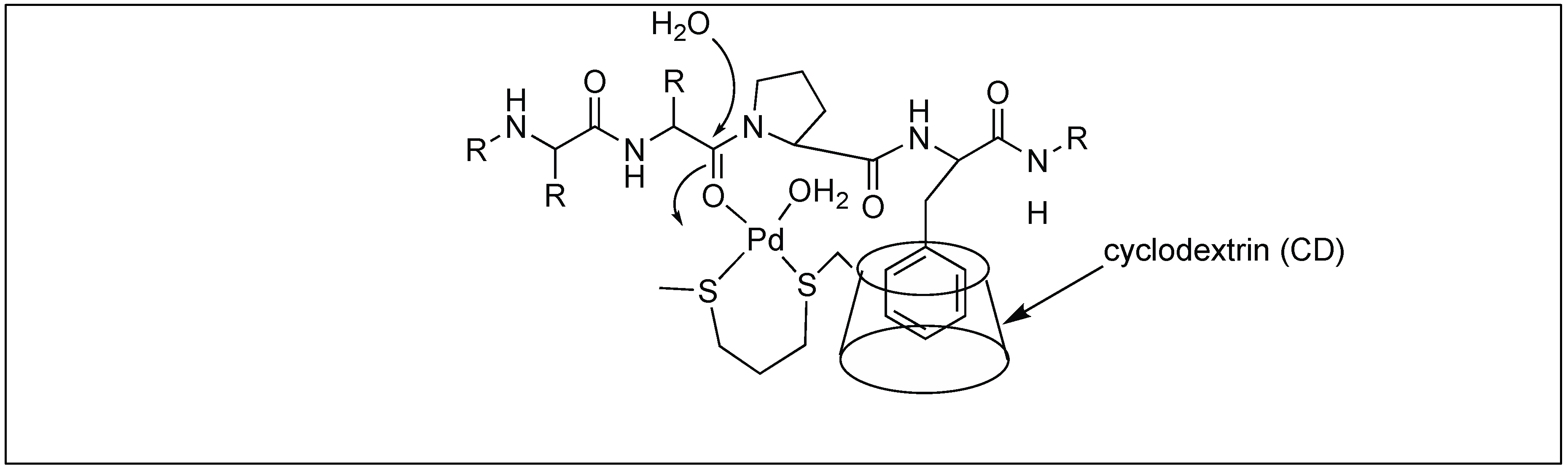
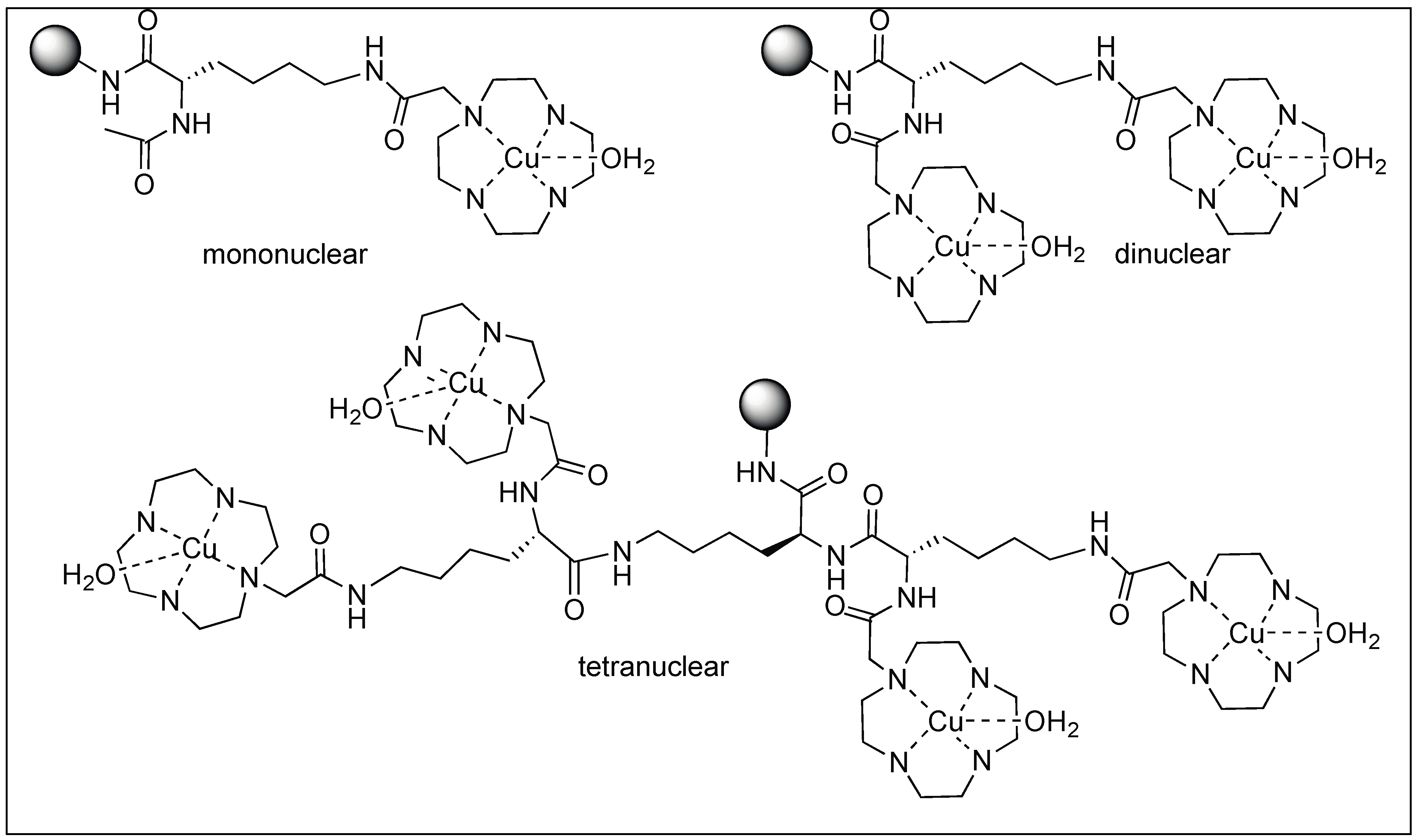
3.1.4. Artificial Metal Proteases
Aldehyde Pendant for the Cleavage of Proteins
Mb-Selective Artificial Protease
PDF-Selective Artificial Protease
AmPs-Selective Artificial Protease
3.2. The Non-Lewis Acid Reaction Mechanisms Based on the N→O Rearrangement
Scandium(III) Triflate-Promoted Serine/Threonine Selective Peptide Bond Cleavage
4. Organic Molecules for Activation of Amide Bonds
4.1. N-Terminal Cleavage of Amide Bonds
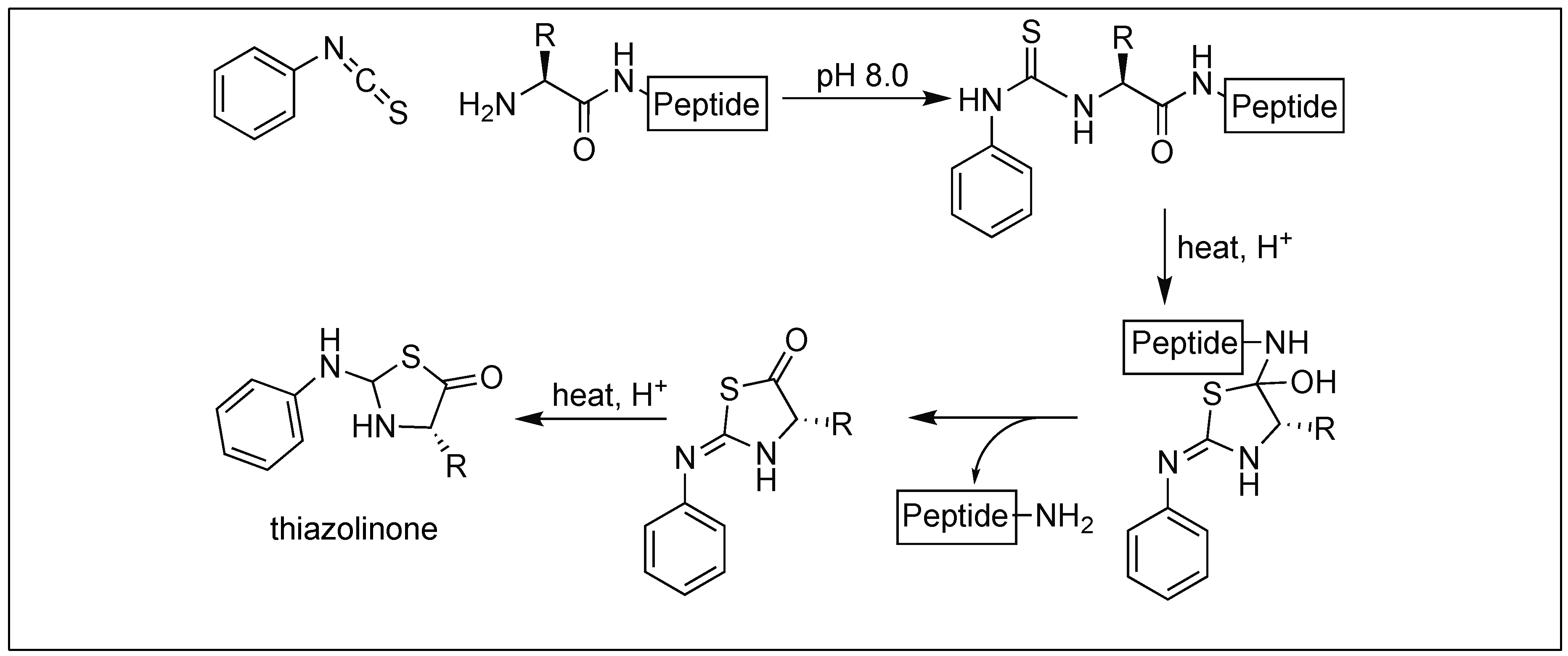
4.1.1. Edman’s Degradation
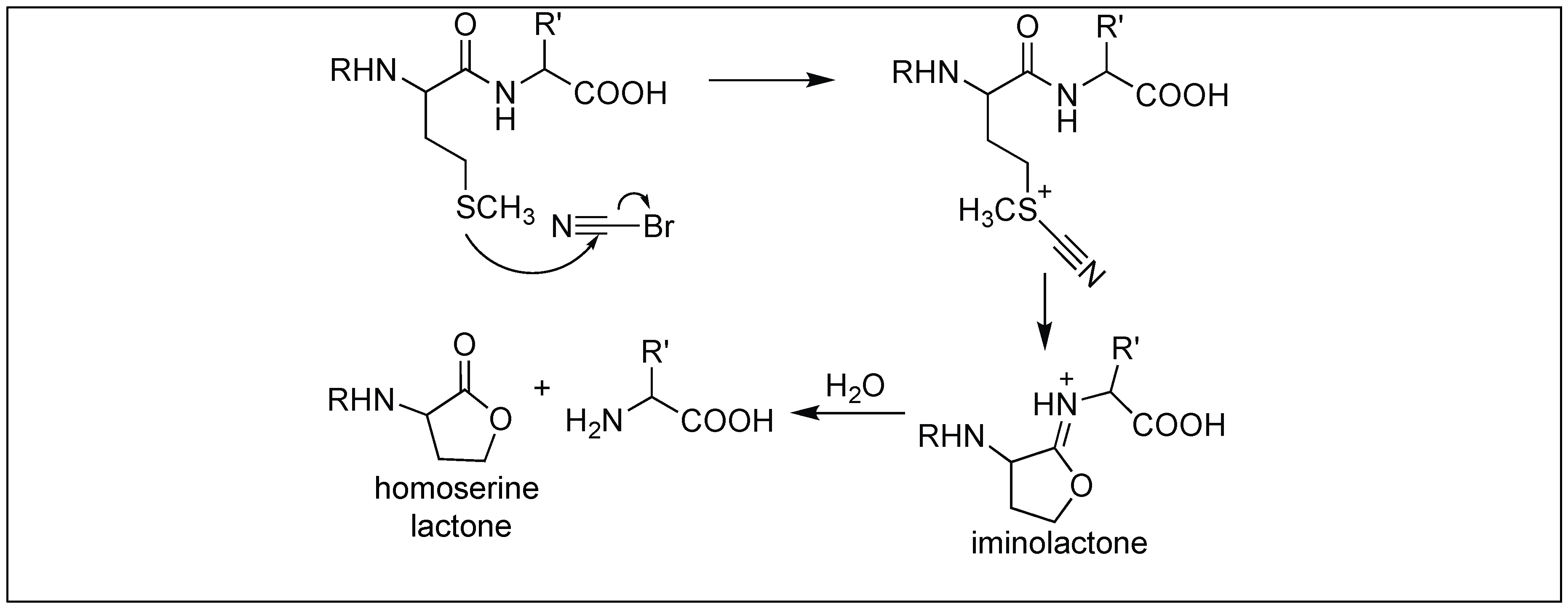
4.1.2. Cyanogen Bromide for Cleavage at Met Residue
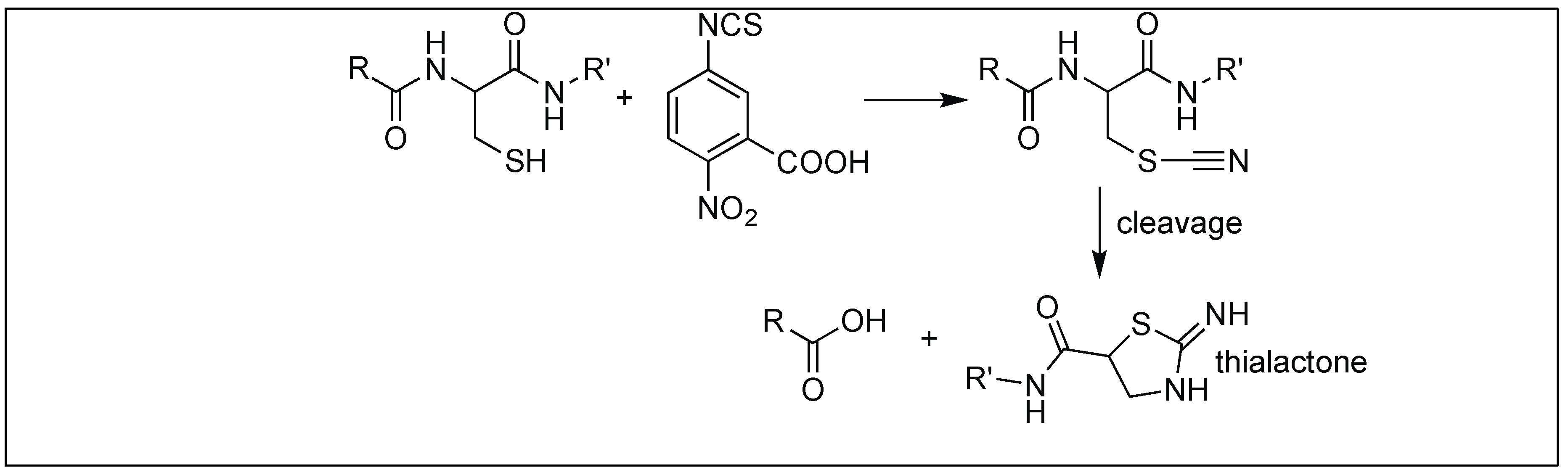
4.1.3. 2-Nitro-5-Thiocyano Benzoic Acid for Cleavage at Cys
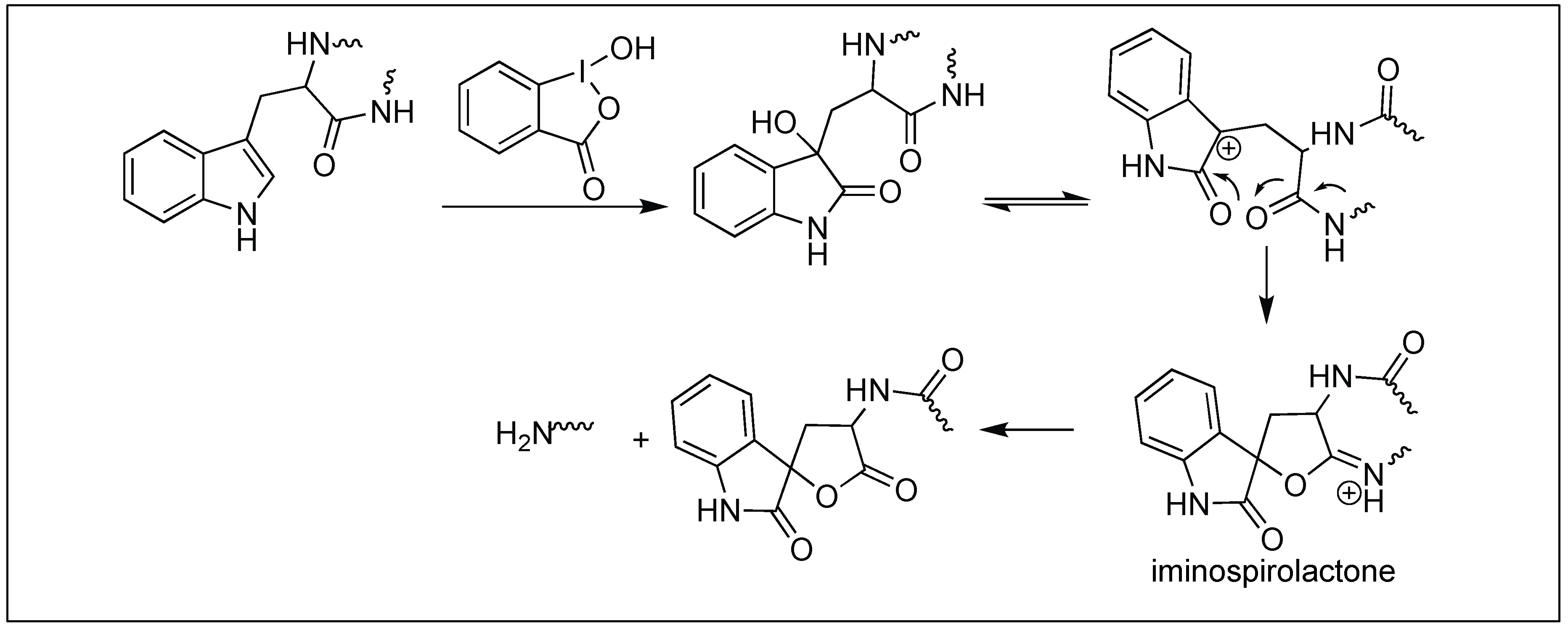
4.1.4. 2-Iodosobenzoic Acid for Cleavage at Trp
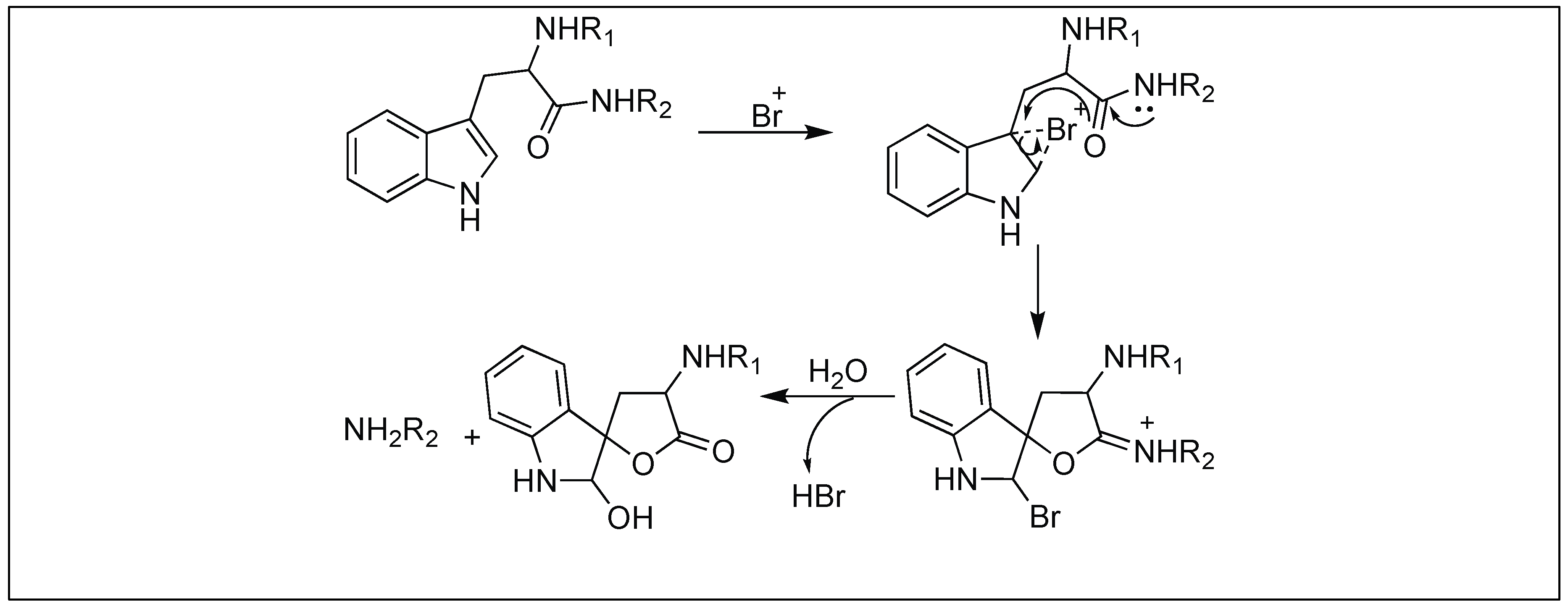
4.1.5. TBC for Cleavage at Trp
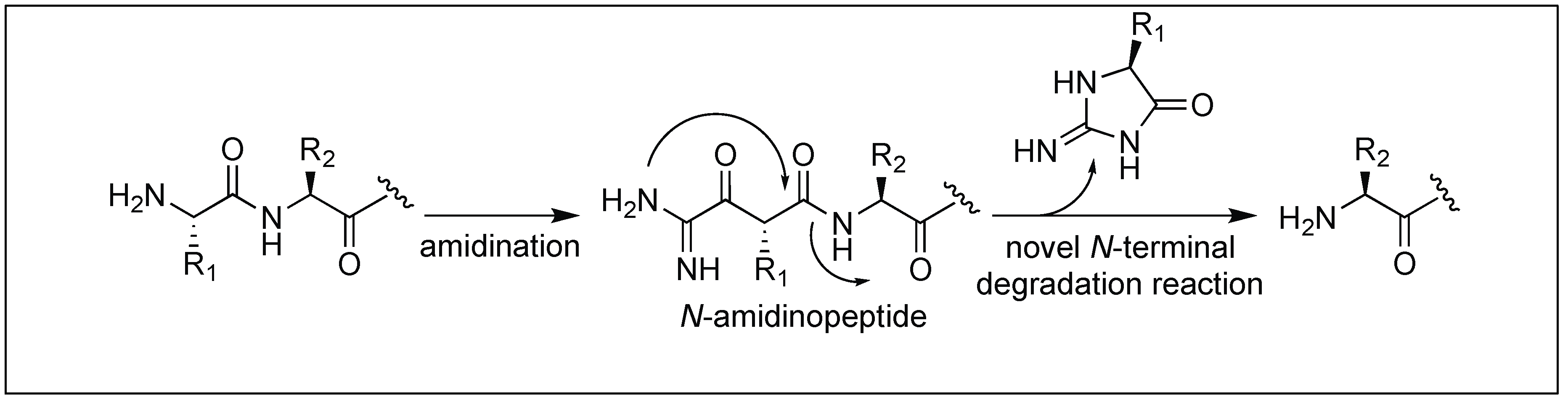
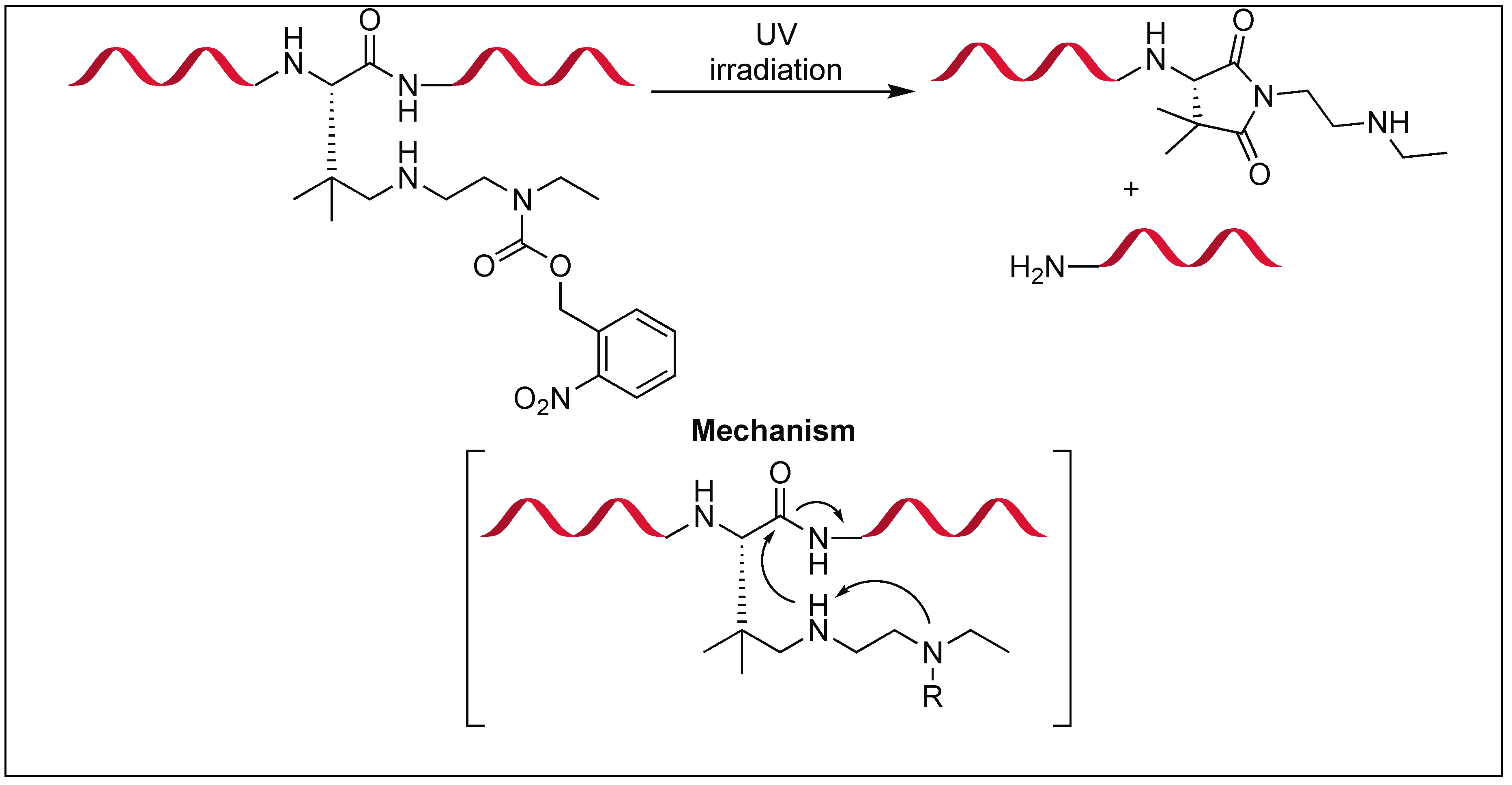
4.2. N-Amidination for Cleavage of the N-Terminal Amide Bond
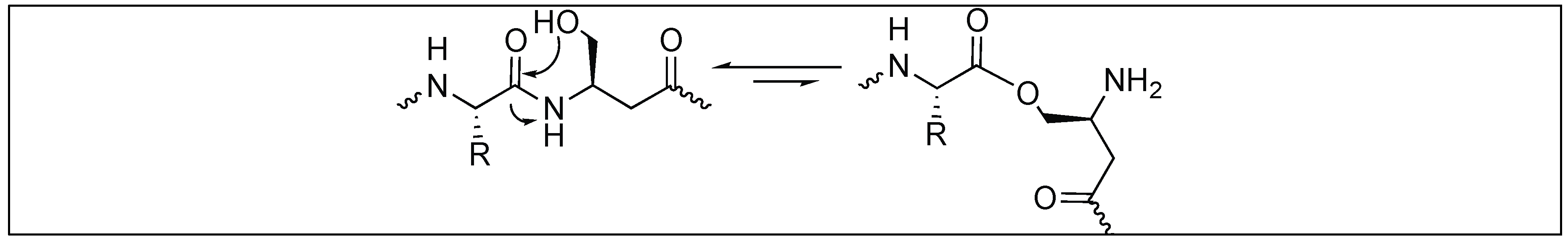
4.3. Lactonization Mediated Cleavage of Amide Bonds
4.4. Hydrogen Peroxide-Induced Amide Bond Cleavage
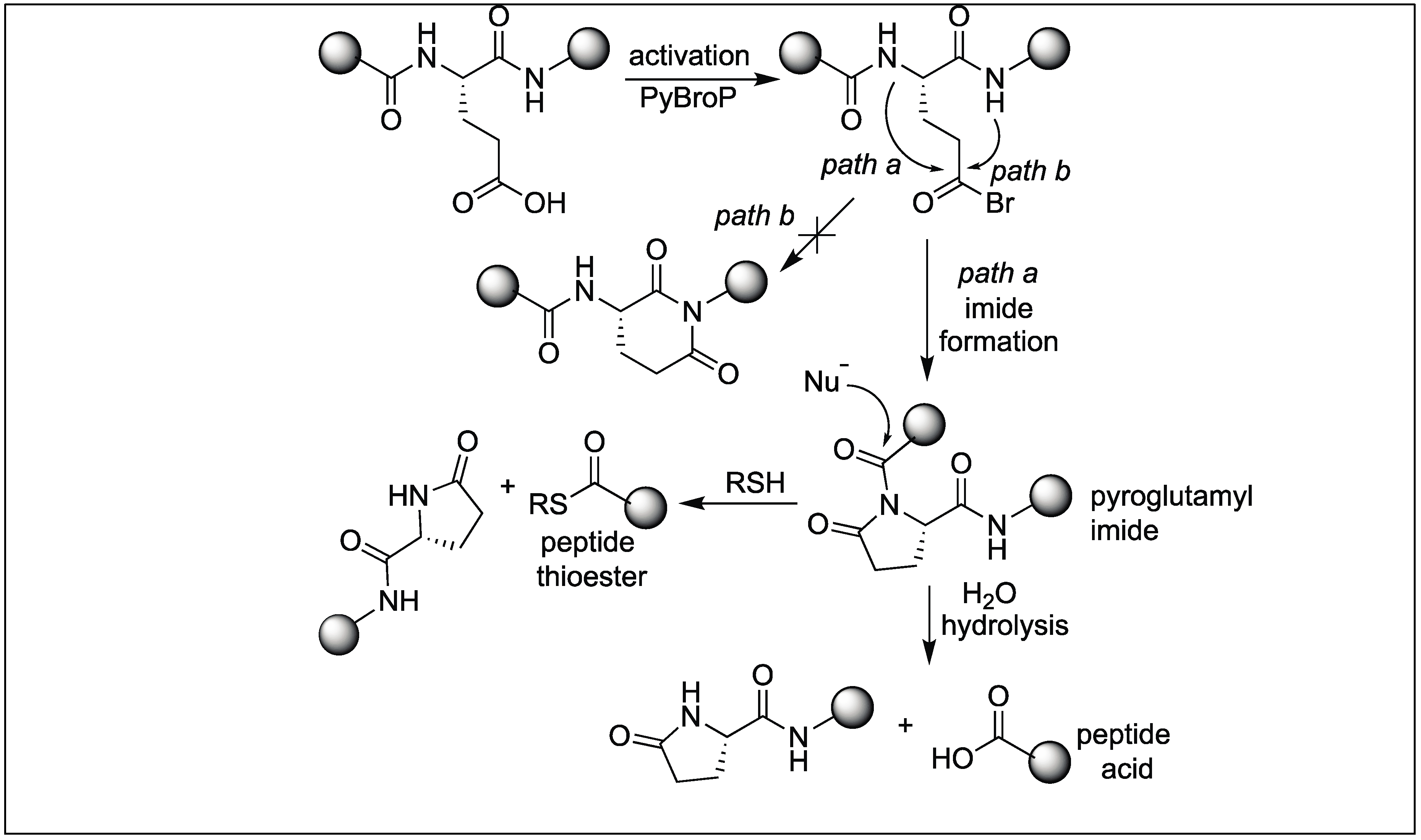
4.5. Glutamic Acid Specific Activation of Amide Bonds
4.6. Asparagine Selective Cleavage of Amide Bonds
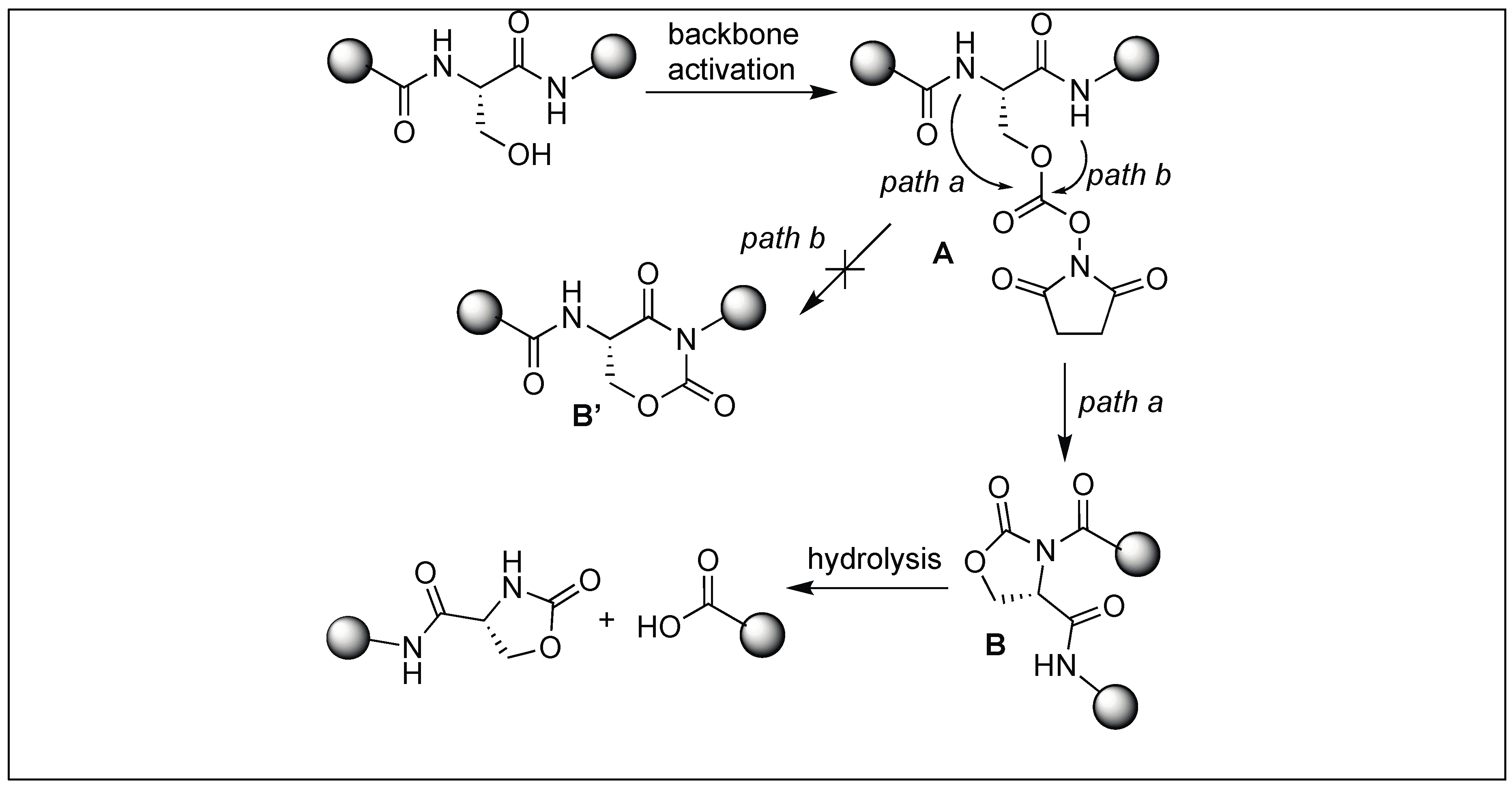
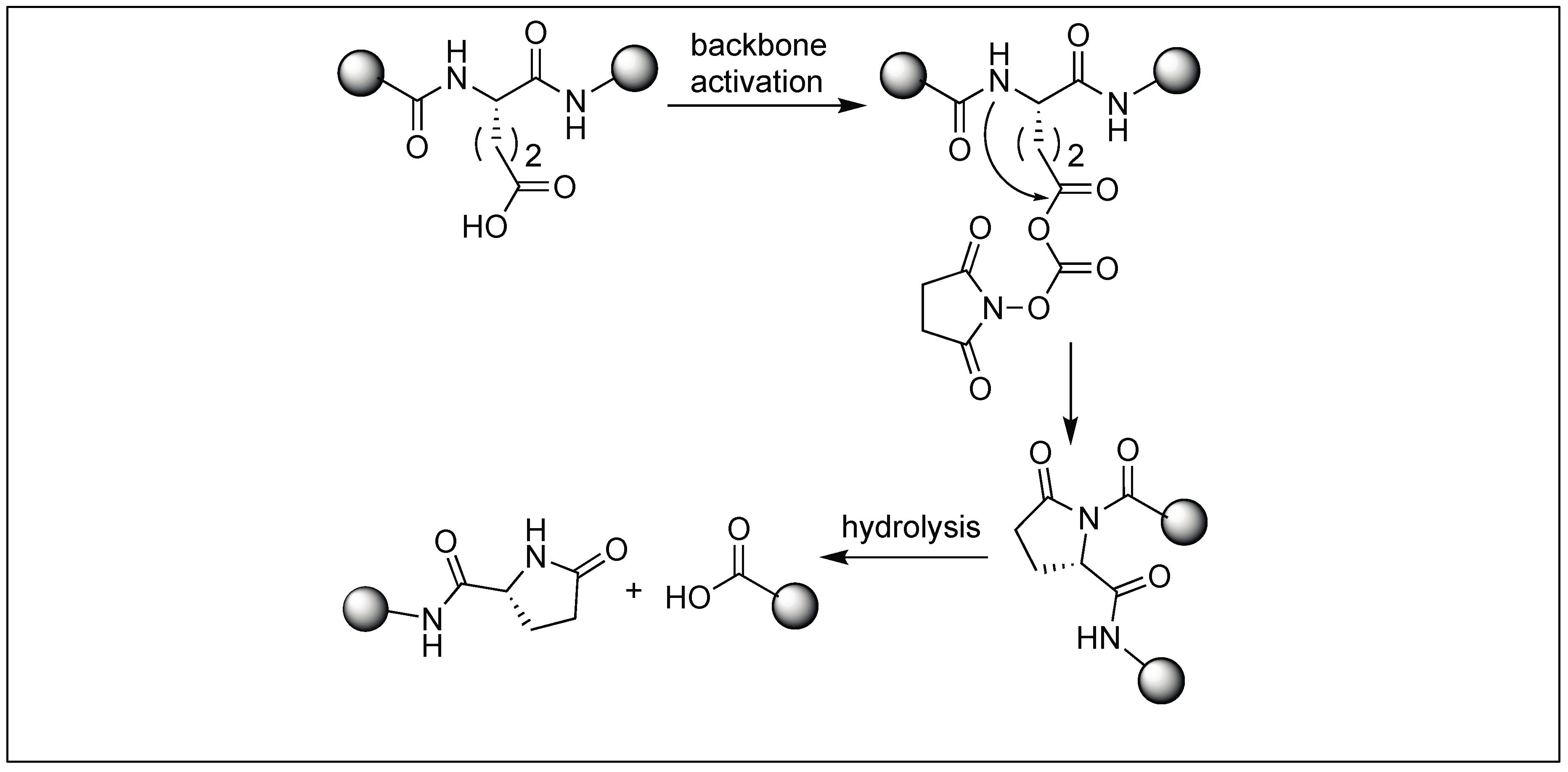
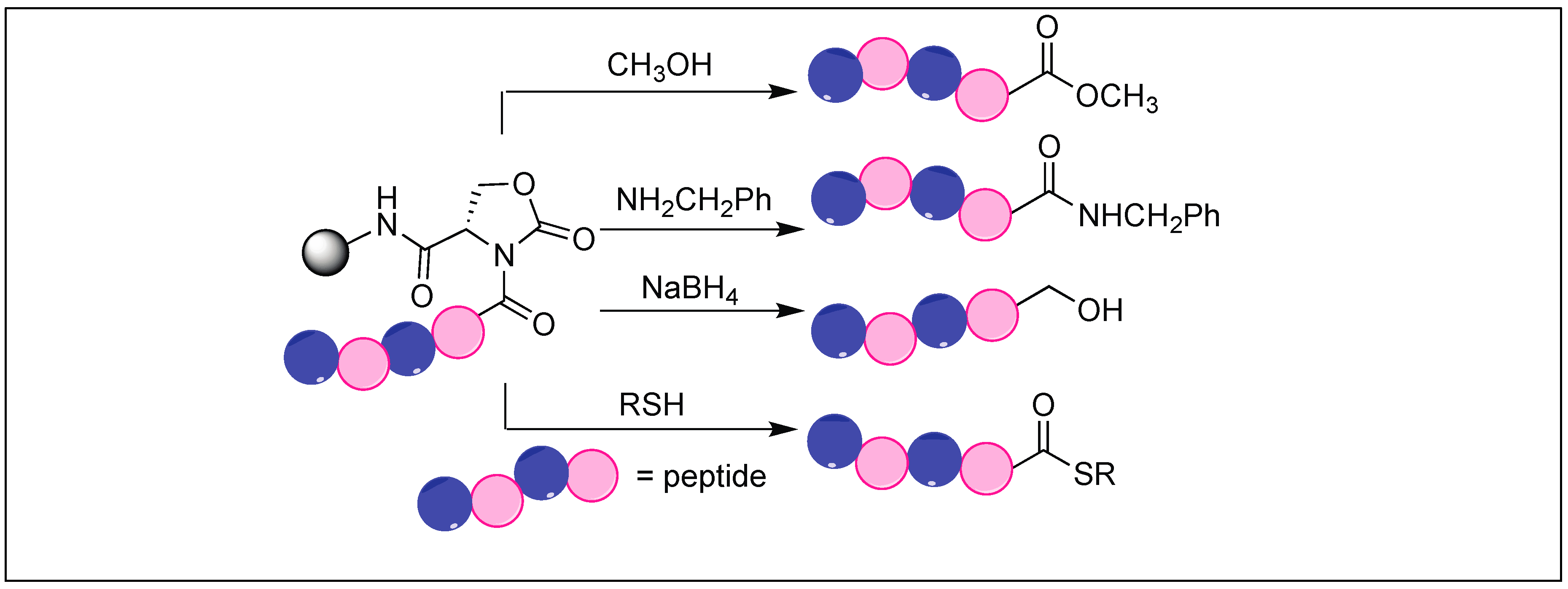
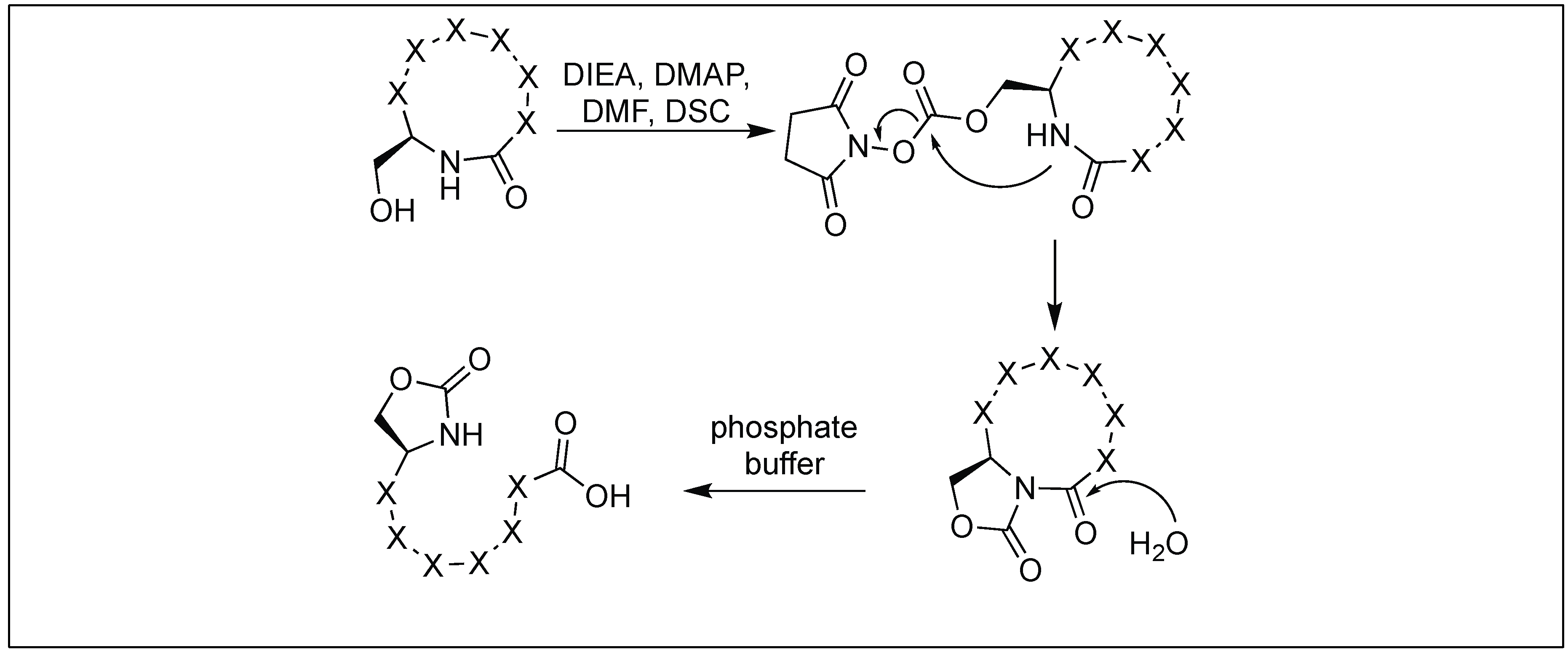
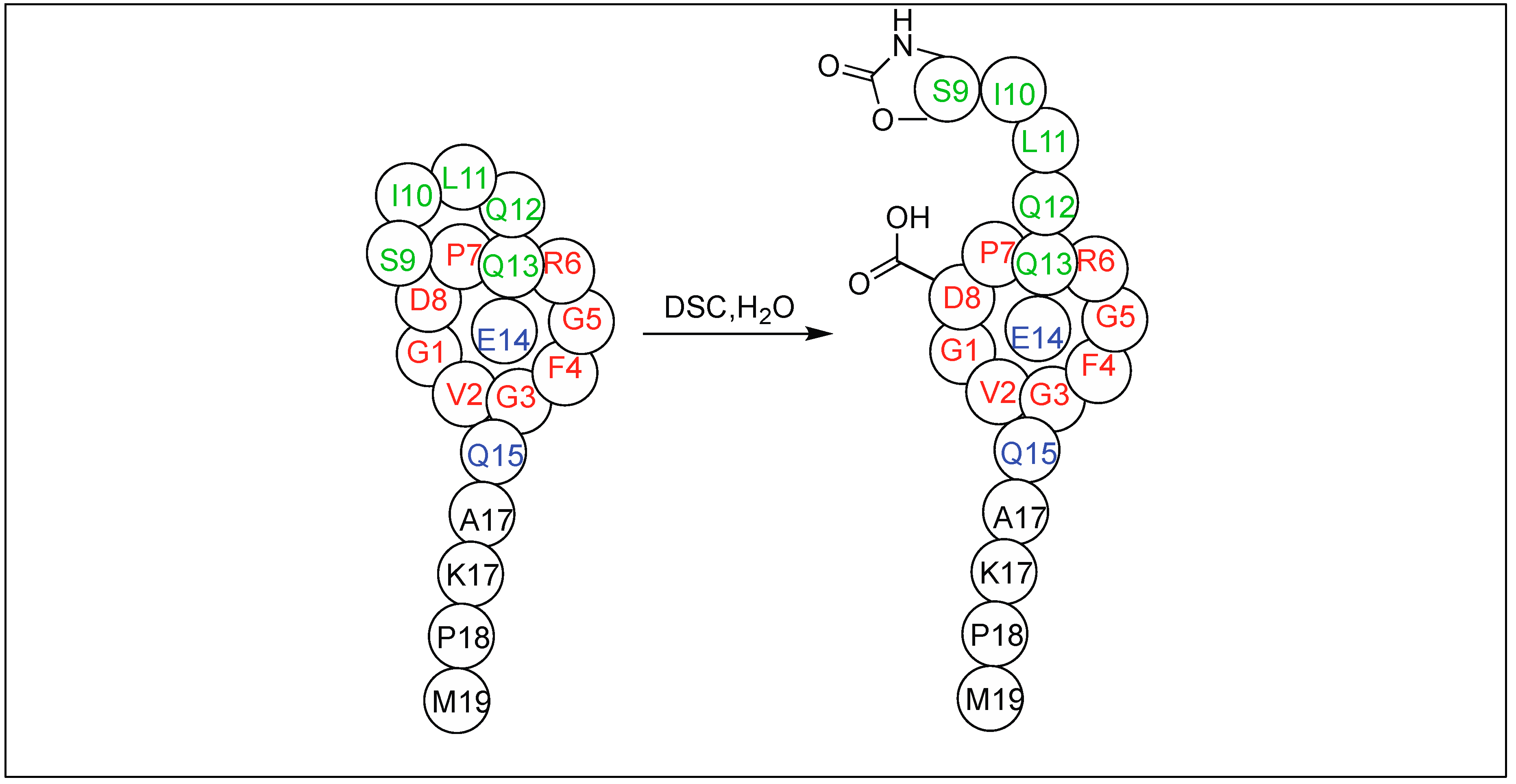
4.7. Cyclic Urethane Mediated Activation of Amide Bonds
4.8. Intein-Inspired Amide Cleavage Chemical Device
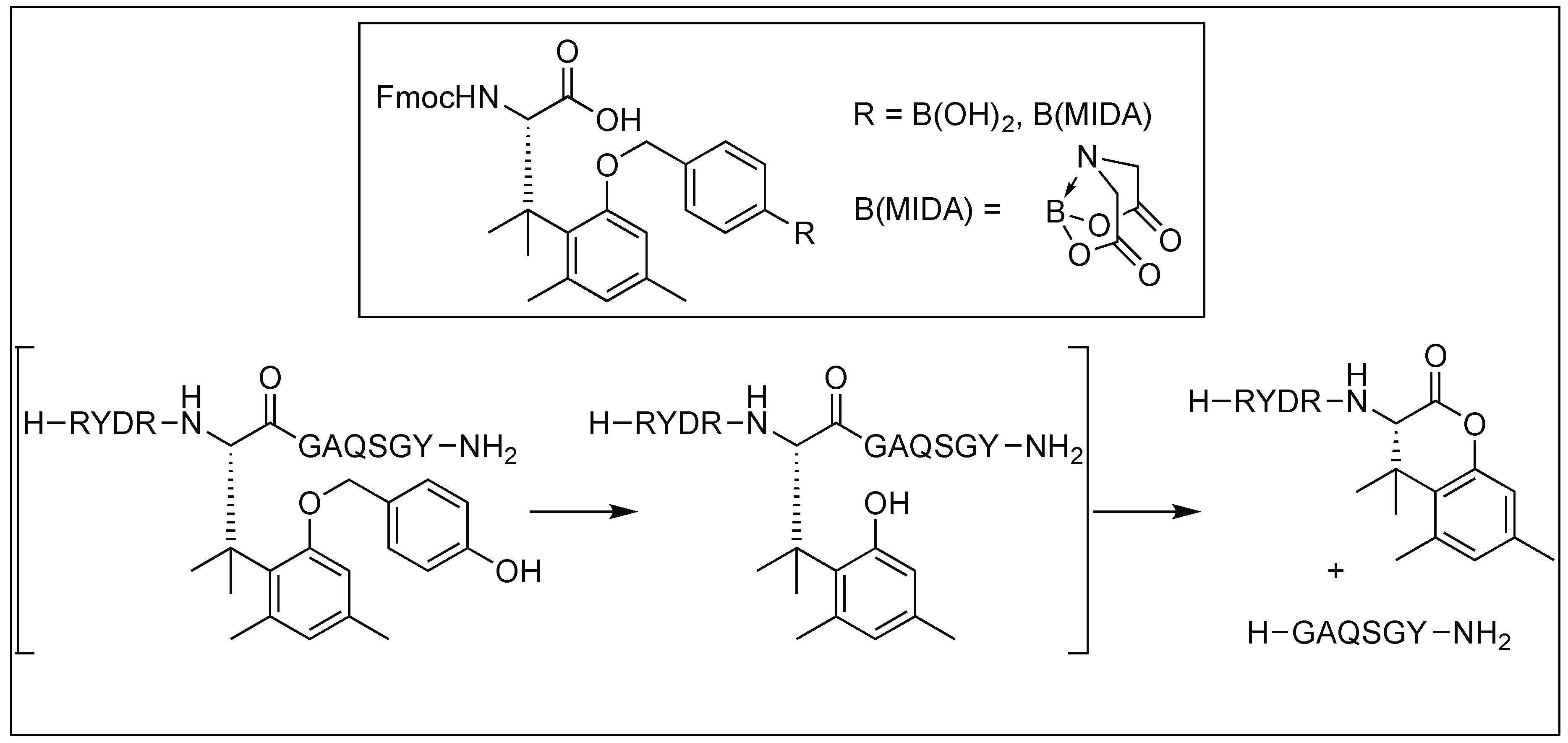
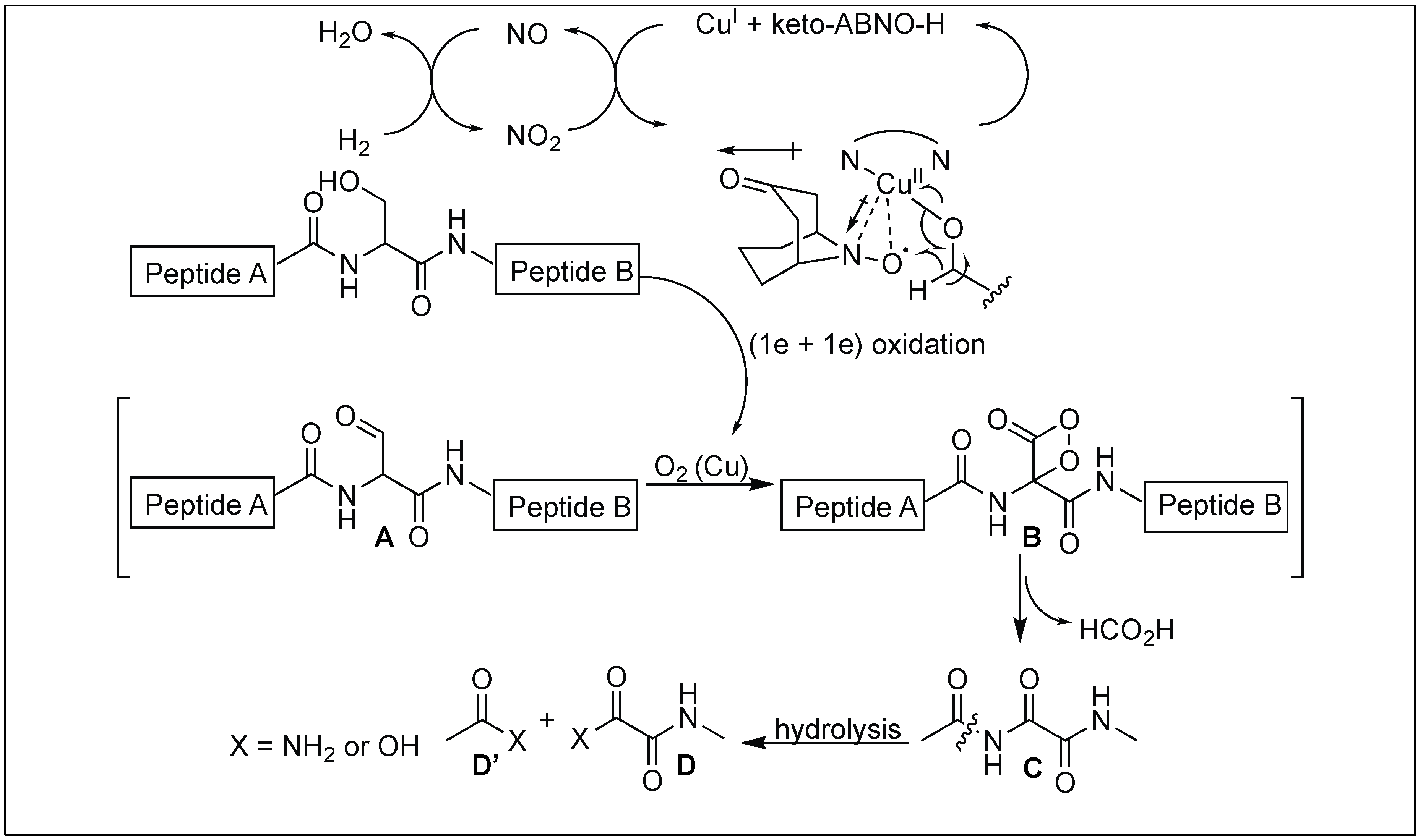
4.9. Serine-Selective Aerobic Cleavage of Peptides
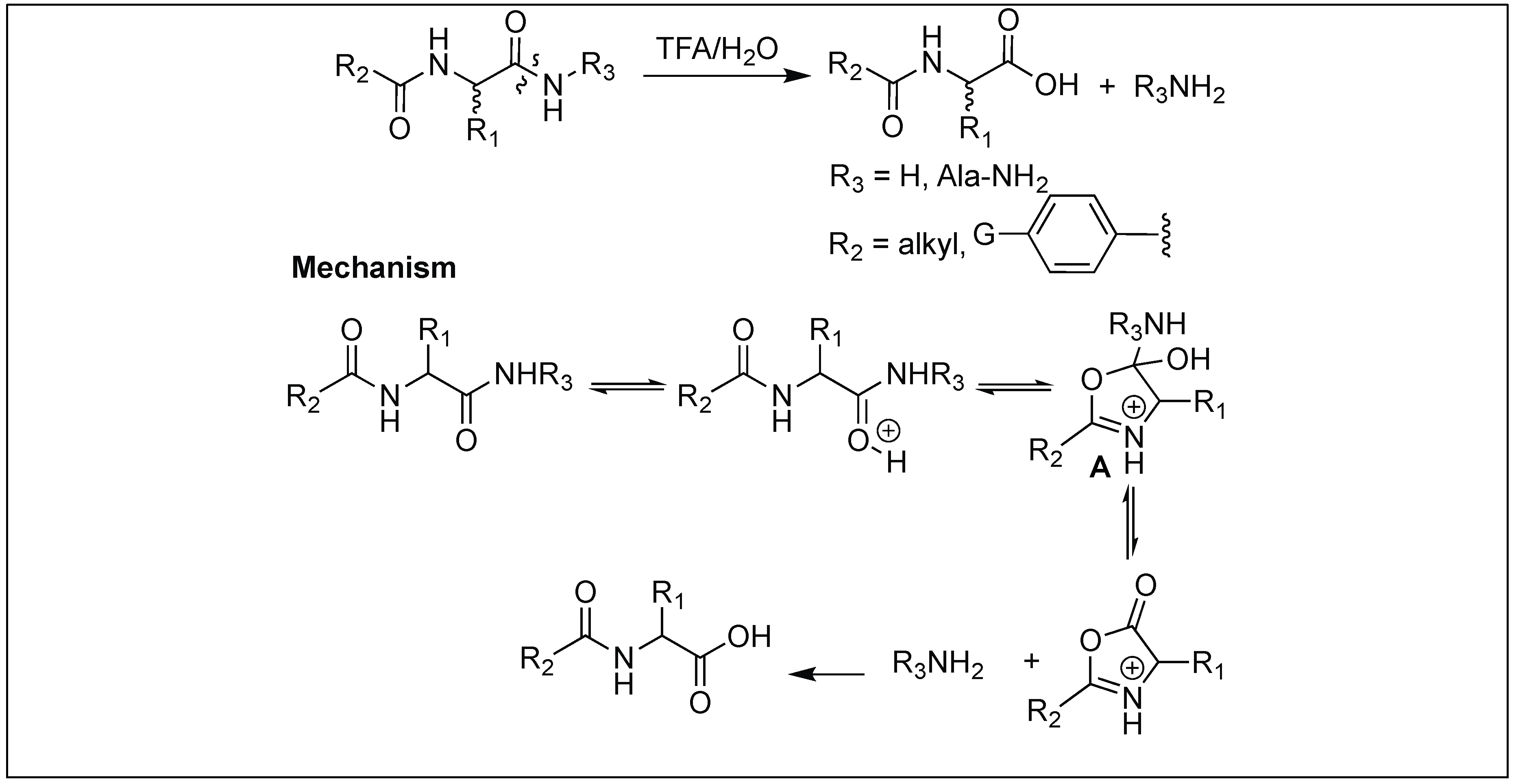
4.10. Hydrolysis of Amide Bonds by the Formation of Oxazolinium Specie: Function of Acyl Protecting Group
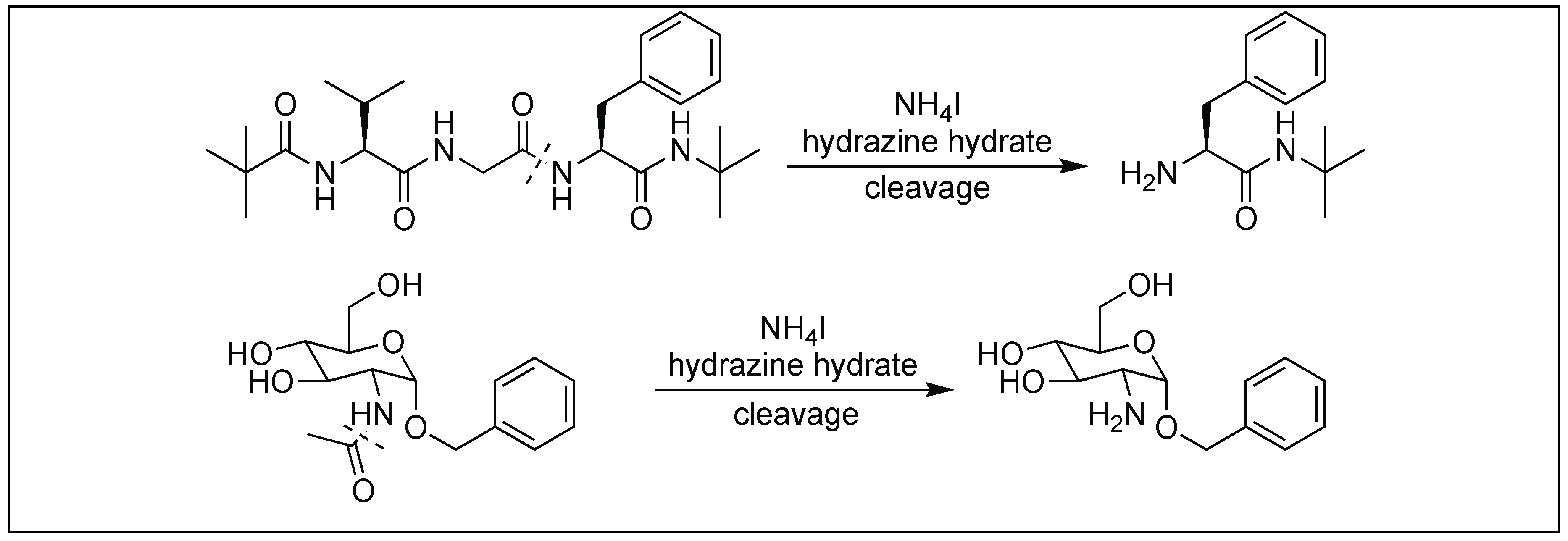
4.11. Hydrazinolysis for the Cleavage of Amide Bonds
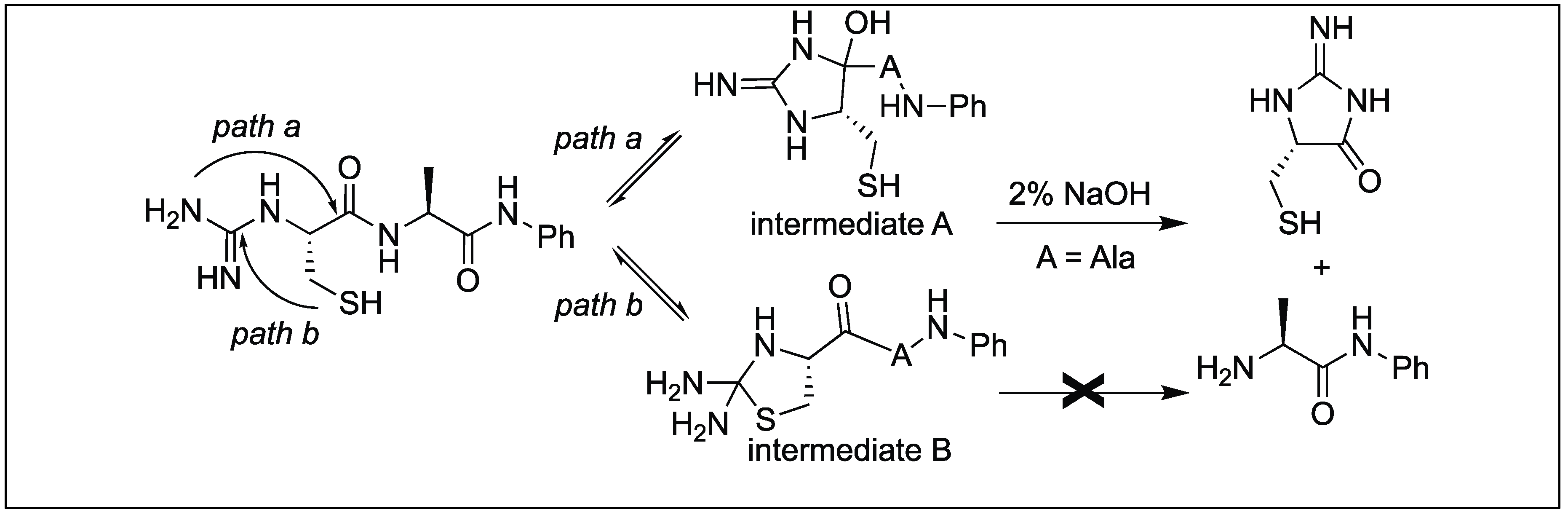
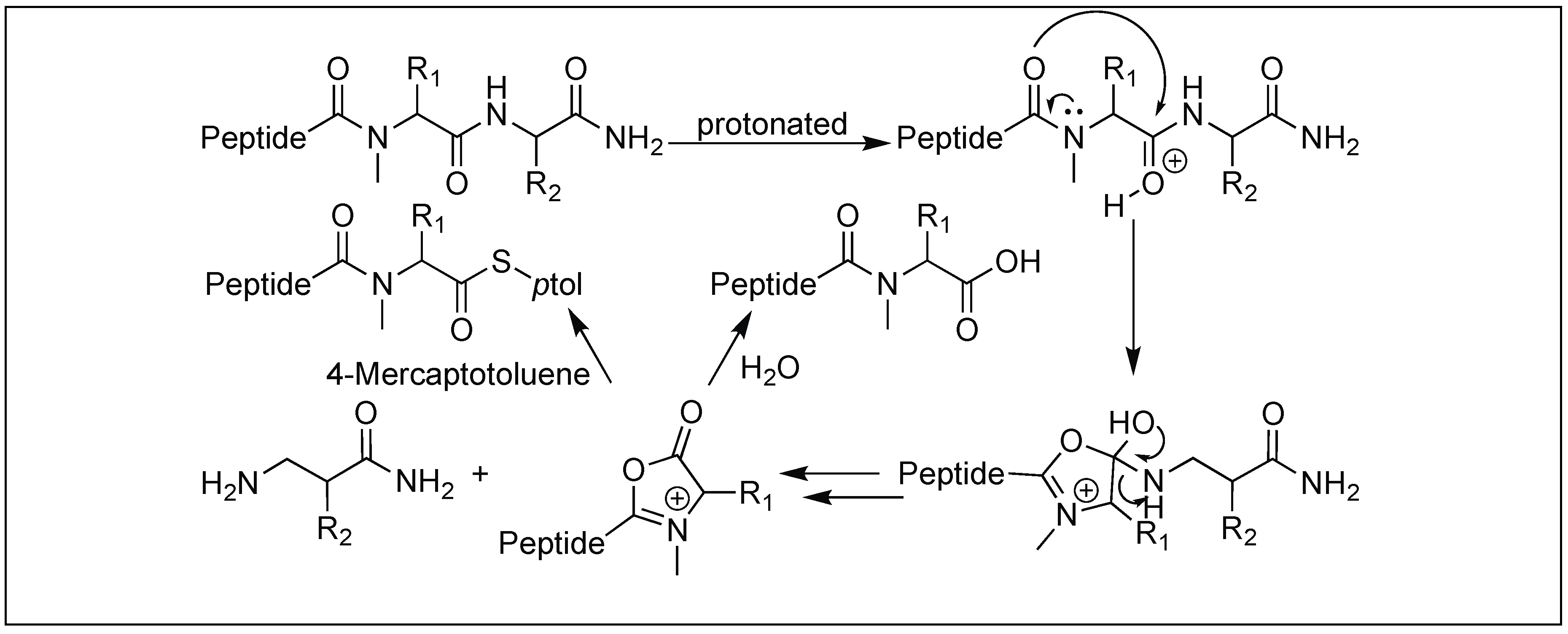
4.12. Amide Bond Cleavage of the N-Methylcysteinyl Peptide
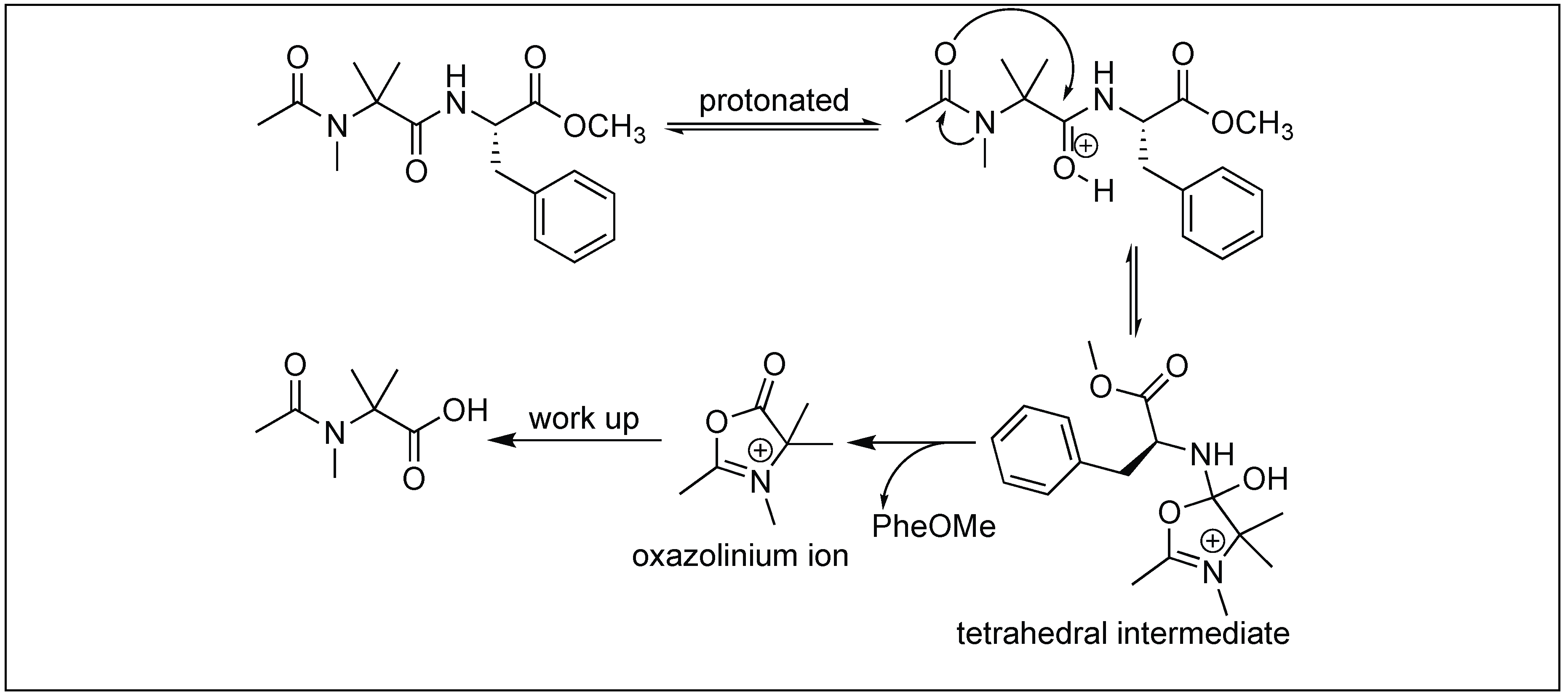
4.13. N-MeAib Induced Unusual Cleavage of Amide Bonds
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. The Amide Linkage: Structural Significance, Shemistry, Biochemistry and Material Science; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Brunton, L.; Chabner, B.; Knollman, B. Goodman and Gilman’s the Pharmacological Basis of Therapeutics; MacGraw-Hill: New York, NY, USA, 2010. [Google Scholar]
- Brown, D.G.; Bostrom, J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 2016, 59, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.B. Amino Acids, Peptides and Proteins in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Kaspar, A.A.; Reichert, J.M. Drug future directions for peptide therapeutics development. Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Thorner, J.; Emr, S.D.; Abelson, J.N. Applications of chimeric genes and hybrid proteins part A: Gene expression and protein purification. Methods Enzymol. 2000, 326, 601–617. [Google Scholar]
- Kemnitz, C.R.; Loewen, M.J. Amide Resonance correlates with a breadth of C-N rotation barriers. J. Am. Chem. Soc. 2007, 129, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Mujika, J.I.; Mercero, J.M.; Lopez, X. Water-promoted hydrolysis of a highly twisted amide: Rate acceleration caused by the twist of the amide bond. J. Am. Chem. Soc. 2005, 127, 4445–4453. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cao, Z. Acid-catalyzed reactions of twisted amides in water solution: Competition between hydration and hydrolysis. Chem.-Eur. J. 2011, 17, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.M.; Hansen, D.E. The pH-rate profile for the hydrolysis of a peptide bond. J. Am. Chem. Soc. 1998, 120, 8910–8913. [Google Scholar] [CrossRef]
- Radzicka, A.; Wolfenden, R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J. Am. Chem. Soc. 1996, 118, 6105–6109. [Google Scholar] [CrossRef]
- Arnau, J.; Lauritzen, C.; Petersen, G.E.; Pedersen, J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Express. Purif. 2006, 48, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, D.; Cho, H.; Schultz, P.G. New strategy for selective protein cleavage. J. Am. Chem. Soc. 1990, 112, 3249–3250. [Google Scholar] [CrossRef]
- Schepartz, A.; Cuenoud, B. Site-specific cleavage of the protein calmodulin using a trifluoperazine-based affinity reagent. J. Am. Chem. Soc. 1990, 112, 3247–3249. [Google Scholar] [CrossRef]
