Role of Extracellular Loops and Membrane Lipids for Ligand Recognition in the Neuronal Adenosine Receptor Type 2A: An Enhanced Sampling Simulation Study

 , , and
, , and
Abstract
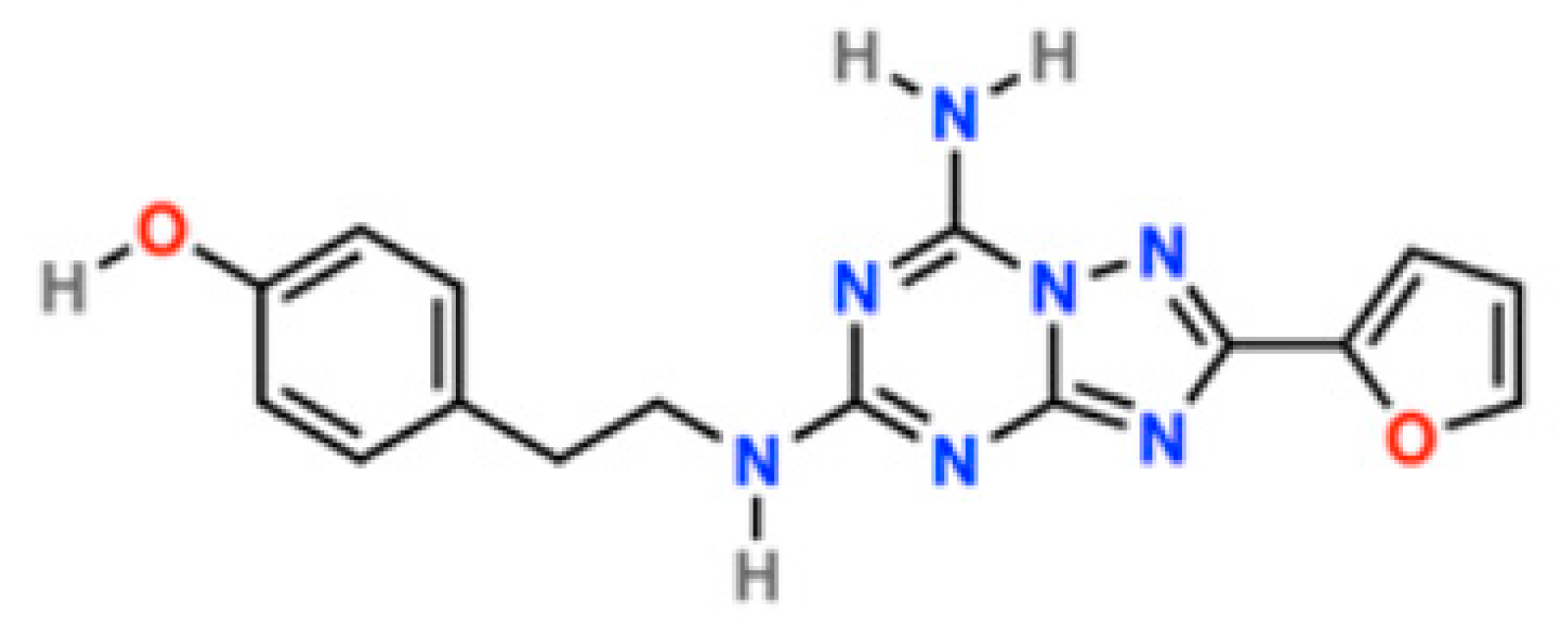
:
1. Introduction
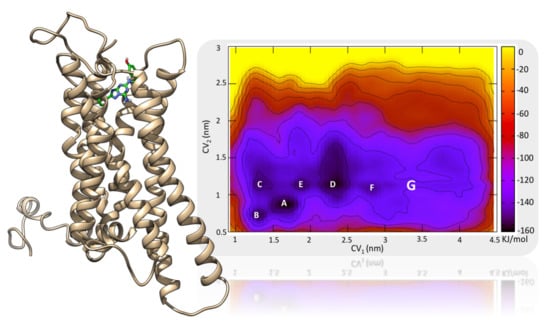
2. Results
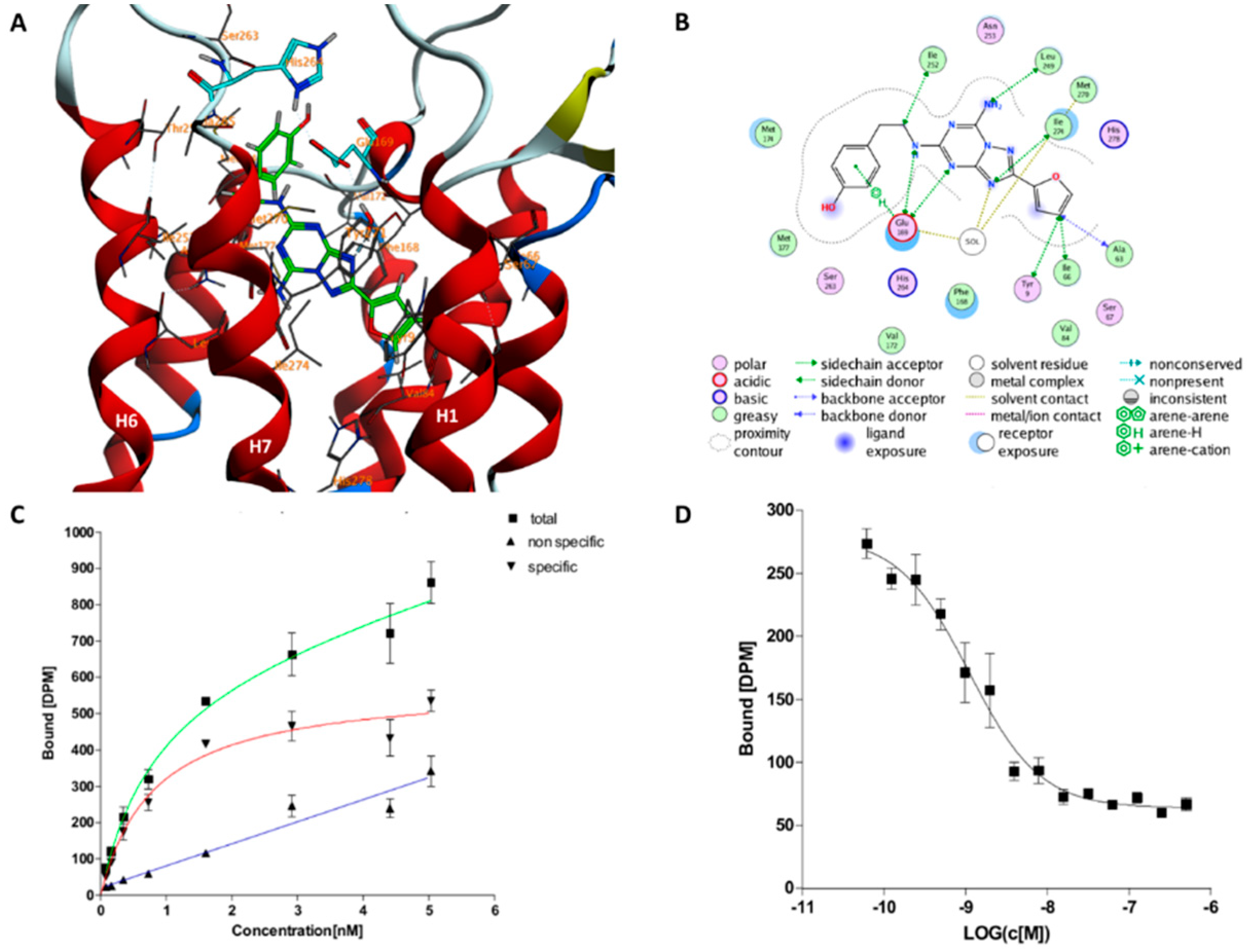
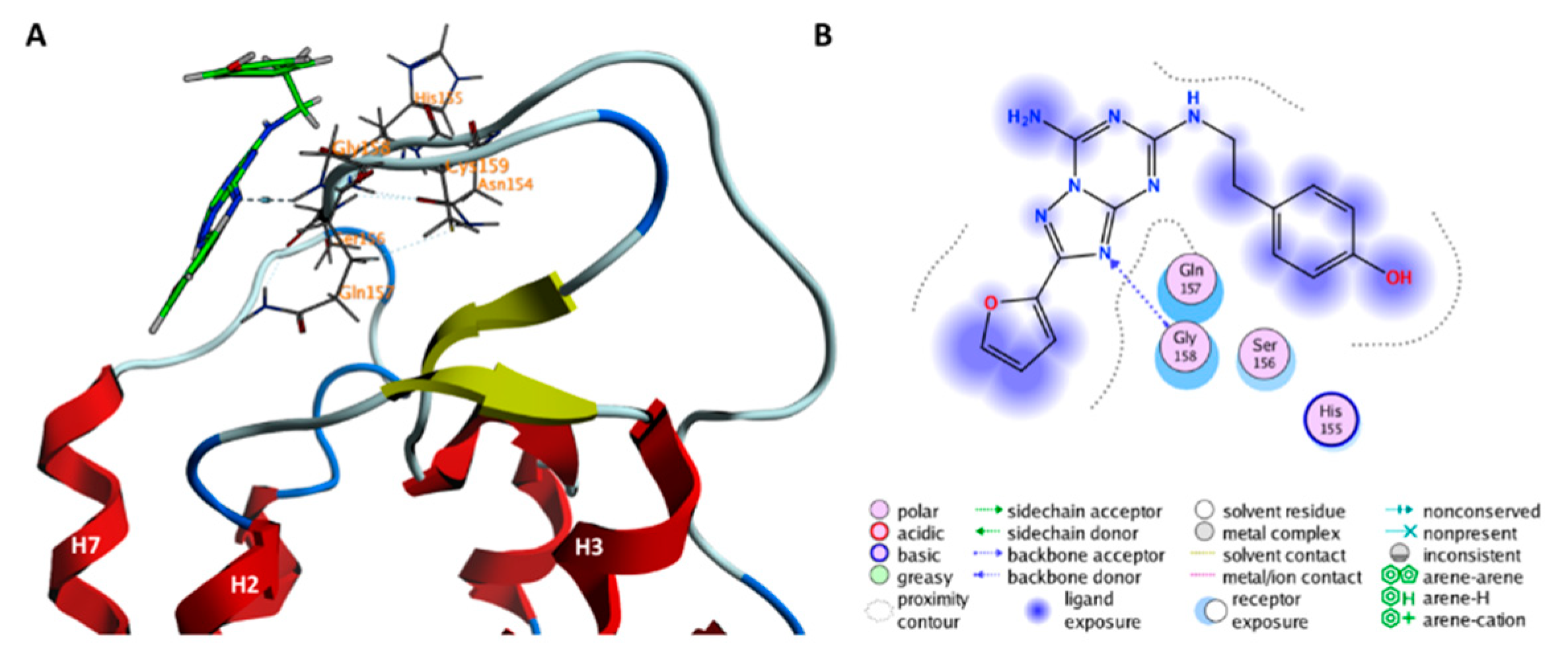
2.1. The Orthosteric Binding Site
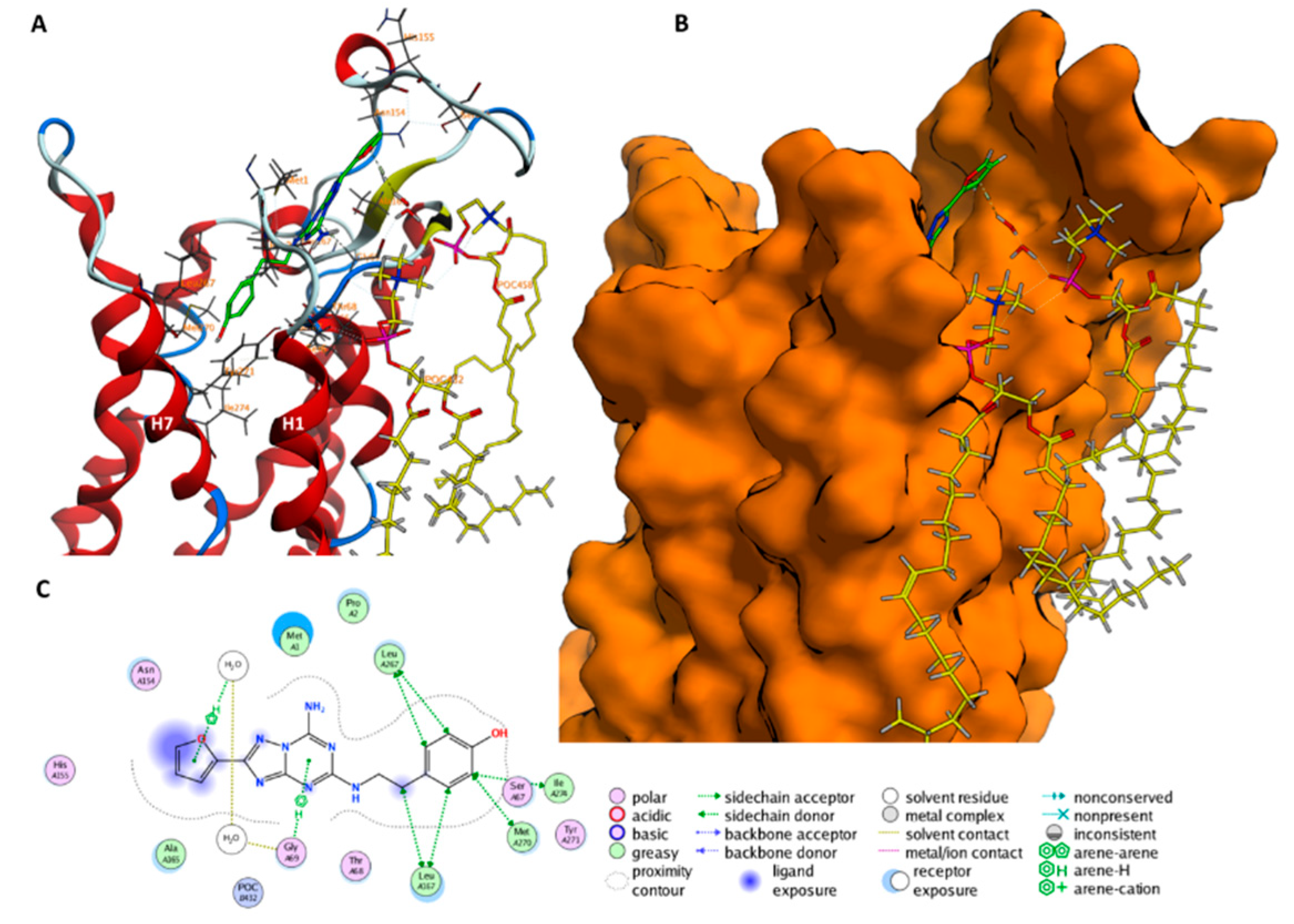
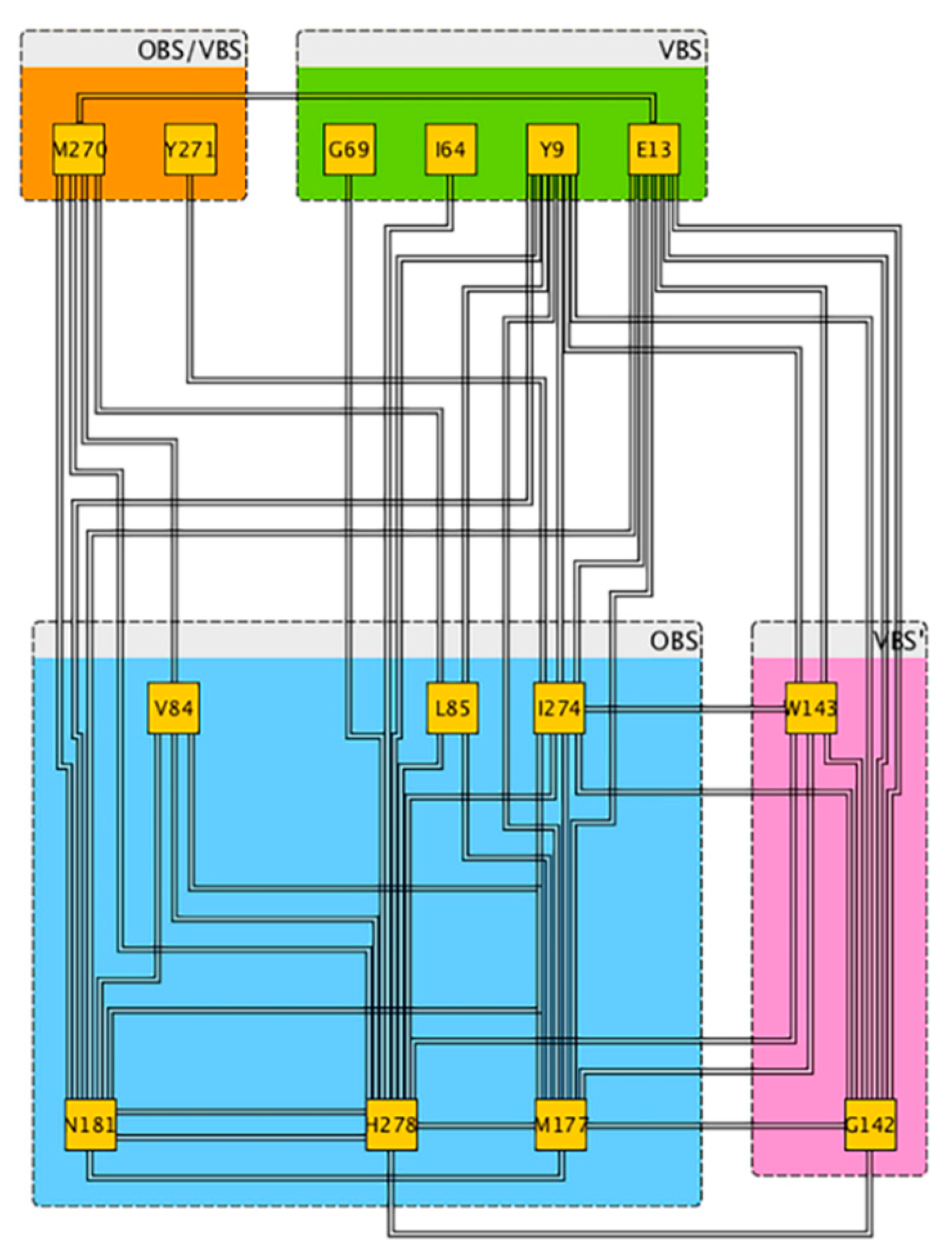
2.2. Role of ECLs in Molecular Recognition