- Tani, K.; Stoltz, B.M. Synthesis and structural analysis of 2-quinuclidonium tetrafluoroborate. Nature 2006, 441, 731. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Bachman, S.; Hashimoto, S.; Eichman, C.C.; Stoltz, B.M. Catalytic anti-Markovnikov transformations of hindered terminal alkenes enabled by aldehyde-selective Wacker-type oxidation. J. Am. Chem. Soc. 2016, 138, 8997–9000. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.J.; Komarov, I.V.; Wothers, P.D.; Feeder, N. The most twisted amide: Structure and reactions. Angew. Chem. Int. Ed. 1998, 37, 785–786. [Google Scholar] [CrossRef]
- Kirby, A.J.; Komarov, I.V.; Feeder, N. Spontaneous, millisecond formation of a twisted amide from the amino acid, and the crystal structure of a tetrahedral intermediate. J. Am. Chem. Soc. 1998, 120, 7101–7102. [Google Scholar] [CrossRef]
- Kirby, A.J.; Komarov, I.V.; Feeder, N. Synthesis, structure and reactions of the most twisted amide. J. Chem. Soc. Perkin Trans. 2001, 2, 522–529. [Google Scholar] [CrossRef]
- Szostak, M.; Aube, J. Chemistry of bridged lactams and related heterocycles. Chem. Rev. 2013, 113, 5701. [Google Scholar] [CrossRef] [PubMed]
- Artacho, J.; Ascic, E.; Rantanen, T.; Karlsson, J.; Wallentin, C.J.; Wang, R.; Wendt, O.F.; Harmata, M.; Snieckus, V.; Warnmark, K. Twisted amide analogues of Troger’s base. Chem. Eur. J. 2012, 18, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Bashore, C.G.; Samardjiev, I.J.; Bordner, J.; Coe, J.W. Twisted amide reduction under Wolff–Kishner conditions: Synthesis of a benzo-1-aza-adamantane derivative. J. Am. Chem. Soc. 2003, 125, 3268. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Sterically controlled Pd-catalyzed chemoselective ketone synthesis via N–C cleavage in twisted amides. Org. Lett. 2015, 17, 4364–4367. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. General olefin synthesis by the palladium catalyzed Heck reaction of amides: Sterically controlled chemoselective N–C activation. Angew. Chem. Int. Ed. 2015, 54, 14518–14522. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Meng, G.; Szostak, M. Synthesis of biaryls through nickel catalyzed Suzuki–Miyaura coupling of amides by carbon–nitrogen bond cleavage. Angew. Chem. Int. Ed. 2016, 55, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Szostak, M. Efficient synthesis of diaryl ketones by nickel-catalyzed Negishi cross-coupling of amides by carbon–nitrogen bond cleavage at room temperature accelerated by a solvent effect. Chem.-Eur. J. 2016, 22, 10420–10424. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Rhodium-catalyzed C–H bond functionalization with amides by double C–H/C–N bond activation. Org. Lett. 2016, 18, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Palladium-catalyzed Suzuki–Miyaura coupling of amides by carbon–nitrogen cleavage: General strategy for amide N–C bond activation. Org. Biomol. Chem. 2016, 14, 5690–5707. [Google Scholar] [CrossRef] [PubMed]
- Hie, L.; Fine Nathel, N.F.; Shah, T.K.; Baker, E.L.; Hong, X.; Yang, Y.F.; Liu, P.; Houk, K.N.; Garg, N.K. Conversion of amides to esters by the nickel-catalysed activation of amide C–N bonds. Nature 2015, 524, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zou, G. Acylative Suzuki coupling of amides: Acyl nitrogen activation via synergy of independently modifiable activating groups. Chem. Commun. 2015, 51, 5089–5092. [Google Scholar] [CrossRef] [PubMed]
- Kraut, J. Serine proteases: Structure and mechanism of catalysis. Annu. Rev. Biochem. 1977, 46, 331–358. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Wells, J.A. Dissecting the catalytic triad of a serine protease. Nature 1988, 332, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine proteases: Modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Otto, H.H.; Schirmeister, T. Cysteine proteases and their inhibitors. Chem. Rev. 1997, 97, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 1999, 27, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, R.; Grochulski, P.; Sivaraman, J.; Menard, R.; Mort, J.S.; Cygler, M. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996, 15, 5492–5503. [Google Scholar] [CrossRef] [PubMed]
- Hooper, N.M. Families of zinc metalloproteases. FEBS Lett. 1994, 354, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hase, C.C.; Finkelstein, R.A. Bacterial extracellular zinc containing metalloproteases. Microbiol. Rev. 1993, 57, 823–837. [Google Scholar] [PubMed]
- Barrett, A. J. Proteinases in Mammalian Cells and Tissues; Elsevier/North-Holland Biomedical Press: Amsterdam, The Netherlands, 1977; pp. 181–208. [Google Scholar]
- Garrett, R.H.; Grisham, C.M. Biochemistry; University of Virginia: Charlottesville, VA, USA, 2010; p. 103. [Google Scholar]
- Hausrath, A.C.; Matthews, B.W. Thermolysin in the absence of substrate has an open conformation. Acta Crystallogr. 2002, 58, 1002–1007. [Google Scholar] [CrossRef] [Green Version]
- Endo, S. Studies on protease produced by thermophilic bacteria. J. Ferment. Technol. 1962, 40, 346. [Google Scholar]
- Kooi, C.; Sokol, P.A. Differentiation of thermolysins and serralysins by monoclonal antibodies. J. Med. Microbiol. 1996, 45, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kreij, A.; Venema, G.; van den Burg, B. Substrate specificity in the highly heterogeneous M4 peptidase family is determined by a small subset of amino acids. J. Biol. Chem. 2000, 275, 31115–31120. [Google Scholar] [CrossRef] [PubMed]
- Titani, K.; Hermodson, M.A.; Ericsson, L.H.; Walsh, K.A.; Neurath, H. Amino-acid sequence of thermolysin. Nat. New Biol. 1972, 238, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Ooshima, H.; Mori, H.; Harano, Y. Synthesis of aspartame precursor by thermolysin solid in organic solvent. Biotechnol. Lett. 1985, 7, 789–792. [Google Scholar] [CrossRef]
- Holmquist, B.; Vallee, B.L. Metal substitutions and inhibition of thermolysin: Spectra of the cobalt enzyme. J. Biol. Chem. 1974, 249, 4601–4607. [Google Scholar] [PubMed]
- Vallee, B.L.; Auld, D.S. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry 1990, 29, 5647–5659. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, D.G.; Monzingo, A.F.; Matthews, B.W. An interactive computer graphics study of thermolysin-catalyzed peptide cleavage and inhibition by N-carboxymethyl peptides. Biochemistry 1984, 23, 5730–5741. [Google Scholar] [CrossRef] [PubMed]
- Pelmenschikov, V.; Blomberg, M.R.A.; Siegbahn, P.E.M. A theoretical study of the mechanism of peptide hydrolysis by thermolysine. J. Biol. Inorg. Chem. 2001, 7, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Kilshtain, A.V.; Warshel, A. On the origin of the catalytic power of carboxypeptidase A and other metalloenzymes. Proteins 2009, 77, 536–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quicho, F.A.; McMurray, C.H.; Lipscomb, W.N. Similarities between the Conformation of Arsanilazotyrosine 248 of Carboxypeptidase Aa in the Crystalline State and in Solution. Proc. Nat. Acad. Sci. USA 1972, 69, 2850–2854. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, C.; Xu, D.; Guo, H. Catalysis of Carboxypeptidase A: Promoted-Water versus Nucleophilic Pathways. J. Phys. Chem. B 2010, 114, 9259–9267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, M.B.; Jiang, J.; Kapuria, V.; Bhuiyan, T.; Janetzko, J.; Zandberg, W.F.; Vocadlo, D.J.; Herr, W.; Walker, S. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 2013, 342, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janetzko, J.; Trauger, S.A.; Lazarus, M.B.; Walker, S. How the glycosyltransferase OGT catalyzes amide bond cleavage. Nat. Chem Biol. 2016, 12, 899–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.S.; Anderson, M.A.; Hoadley, K.A.; Keck, J.L.; Cleland, W.W.; Denu, J. M. Structural and kinetic isotope effect studies of nicotinamidase (Pnc1) from saccharomyces cerevisiae. Biochemistry 2012, 51, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, P.K.; Rao, V.A.; Zemla, A.; Cameron, S.; Hunter, W.N. Specificity and mechanism of acinetobacter baumanii nicotinamidase: Implications for activation of the front-line tuberculosis drug pyrazinamide. Angew. Chem. Int. Ed. 2009, 48, 9176–9179. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Taylor, A.B.; McAlister-Henn, L.; Hart, P.J. Crystal structure of the yeast nicotinamidase Pnc1p. Arch. Biochem. Biophys. 2007, 461, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Wang, W.; Kim, R.; Yakota, H.; Nguyen, H.; Kim, S.H. Crystal structure and mechanism of catalysis of a pyrazinamidase from Pyrococcus horikoshii. Biochemistry 2001, 40, 14166–14172. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Zhang, Y.; Abdelwahed, S.; Ealick, S.E.; Begley, T.P. Catalysis of a flavoenzyme-mediated amide hydrolysis. J. Am. Chem. Soc. 2010, 132, 5550–5551. [Google Scholar] [CrossRef] [PubMed]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459. [Google Scholar] [PubMed]
- Vaughn, H.L.; Robbins, M.D. Rapid procedure for the hydrolysis of amides to acids. J. Org. Chem. 1975, 40, 1187. [Google Scholar] [CrossRef]
- Gao, C.; Lavey, B.J.; Lo, C.L.; Datta, A.; Wentworth, P., Jr.; Janda, K.D. Direct selection for catalysis from combinatorial antibody libraries using a boronic acid probe: Primary amide bond hydrolysis. J. Am. Chem. Soc. 1998, 120, 2211–2217. [Google Scholar] [CrossRef]
- Martin, M.T.; Angeles, T.S.; Sugasawara, R.; Aman, N.I.; Napper, A.D.; Darsley, M.J.; Sanchez, R.I.; Booth, P.; Titmas, R.C. Antibody-catalyzed hydrolysis of an unsubstituted amide. J. Am. Chem. Soc. 1994, 116, 6508–6512. [Google Scholar] [CrossRef]
- Dai, X.; de Mesmaeker, A.; Joyce, G.F. Cleavage of an amide bond by a ribozyme. Science 1995, 267, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Wezynfeld, N.E.; Frączyk, T.; Bal, W. Metal assisted peptide bond hydrolysis: Chemistry, biotechnology and toxicological implications. Coord. Chem. Rev. 2016, 327–328, 166–187. [Google Scholar] [CrossRef] [Green Version]
- Yashiro, M.; Sonobe, Y.; Yamamura, A.; Takarada, T.; Komiyama, M.; Fujii, Y. Metal-ion-assisted hydrolysis of dipeptides involving a serine residue in a neutral aqueous solution. Org. Biomol. Chem. 2003, 1, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H. Model studies of metalloenzymes involving metal-ions as Lewis acid catalysts. Acc. Chem. Res. 1992, 25, 273–279. [Google Scholar] [CrossRef]
- Wybon, C.C.D.; Mensch, C.; Hollanders, K.; Gadais, C.; Herrebout, W.A.; Ballet, S.; Maes, B.U.W. Zn-catalyzed tert-Butyl nicotinate-directed amide cleavage as a biomimic of metallo-exopeptidase activity. ACS Catal. 2018, 8, 203–218. [Google Scholar] [CrossRef]
- Yashiro, M.; Takarada, T.; Miyama, S.; Komiyama, M. Cerium(IV)–cyclodextrin complex for peptide hydrolysis in neutral homogeneous solutions. J. Chem. Soc. Chem. Commun. 1994, 15, 1757–1758. [Google Scholar] [CrossRef]
- Kita, Y.; Nishii, Y.; Higuchi, T.; Mashima, K. Zinc-catalyzed amide cleavage and esterification of beta-hydroxyethylamides. Angew. Chem. Int. Ed. 2012, 51, 5723–5726. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.H.; Stroobants, K.; Parac-Vogt, T.N. Hydrolysis of serine-containing peptides at neutral pH promoted by [MoO4]2–oxyanion. Inorg. Chem. 2011, 50, 12025–12033. [Google Scholar] [CrossRef] [PubMed]
- Absillis, G.; Parac-Vogt, T.N. hydrolytic activity of vanadate toward serine-containing peptides studied by kinetic experiments and DFT theory. Inorg. Chem. 2012, 51, 9902–9910. [Google Scholar] [CrossRef] [PubMed]
- Cartuyvels, E.; Absillis, G.; Parac-Vogt, T.N. Questioning the paradigm of metal complex promoted phosphodiester hydrolysis: [Mo7O24]6−polyoxometalate cluster as an unlikely catalyst for the hydrolysis of a DNA model substrate. Chem. Commun. 2008, 85–87. [Google Scholar] [CrossRef]
- Absillis, G.; Cartuyvels, E.; Van Deun, R.; Parac-Vogt, T.N. Hydrolytic cleavage of an RNA-Model phosphodiester catalyzed by a highly negatively charged polyoxomolybdate [Mo7O24]6−Cluster. J. Am. Chem. Soc. 2008, 130, 17400–17408. [Google Scholar] [CrossRef] [PubMed]
- Absillis, G.; Van Deun, R.; Parac-Vogt, T.N. Polyoxomolybdate promoted hydrolysis of a DNA-model phosphoester studied by NMR and EXAFS spectroscopy. Inorg. Chem. 2011, 50, 11552–115560. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Yashiro, M.; Komiyama, M. Catalytic hydrolysis of peptides by cerium(IV). Chem. Eur. J. 2000, 6, 3906–3913. [Google Scholar] [CrossRef]
- Kassai, M.; Ravi, R.G.; Shealy, S.J.; Grant, K.B. Unprecedented acceleration of zirconium(IV)-assisted peptide hydrolysis at neutral pH. Inorg. Chem. 2004, 43, 6130–6132. [Google Scholar] [CrossRef] [PubMed]
- Absillis, G.; Parac-Vogt, T.N. Peptide bond hydrolysis catalyzed by the Wells−Dawson Zr(α2-P2W17O61)2 polyoxometalate. Inorg. Chem. 2012, 51, 9902–9910. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.H.; Stroobants, K.; Moelants, E.; Proost, P.; Parac-Vogt, T.N. Selective hydrolysis of hen egg white lysozyme at Asp-X peptide bonds promoted by oxomolybdate. J. Inorg. Biochem. 2014, 136, 73–80. [Google Scholar]
- Ly, H.G.T.; Fu, G.; Kondinski, A.; Bueken, B.; De Vos, D.; Parac-Vogt, T.N. Superactivity of MOF-808 toward peptide bond hydrolysis. J. Am. Chem. Soc. 2018, 140, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Meriwether, L.; Westheimer, F.H. Metal ion promoted hydrolysis of glycine amide and of phenylalanylglycine amide. J. Am. Chem. Soc. 1956, 78, 5119. [Google Scholar] [CrossRef]
- Bamann, E.; Hass, J.G.; Trapmann, H. Metallionenkatalytische zerlegung der (-CO-NH-)-bindung in peptiden und N-Acetyl-aminosäuren. Arch. Pharm. 1961, 294, 569–580. [Google Scholar] [CrossRef]
- Collman, J.P.; Buckingham, D.A. Hydrolytic cleavage of N-terminal peptide bonds by a cobalt chelate. J. Am. Chem. Soc. 1963, 85, 3039–3040. [Google Scholar] [CrossRef]
- Buckingham, D.A.; Coliman, J.P.; Happer, D.A.R.; Marzilli, L.G. Hydrolysis of N-terminal peptide bonds and amino acid derivatives by the β-hydroxoaquotriethylenetetraminecobalt(III) ion. J. Am. Chem. Soc. 1967, 89, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.W.; Creaser, E.H. Qualitative determination of N-terminal amino acids of peptides and proteins with cobalt (3) chelates. Bfochem. J. 1973, 135, 507–511. [Google Scholar] [CrossRef]
- Kimura, E. Sequential hydrolysis of peptides with β-hydroxoaquo triethylenetetraminecobalt(III) ion. Inorg. Chem. 1974, 13, 951–954. [Google Scholar] [CrossRef]
- Erxleben, A. Interaction of molybdocene dichloride with cysteine-containing peptides: Coordination, regioselective hydrolysis, and intramolecular aminolysis. Inorg. Chem. 2005, 44, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Burgeson, I.E.; Kostic’, N.M. Selective hydrolysis of unactivated peptide bonds, promoted by platinum(II) complexes anchored to amino acid side chains. Inorg. Chem. 1991, 30, 4299–4305. [Google Scholar] [CrossRef]
- Rajkovic, S.; Zivkovic, M.D.; Djuran, M.I. Reactions of dinuclear platinum(II) complexes with peptides. Curr. Protein Pept. Sci. 2016, 17, 95–105. [Google Scholar] [CrossRef] [PubMed]
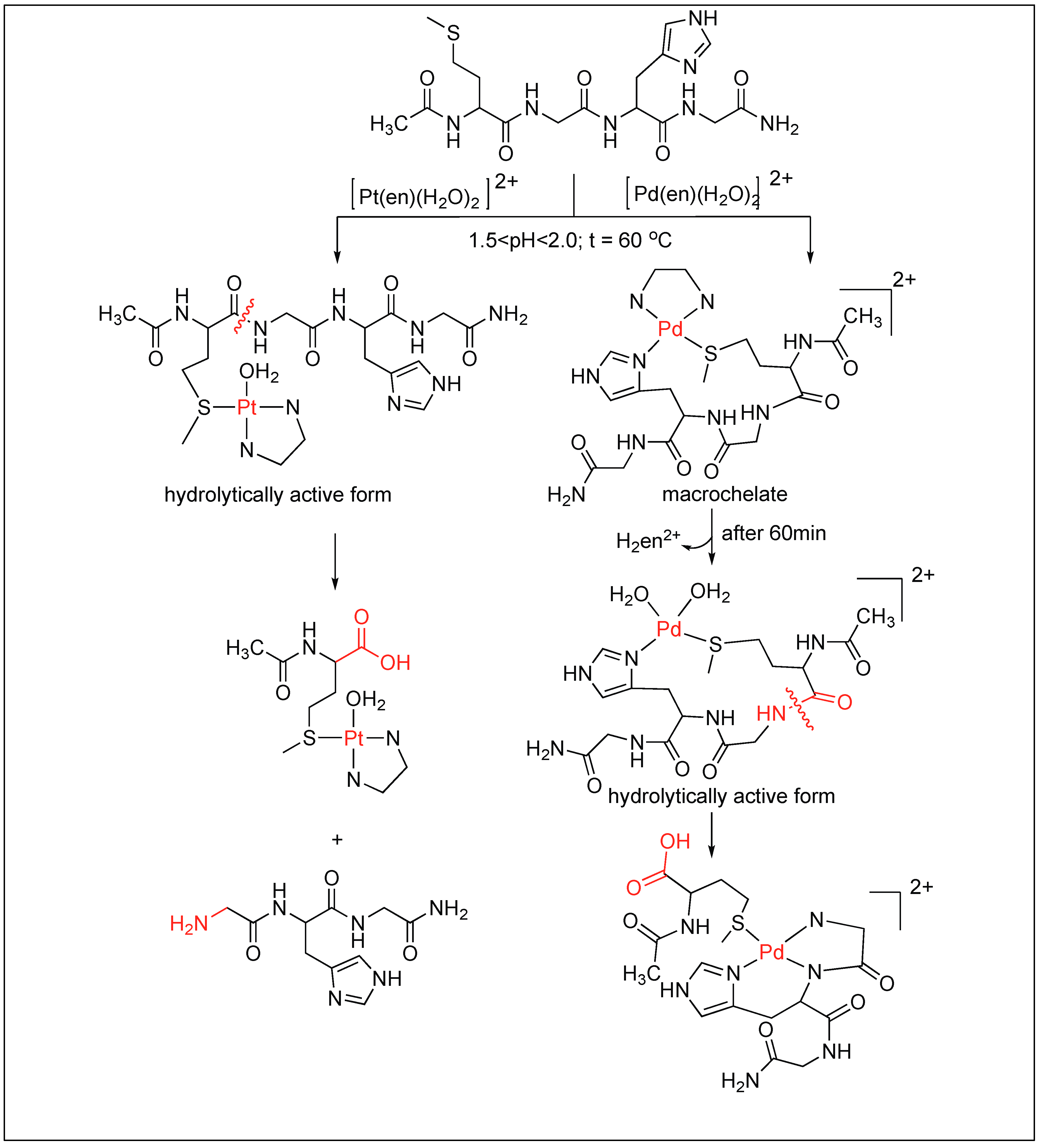
- Parac, T.N.; Kostic, N.M. New selectivity and turnover in peptide hydrolysis by metal complexes. A palladium(II) aqua complex catalyzes cleavage of peptides next to the histidine residue. J. Am. Chem. Soc. 1996, 118, 51–58. [Google Scholar] [CrossRef]
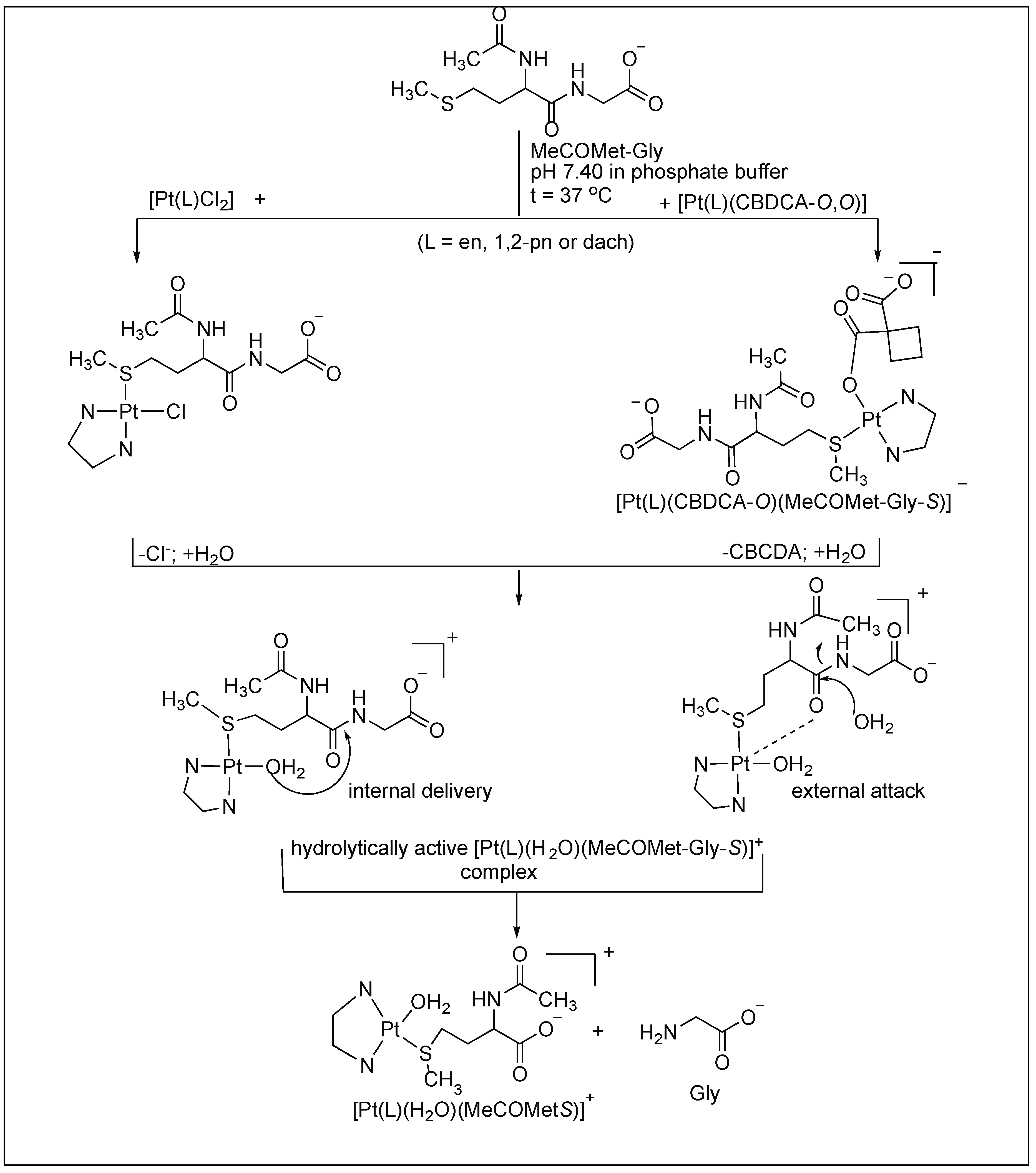
- Živković, M.D.; Rajković, S.; Glišić, B.D.; Drašković, N.S.; Djuran, M.I. Hydrolysis of the amide bond in histidine- and methionine-containing dipeptides promoted by pyrazine and pyridazine palladium(II)-aqua dimers: Comparative study with platinum(II) analogues. Bioorg. Chem. 2017, 72, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Rajkovic, S.; Asanin, D.P.; Zivkovic, M.D.; Djuran, M.I. Synthesis of different pyrazine-bridged platinum(II) complexes and 1H NMR study of their catalytic abilities in the hydrolysis of the Nacetylated l-methionylglycine. Polyhedron 2013, 65, 42–47. [Google Scholar] [CrossRef]
- Asanin, D.P.; Zivkovic, M.D.; Rajkovic, S.; Warzajtis, B.; Rychlewska, U.; Djuran, M.I. Crystallographic evidence of anion···π interactions in the pyrazine bridged {[Pt(en)Cl]2(μ-pz)}Cl2 complex and a comparative study of the catalytic ability of mononuclear and binuclear platinum(II) complexes in the hydrolysis of N-acetylated l-methionylglycine. Polyhedron 2013, 51, 255–262. [Google Scholar]
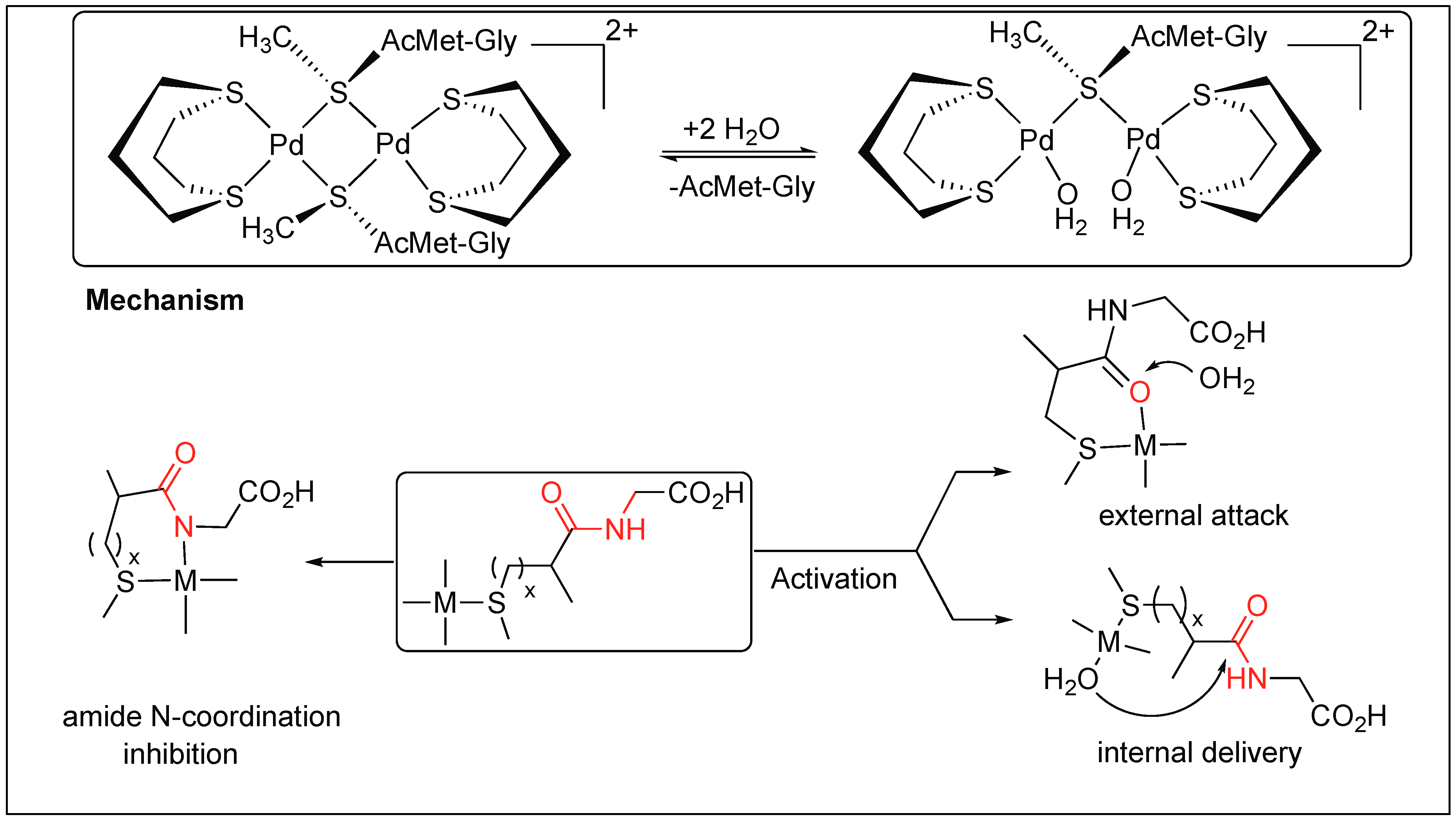
- Živković, M.D.; Asanin, D.P.; Rajkovic, S.; Djuran, M.I. Hydrolysis of the amide bond in N-acetylated l-methionylglycine catalyzed by various platinum(II) complexes under physiologically relevant conditions. Polyhedron 2011, 30, 947–952. [Google Scholar] [CrossRef]
- Rajkovic, S.; Zivkovic, M.D.; Kallay, C.; Sóvágó, I.; Djuran, M.I. A study of the reactions of a methionine- and histidine-containing tetrapeptide with different Pd(II) and Pt(II) complexes: Selective cleavage of the amide bond by platination of the peptide and steric modification of the catalyst. Dalton Trans. 2009, 39, 8370–8377. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kostie, N.M. Toward artificial metalloproteases: Mechanisms by which platinum(II) and palladium(II) complexes promote selective, fast hydrolysis of unactivated amide bonds in peptides. Inorg. Chem. 1992, 31, 3994–4001. [Google Scholar] [CrossRef]
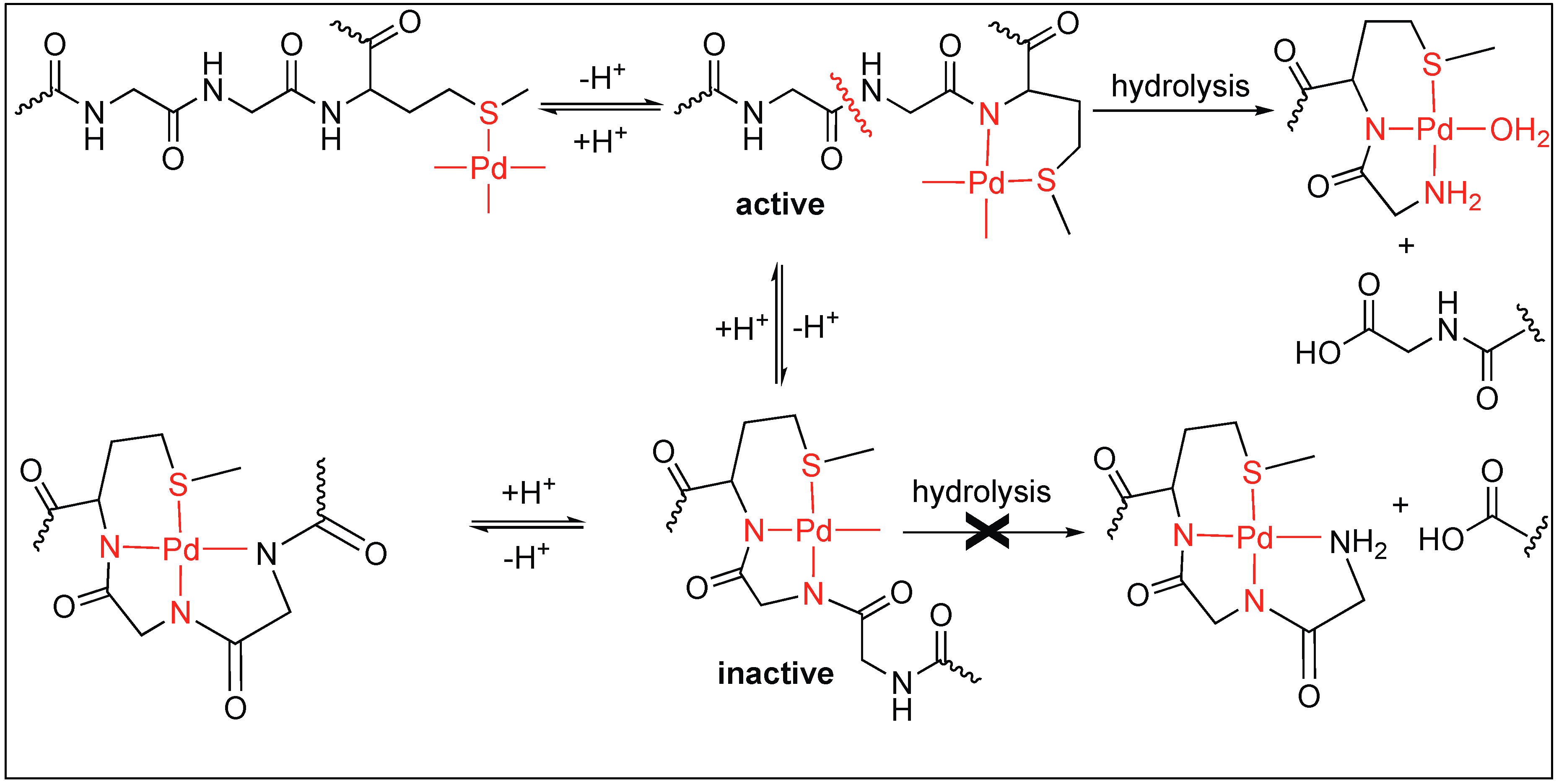
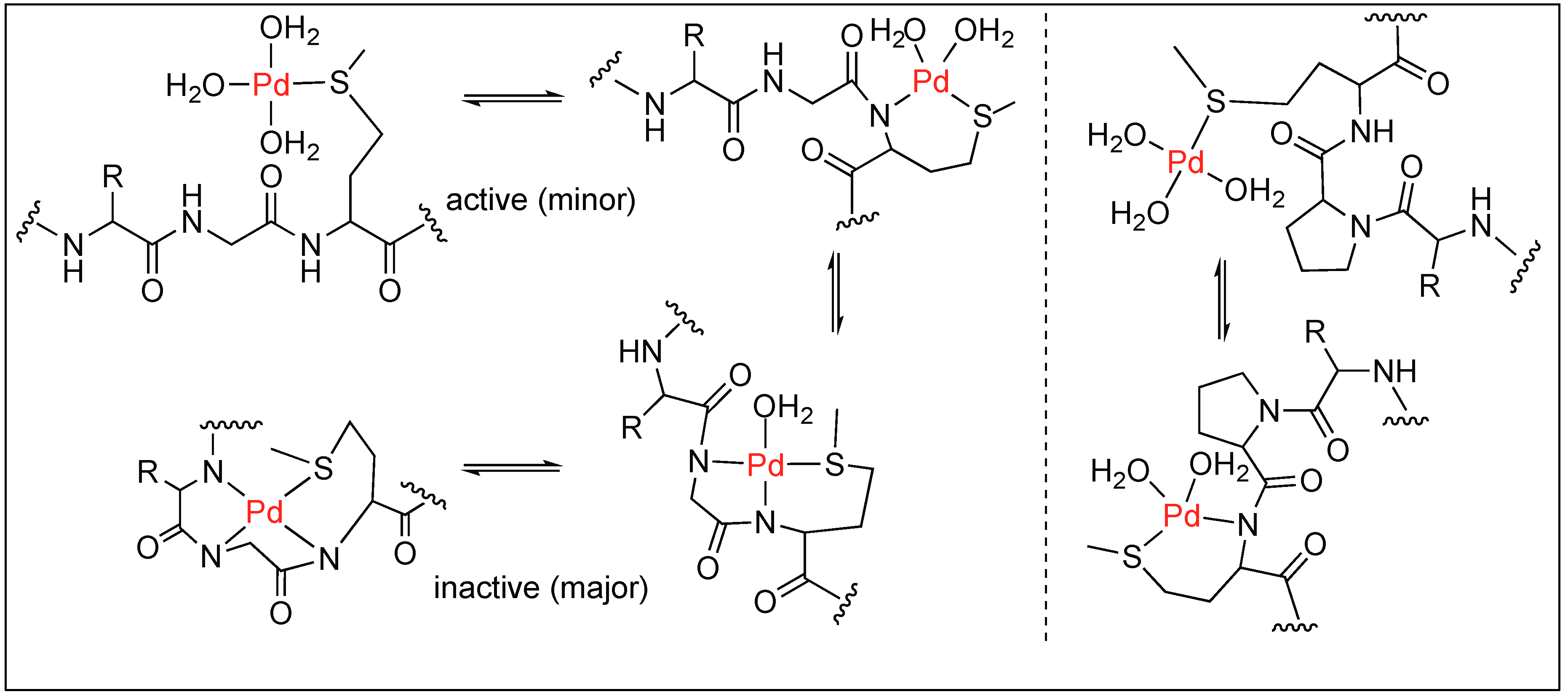
- Milovic, N.M.; Kostic, N.M. Palladium(II) complexes, as synthetic peptidases, regioselectively cleave the second peptide bond “upstream” from methionine and histidine side chains. J. Am. Chem. Soc. 2002, 124, 4759–4769. [Google Scholar] [CrossRef] [PubMed]
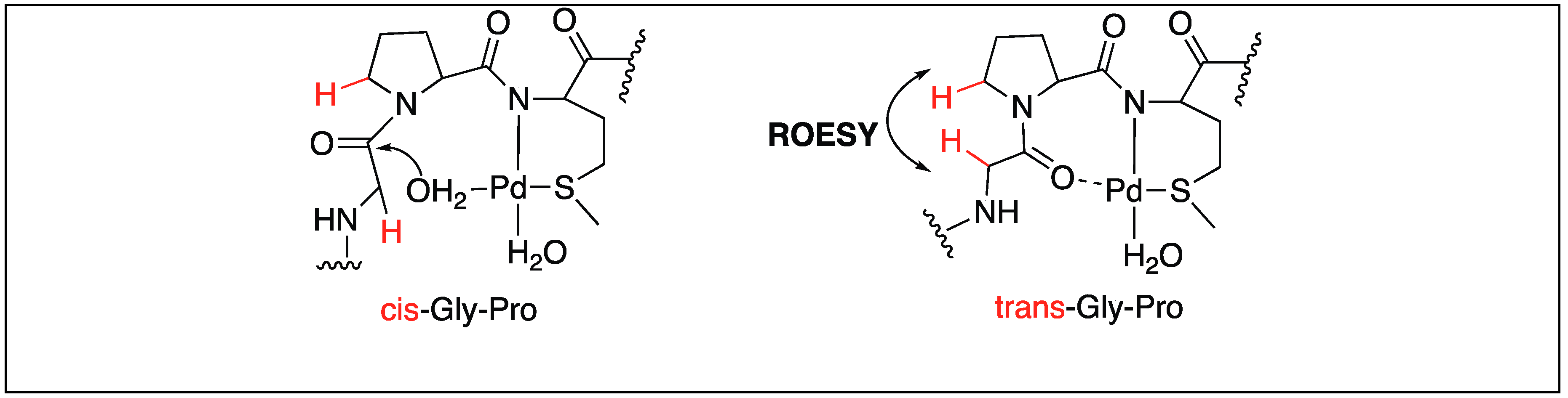
- Milovic, N.; Kostic, N. Palladium(II) complex as a sequence-specific peptidase: Hydrolytic cleavage under mild conditions of X-Pro peptide bonds in X-Pro-Met and X-Pro-His segments. J. Am. Chem. Soc. 2003, 125, 781–789. [Google Scholar] [CrossRef] [PubMed]
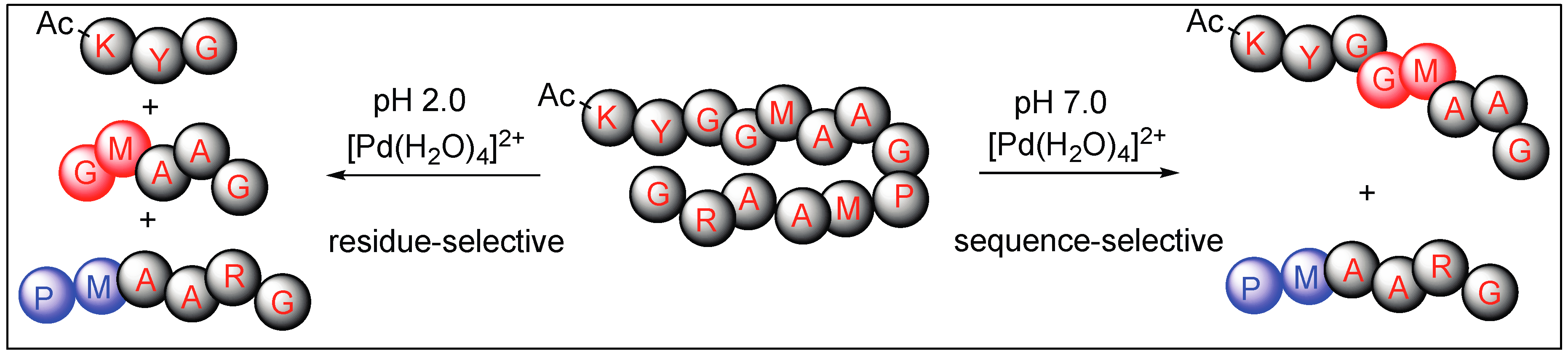
- Milovic, N.M.; Kostic, N.M. Interplay of terminal amino group and coordinating side chains in directing regioselective cleavage of natural peptides and proteins with palladium(II) complexes. Inorg. Chem. 2002, 41, 7053–7063. [Google Scholar] [CrossRef] [PubMed]
- Milovic, N.M.; Badjic, J.D.; Kostic, N.M. Conjugate of palladium(II) complex and β-cyclodextrin acts as a biomimetic peptidase. J. Am. Chem. Soc. 2004, 126, 696–697. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.E.; Chae, P.S.; Kim, J.E.; Jeong, E.J.; Suh, J. Degradation of myoglobin by polymeric artificial metalloproteases containing catalytic modules with various catalytic group densities: Site selectivity in peptide bond cleavage. J. Am. Chem. Soc. 2003, 125, 14580–14589. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Lee, B.J.; Kim, H.; Suh, J. Artificial metalloprotease with active site comprising aldehyde group and Cu(II)Cyclen complex. J. Am. Chem. Soc. 2005, 127, 9593–9602. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.W.; Son, S.J.; Yoo, C.E.; Hong, I.S.; Suh, J. Toward protein-cleaving catalytic drugs: Artificial protease selective for myoglobin. Bioorg. Med. Chem. 2003, 11, 2901–2910. [Google Scholar] [CrossRef]
- Kim, H.; Jang, B.; Cheon, Y.; Suh, M.; Suh, J. Proteolytic activity of Co(III) complex of 1-oxa-4,7,10-triazacyclododecane: A new catalytic center for peptide-cleavage agents. J. Biol. Inorg. Chem. 2009, 14, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Chae, P.S.; Kim, M.-S.; Jeung, C.-S.; Lee, S.D.; Park, H.; Lee, S.; Suh, J. Peptide-cleaving catalyst selective for peptide deformylase. J. Am. Chem. Soc. 2005, 127, 2396–2397. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Chei, W.S.; Lee, T.Y.; Kim, M.G.; Yoo, S.H.; Jeong, K.; Ahn, J.Y. Cleavage agents for soluble oligomers of human islet amyloid polypeptide. J. Biol. Inorg. Chem. 2008, 13, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Suh, J. Target-selective peptide-cleaving catalysts as a new paradigm in drug design. Chem. Soc. Rev. 2008, 38, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Suh, J. Progress in designing artificial proteases: A new therapeutic option for amyloid diseases. Asian J. Org. Chem. 2014, 3, 18–32. [Google Scholar] [CrossRef]
- De Oliveira, M.C.B.; Scarpellini, M.; Neves, A.; Terenzi, H.; Bortoluzzi, A.J.; Szpoganics, B.; Greatti, A.; Mangrich, A.S.; de Souza, E.M.; Fernandez, P.M.; et al. Hydrolytic protein cleavage mediated by unusual mononuclear copper(II) complexes: X-ray structures and solution studies. Inorg. Chem. 2005, 44, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Kopera, E.; Krężel, A.; Protas, A.M.; Belczyk, A.; Bonna, A.; Wysłouch-Cieszynska, A.; Poznanski, J.; Bal, W. Sequence-specific Ni(II)-dependent peptide bond hydrolysis for protein engineering: Reaction conditions and molecular mechanism. Inorg. Chem. 2010, 49, 6636–6645. [Google Scholar] [CrossRef] [PubMed]
- Ariani, H.H.; Polkowska-Nowakowska, A.; Bal, W. Effect of D-amino acid substitutions on Ni(II)-assisted peptide bond hydrolysis. Inorg. Chem. 2013, 52, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Sohma, Y.; Kanai, M. Scandium(III) triflate-promoted serine/threonine-selective peptide bond cleavage. Chem. Commun. 2017, 53, 3311–3314. [Google Scholar] [CrossRef] [PubMed]
- Edman, P.; Begg, G.A. Protein sequenator. Eur. J. Biochem. 1967, 1, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Gross, E. The cyanogen bromide reaction. Methods Enzymol. 1967, 11, 238. [Google Scholar]
- Degani, Y.; Patchornik, A. Cyanylation of sulfhydryl groups by 2-nitro-5-thiocyanobenzoic acid. High-yield modification and cleavage of peptides at cysteine residues. Biochemistry 1974, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, W.C.; Smith, P.K.; Hermodson, M.A. Fragmentation of proteins with o-iodosobenzoic acid: Chemical mechanism and identification of o-iodoxybenzoic acid as a reactive contaminant that modifies tyrosyl residues. Biochemistry 1981, 20, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Burstein, Y.; Patchornik, A. Selective chemical cleavage of tryptophanyl peptide bonds in peptides and proteins. Biochemistry 1972, 11, 4641–4650. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.A. Novel N-terminal degradation reaction of peptides via N-amidination. Bioorg. Med. Chem. Lett. 2016, 26, 1690. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Denda, M.; Maeda, N.; Kita, M.; Komiya, C.; Tanaka, T.; Nomura, W.; Tamamura, H.; Sato, Y.; Yamauchi, A.; et al. Development of a traceable linker containing a thiol-responsive amino acid for the enrichment and selective labelling of target proteins. Org. Biomol. Chem. 2014, 12, 3821–3826. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Maeda, N.; Komiya, C.; Tanaka, T.; Denda, M.; Ebisuno, K.; Nomura, W.; Tamamura, H.; Sato, Y.; Yamauchi, A.; et al. Development of a fluoride-responsive amide bond cleavage device that is potentially applicable to a traceable linker. Tetrahedron 2014, 70, 5122–5127. [Google Scholar] [CrossRef] [Green Version]
- Shigenaga, A.; Ogura, K.; Hirakawa, H.; Yamamoto, J.; Ebisuno, K.; Miyamoto, L.; Ishizawa, K.; Tsuchiya, K.; Otaka, A. Development of a reduction-responsive amino acid that induces peptide bond cleavage in hypoxic cells. ChemBioChem 2012, 13, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, A.; Hirakawa, H.; Yamamoto, J.; Ogura, K.; Denda, M.; Yamaguchi, K.; Tsuji, D.; Itoh, K.; Otaka, A. Design and synthesis of caged ceramide: UV-responsive ceramide releasing system based on UV-induced amide bond cleavage followed by O–N acyl transfer. Tetrahedron 2011, 67, 3984–3990. [Google Scholar] [CrossRef]
- Shigenaga, A.; Yamamoto, J.; Sumikawa, Y.; Furuta, T.; Otaka, A. Development and photo-responsive peptide bond cleavage reaction of two-photon near-infrared excitation-responsive peptide. Tetrahedron Lett. 2010, 51, 2868–2871. [Google Scholar] [CrossRef]
- Shigenaga, A.; Yamamoto, J.; Hirakawa, H.; Ogura, K.; Maeda, N.; Morishita, K.; Otaka, A. Development of thiol-responsive amide bond cleavage device and its application for peptide nucleic acid-based DNA releasing system. Tetrahedron Lett. 2010, 51, 2525. [Google Scholar] [CrossRef]
- Shigenaga, A.; Tsuji, D.; Nishioka, N.; Tsuda, S.; Itoh, K.; Otaka, A. Synthesis of a stimulus-responsive processing device and its application to a nucleocytoplasmic shuttle peptide. ChemBioChem 2007, 8, 1929–1931. [Google Scholar] [CrossRef] [PubMed]
- Kita, M.; Yamamoto, J.; Morisaki, T.; Komiya, C.; Inokuma, T.; Miyamoto, L.; Tsuchiya, K.; Shigenaga, A.; Otaka, A. Design and synthesis of a hydrogenperoxide-responsive amino acid that induces peptide bond cleavage after exposure to hydrogen peroxide. Tetrahedron Lett. 2015, 56, 4228–4231. [Google Scholar] [CrossRef]
- Nalbone, J.M.; Lahankar, N.; Buissereth, L.; Raj, M. Glutamic acid selective chemical cleavage of peptide bonds. Org. Lett. 2016, 18, 1186–1189. [Google Scholar] [CrossRef] [PubMed]
- Tofteng, A.P.; Sørensen, K.K.; Conde-Frieboes, K.W.; HoegJensen, T.; Jensen, K.J. Fmoc solid-phase synthesis of C-terminal peptide thioesters by formation of a backbone pyroglutamyl imide moiety. Angew. Chem. Int. Ed. 2009, 48, 7411–7414. [Google Scholar] [CrossRef] [PubMed]
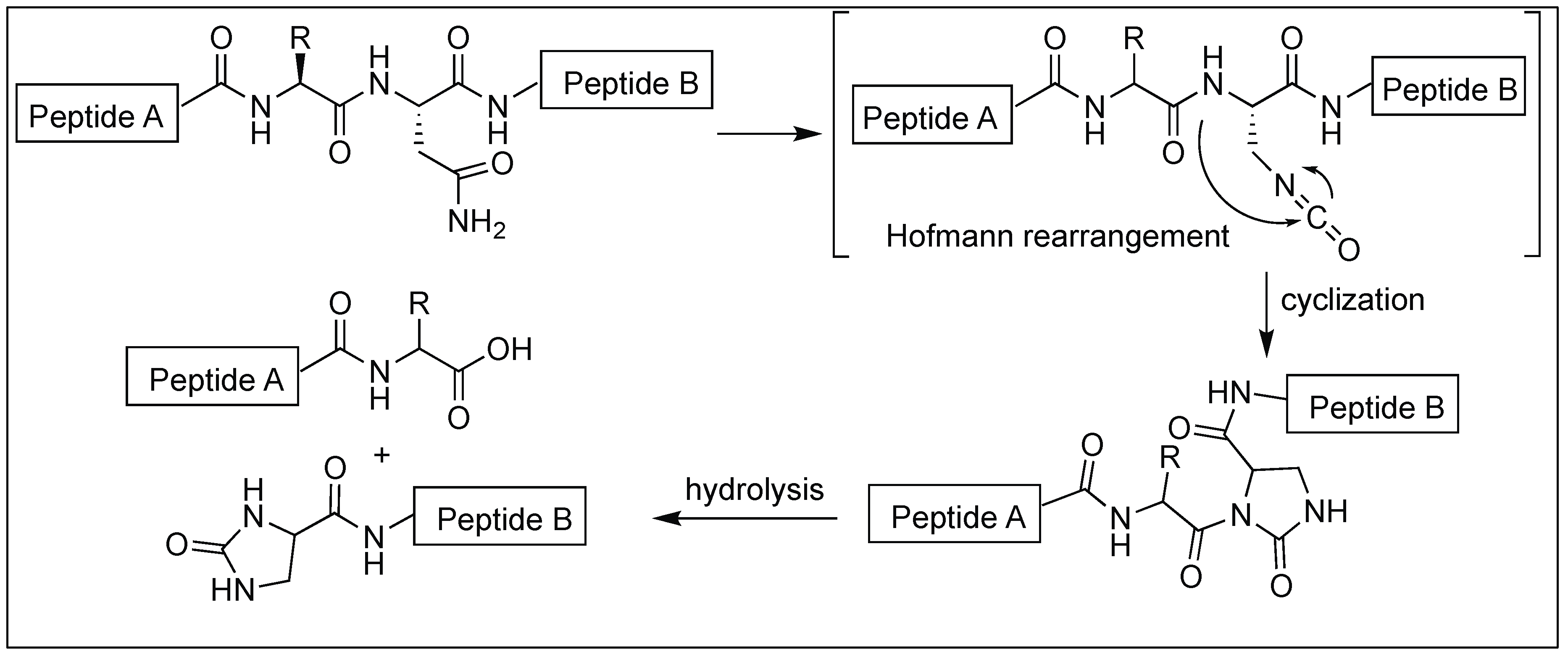
- Tanabe, K.; Taniguchi, A.; Matsumoto, T.; Oisaki, K.; Sohma, Y.; Kanai, M. Asparagine-selective cleavage of peptide bonds through hypervalent iodine-mediated Hofmann rearrangement in neutral aqueous solution. Chem. Sci. 2014, 5, 2747–2753. [Google Scholar] [CrossRef]
- Elashal, H.E.; Raj, M. Site-selective chemical cleavage of peptide bonds. Chem. Commun. 2016, 52, 6304–6307. [Google Scholar] [CrossRef] [PubMed]
- Elashal, H.E.; Cohen, R.D.; Raj, M. Fmoc solid-phase synthesis of C-terminal modified peptides by formation of a backbone cyclic urethane moiety. Chem. Commun. 2016, 52, 9699–9702. [Google Scholar] [CrossRef] [PubMed]
- Elashal, H.E.; Sim, Y.E.; Raj, M. Serine promoted synthesis of peptide thioester precursor on solid support for native chemical ligation. Chem. Sci. 2017, 8, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Elashal, H.E.; Cohen, R.D.; Elashal, H.E.; Raj, M. Oxazolidinone-mediated sequence determination of one-bead one-compound cyclic peptide libraries. Org. Lett. 2018, 20, 2374–2377. [Google Scholar] [CrossRef] [PubMed]
- Elashal, H.E.; Cohen, R. D.; Elashal, H.E.; Zong, C.H.; Link, A.J.; Raj, M. Cyclic and lasso peptides: Sequence determination, topology analysis, and rotaxane formation. Angew. Chem. Int. Ed. 2018, 57, 6150–6154. [Google Scholar] [CrossRef] [PubMed]
- Komiya, C.; Aihara, K.; Morishita, K.; Ding, H.; Inokuma, T.; Shigenaga, A.; Otaka, A. Development of an intein-inspired amide cleavage chemical device. J. Org. Chem. 2016, 81, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Piizzi, G. Gem-Disubstituent effect: Theoretical basis and synthetic applications. Chem. Rev. 2005, 105, 1735–1766. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, S.M. The gem-dimethyl effect revisited. J. Org. Chem. 2008, 73, 2466–2468. [Google Scholar] [CrossRef] [PubMed]
- Beesley, R.M.; Ingold, C.K.; Thorpe, J.F. CXIX—The formation and stability of spiro-compounds. Part I. spiro-compounds from cyclohexane. J. Chem. Soc. Trans. 1915, 107, 1080–1106. [Google Scholar] [CrossRef]
- Bochet, C.G. Photolabile protecting groups and linkers. J. Chem. Soc. Perkin Trans. 2001, 2, 125–142. [Google Scholar]
- Brieke, C.; Rohrbach, F.; Gottschalk, A.; Mayer, G.; Heckel, A. Light-controlled tools. Angew. Chem. Int. Ed. 2012, 51, 8446–8476. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Tanabe, K.; Sasaki, D.; Sohma, Y.; Oisaki, K.; Kanai, M. Serine-selective aerobic cleavage of peptides and a protein using a water-soluble copper–organoradical conjugate. Angew. Chem. Int. Ed. 2014, 53, 6501–6505. [Google Scholar] [CrossRef] [PubMed]
- Samaritoni, J.G.; Copes, A.T.; Crews, D.K.; Glos, C.; Thompson, A.L.; Wilson, C.; O’Donnell, M.J.; Scott, W.L. Unexpected hydrolytic instability of N-acylated amino acid amides and peptides. J. Org. Chem. 2014, 79, 3140–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Noshita, M.; Mukai, Y.; Morimoto, H.; Ohshima, T. Cleavage of unactivated amide bonds by ammonium salt-accelerated hydrazinolysis. Chem. Commun. 2014, 50, 12623–12625. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Hemu, X.; Liu, D.X.; Tam, J.P. Selective bi-directional amide bond cleavage of N-methylcysteinyl peptide. Eur. J. Org. Chem. 2014, 20, 4370–4380. [Google Scholar] [CrossRef]
- Creighton, C.J.; Romoff, T.T.; Bu, J.H.; Goodman, M. Mechanistic studies of an unusual amide bond scission. J. Am. Chem. Soc. 1999, 121, 6786–6791. [Google Scholar] [CrossRef]

























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Enzyme | Method of Hydrolysis | Point of Cleavage | Ref. |
|---|---|---|---|---|
| 1 | Serine and Cysteine Proteases | Oxyanion binding hole with catalytic triad | - | [32,33,34,35,36,37] |
| 2 | Metallo-endopeptidase | Thermolysine with Zn2+ binds to His 142, His 146, Glu 166 | Internal peptide bonds on the N-terminal side of large hydrophobic amino acids | [38,39,40,41,42,43,44,45,46,47,48,49,50,51] |
| 3 | Metalloexopeptidase | Carboxypeptidase A with Zn2+ through Lewis acid activation | C-terminus comprising large hydrophobic amino acids | [52,53,54] |
| 4 | O-GlcNAc transferase | Glycosylation followed by enzyme catalyzed pyroglutamate formation | N-terminal glutamic acid | [55,56] |
| 5 | Nicotinamidase | Enzyme Chelation to Zn2+ and catalytic triad | Nicotinamide | [57,58,59,60] |
| 6 | Flavoenzyme | Flavin hydroperoxide intiated oxidative mechanism | Unactivated amide bond in uracil | [61,62,63] |
| 7 | Antibody Fab-BL 125 | Catalyzes unactivated primary amide bond hydrolysis | Primary amide bond of l-isomer of peptides | [64,65] |
| 8 | RNA | Mg2+ catalyzed mechanism | Unactivated alkyl amide of DNA analog | [66] |
| Entry | Metal Complex | Method of Hydrolysis | Point of Cleavage | Ref. |
|---|---|---|---|---|
| 1 | Simple metal ions | Lewis acidity of metal ion | C-terminal of peptide | [67,68,69,70,71,72] |
| 2 | Zr POMs, Zr MOF-808 | Lewis acidity of metal ion | C-terminal of peptide | [73,74,75,76,77,78,79,80,81,82] |
| 3 | Mo(VI) | Lewis acidity of metal ion and formation of 5 membered ring | C-terminal side of Asp | [81,82] |
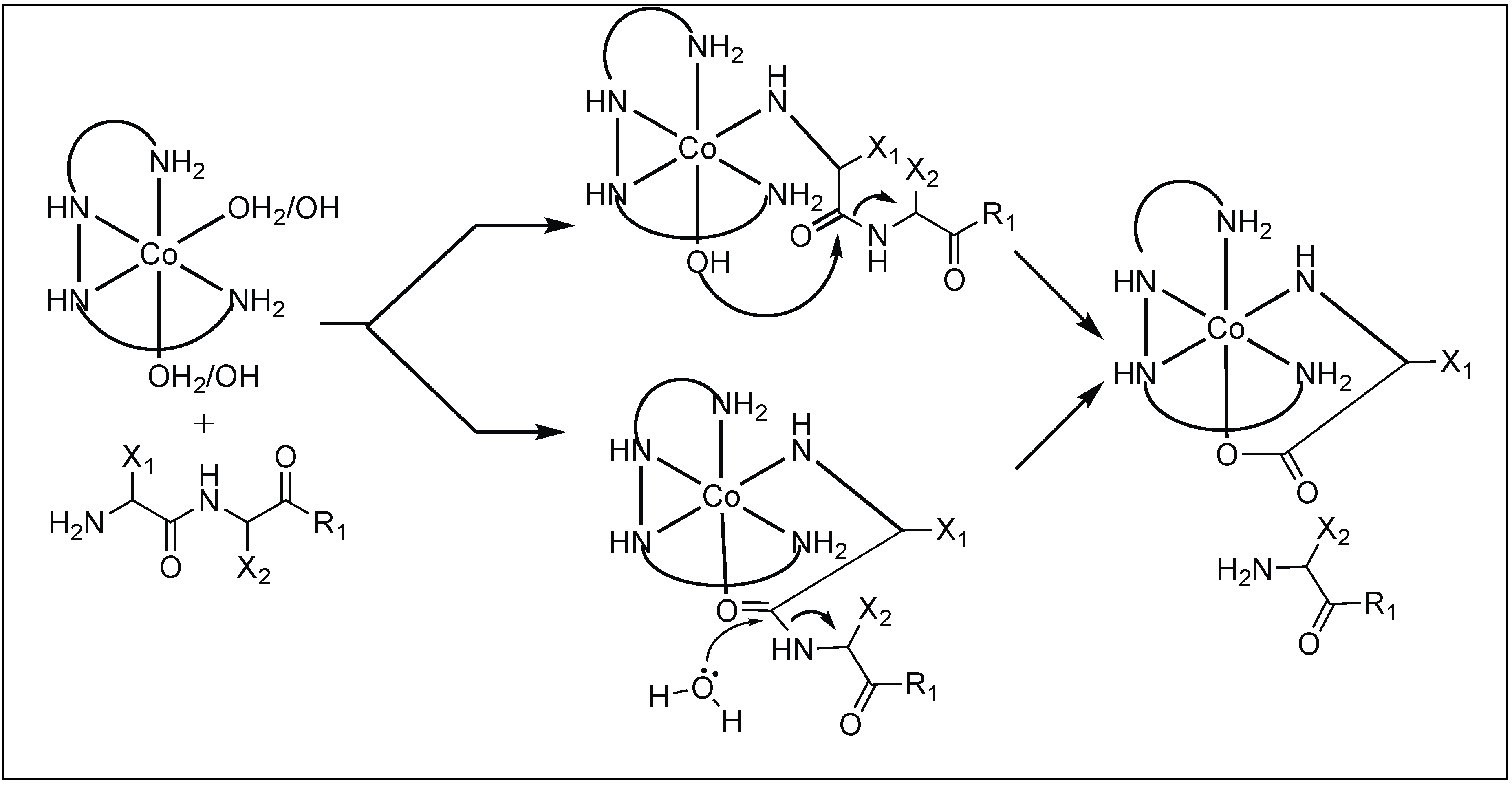
| 4 | Co(III) | N-terminal amine intiated tertiary complex | C-terminal of peptide | [83,84,85,86,87,88] |
| 5 | Mo(II) | Favorable six-membered chelate ring | C-terminal side of Cys | [89] |
| 6 | Pd(II), Pt(II) | Carboxylic group of amino acid and side chain of amino acid anchoring metal complex | C-terminal side of Met, His, Cys and S-MeCys | [90,91,92,93,94,95,96,97,98] |
| 7 | Pd(0) | Methionine side chain anchoring metal complex | Second amide bond upstream from Met | [99,100,101,102] |
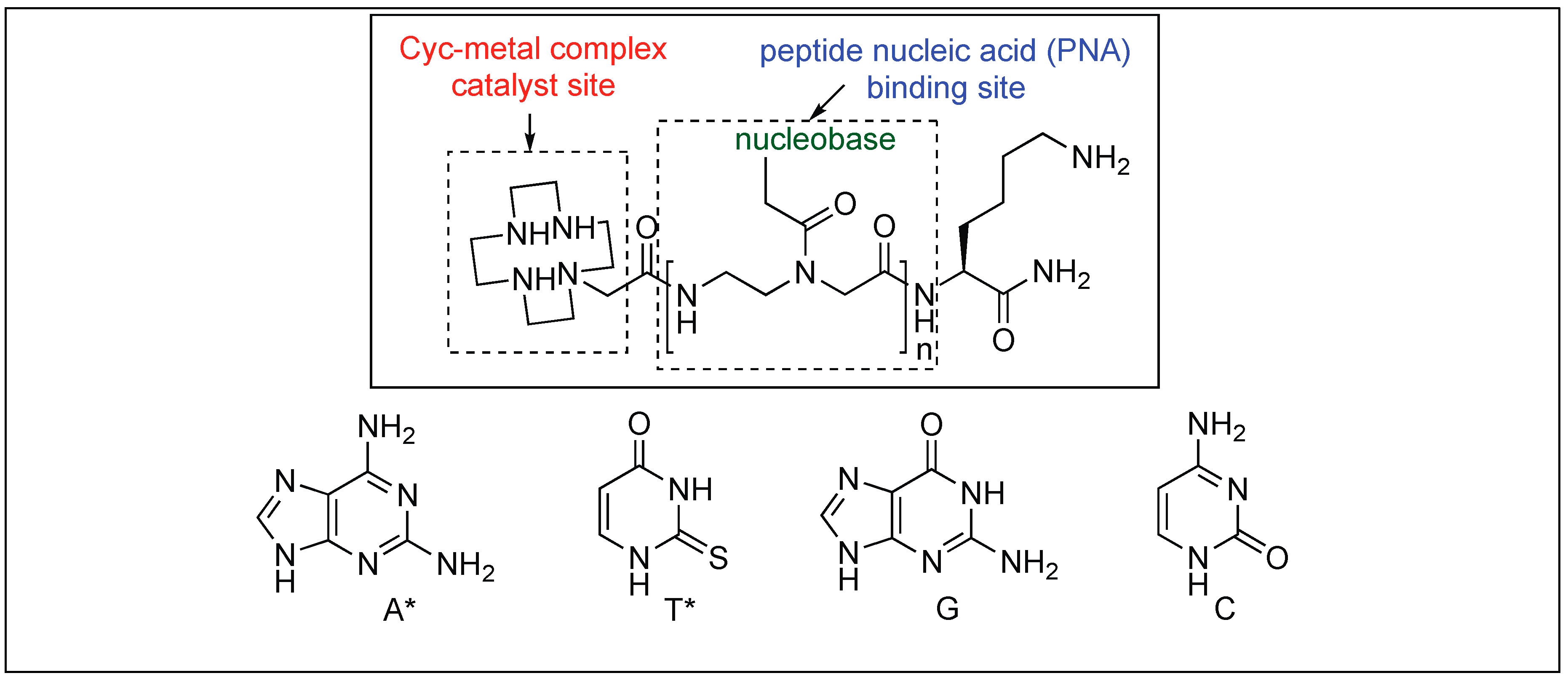
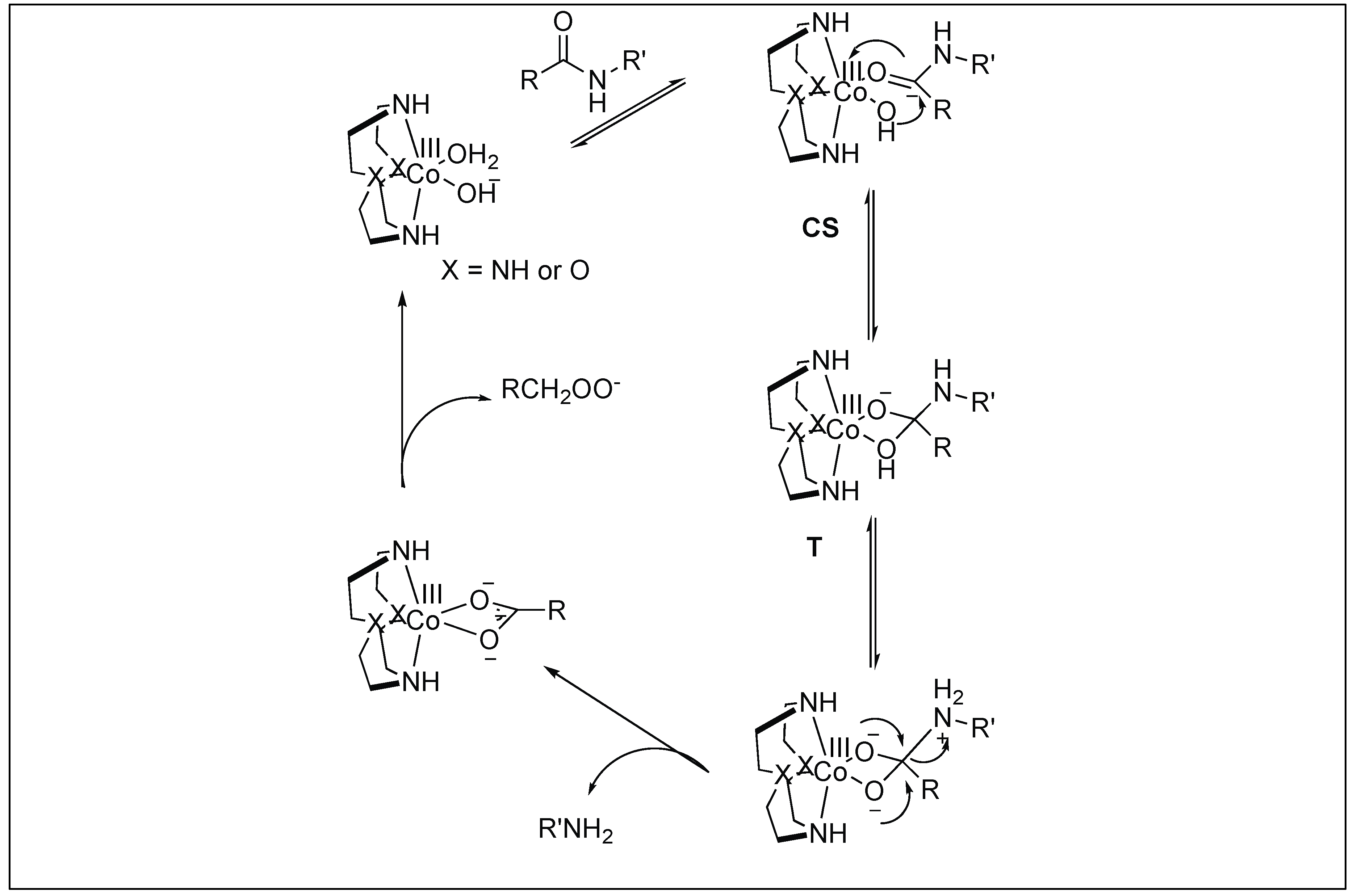
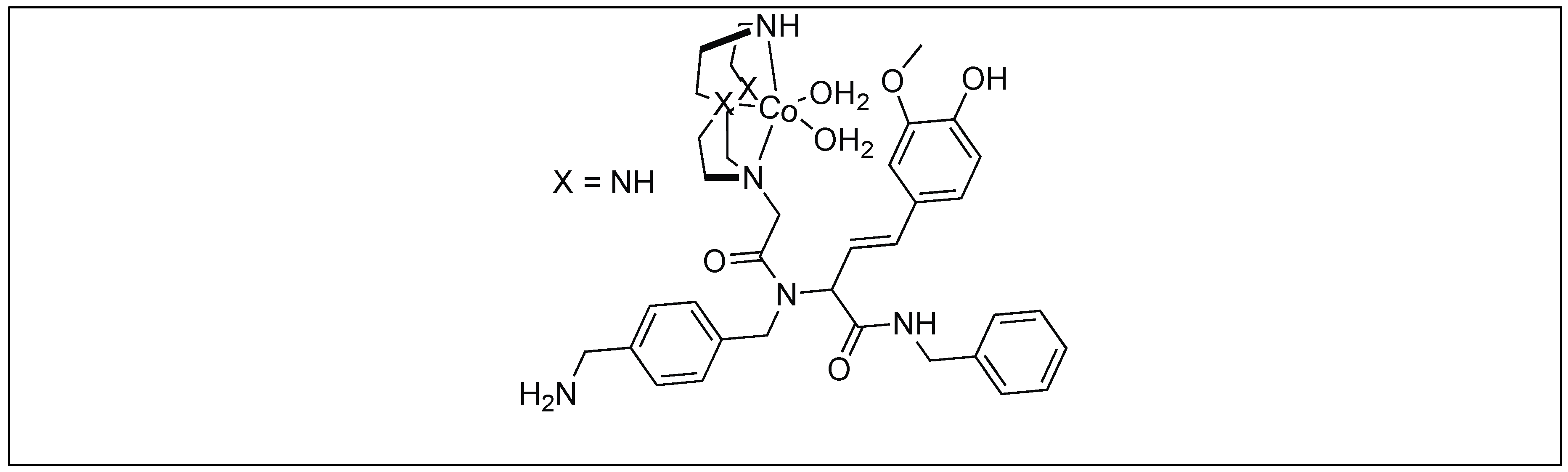
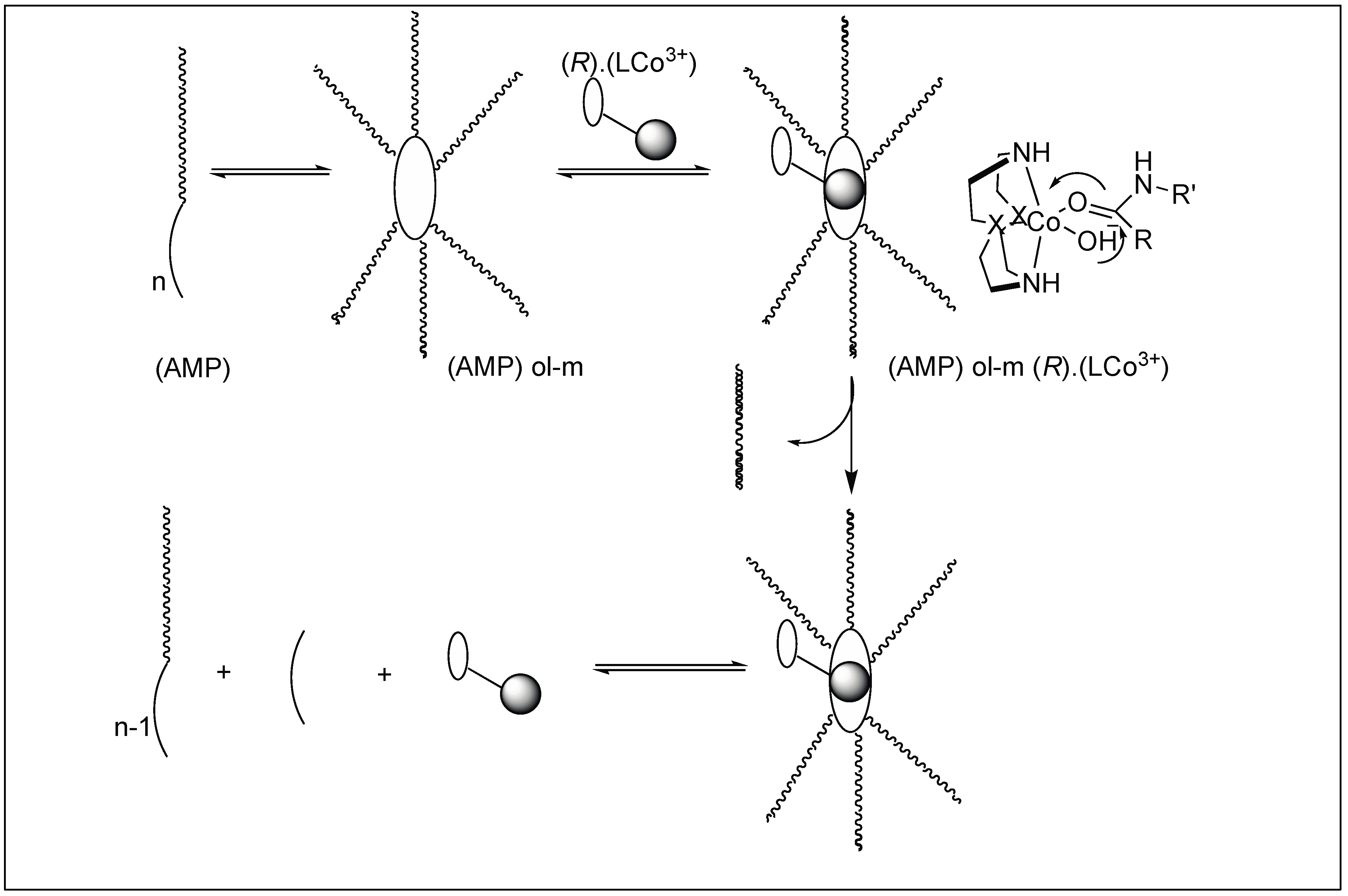
| 8 | Co(III) and Cu(II) | Lewis acidity activation and PNA for selectivity towards a particular protein | Mb = Leu 89 − Ala 90 | [103,104,105,106,107,108,109,110,111] |
| PDF = Gln 152 − Arg 153 | ||||
| BSA = Solvent exposed portion | ||||
| 9 | Ni(II) | Non Lewis acid based N,O acyl rearrangement | N-terminal side of Ser/Thr | [112,113] |
| 10 | Sc(III) | Lewis acid based N,O acyl rearrangement | N-terminal side of Ser/Thr | [114] |
| Entry | Organic Molecules | Method of Hydrolysis | Point of Cleavage | Ref. |
|---|---|---|---|---|
| 1 | Phenyl isothiocyanate | Through 5 membered cyclic phenylisothiocyanate intermediate | N-terminal side of peptide | [115] |
| 2 | Cyanogen bromide | Through 5 membered iminolactone | C-terminal side of Met | [116] |
| 3 | 2-Nitro-5-thiocyano benzoic acid | Through 5 membered thialactone | N-terminal side of Cys | [117] |
| 4 | 2-iodosobenzoic acid | Through iminospirolactone | C-terminal side of Trp | [118,119] |
| 5 | TBC | Through Oxindole | C-terminal side of Trp | [118,119] |
| 6 | N-amidination | Through 5 membered cyclic amidine ring | N-terminal side of peptide | [120] |
| 7 | Protecting goups (Table 4) | Lactonization strategy | C-terminal side of peptide | [121,122,123,124,125,126,127] |
| 8 | H2O2 responsive protecting groups | Lactonization strategy | C-terminal side of peptide | [128] |
| 9 | PyBroP | Glutamic acid selective activation through pyroglutamyl imide | N-terminal side of Glu | [129,130] |
| 10 | DIB | Hofmann rearrangement mediated N-acylurea intermediate | N-terminal side of Asn | [131] |
| 11 | DSC | Cyclic urethane amide activation | N-terminal side of Ser, Thr, Cys and Glu | [132,133,134,135,136] |
| 12 | o-NBnoc | A photo responsive amide cleavage through succinimide ring | C-terminal side of Asn | [137,138,139,140,141,142] |
| 13 | Cu-organo radical conjugate | Aerobic chemoselective oxidation of Ser followed by oxalimide formation | N-terminal side of Ser | [143] |
| 14 | TFA/H2O | N-acyl group mediated oxazolinium specie | C-terminal side of peptide | [144] |
| 15 | Hydrazine hydrate | Hydrazinolysis accelarated by addition of ammonium salts | N-terminal of peptide | [145] |
| 16 | N-Mecysteine | Through oxazolinium ion | C-terminal side of N-MeCys | [146] |
| 17 | Acylated N-MeAib | Through oxazolinium ion | C-terminal side of N-MeAib | [147] |

| Reagent/Condition | PG | R1 | R2 | R3 |
|---|---|---|---|---|
| Ultraviolet Near-infrared Fluoride hypoxia |  | NO2 | H | H |
| NO2 | OMe | OMe | ||
| H | OTBDPS | H | ||
| H | NO2 | H | ||
| Thiol | 4-nitrobenzenesulfonyl | - | - | - |
| Phosohatase | phosphate | - | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahesh, S.; Tang, K.-C.; Raj, M. Amide Bond Activation of Biological Molecules. Molecules 2018, 23, 2615. https://doi.org/10.3390/molecules23102615
Mahesh S, Tang K-C, Raj M. Amide Bond Activation of Biological Molecules. Molecules. 2018; 23(10):2615. https://doi.org/10.3390/molecules23102615
Chicago/Turabian StyleMahesh, Sriram, Kuei-Chien Tang, and Monika Raj. 2018. "Amide Bond Activation of Biological Molecules" Molecules 23, no. 10: 2615. https://doi.org/10.3390/molecules23102615
APA StyleMahesh, S., Tang, K. -C., & Raj, M. (2018). Amide Bond Activation of Biological Molecules. Molecules, 23(10), 2615. https://doi.org/10.3390/molecules23102615