2.3. An Access Control Binding Site
2.4. Allosterism
3. Discussion
4. Materials and Methods
4.1. System Preparation and MD Simulations
4.2. Metadynamics Simulations
4.3. Experimental Affinity Testing
4.3.1. Cell Culture
4.3.2. Experimental Binding Affinity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Kroeze, W.K.; Sheffler, D.J.; Roth, B.L. G-protein-coupled receptors at a glance. J. Cell Sci. 2003, 116, 4867–4869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöneberg, T.; Schulz, A.; Biebermann, H.; Hermsdorf, T.; Römpler, H.; Sangkuhl, K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol. Ther. 2004, 104, 173–206. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.S.; Weaver, D.R.; Rivkees, S.A.; Peterfreund, R.A.; Pollack, A.E.; Adler, E.M.; Reppert, S.M. Molecular cloning of the rat A2 adenosine receptor: Selective co-expression with D2 dopamine receptors in rat striatum. Brain Res. Mol. Brain Res. 1992, 14, 186–195. [Google Scholar] [CrossRef]
- Xu, K.; Bastia, E.; Schwarzschild, M. Therapeutic potential of adenosine A(2A) receptor antagonists in Parkinson’s disease. Pharmacol. Ther. 2005, 105, 267–310. [Google Scholar] [CrossRef] [PubMed]
- Gimpl, G. Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 2016, 199, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.C.; van Westen, G.J.P.; Li, Q.; IJzerman, A.P. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 2011, 32, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed]
- Avlani, V.A.; Gregory, K.J.; Morton, C.J.; Parker, M.W.; Sexton, P.M.; Christopoulos, A. Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J. Biol. Chem. 2007, 282, 25677–25686. [Google Scholar] [CrossRef] [PubMed]
- Granier, S.; Kobilka, B. A new era of GPCR structural and chemical biology. Nat. Chem. Biol. 2012, 8, 670–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Sandal, M.; Behrens, M.; Brockhoff, A.; Musiani, F.; Giorgetti, A.; Carloni, P.; Meyerhof, W. Evidence for a transient additional ligand binding site in the TAS2R46 bitter taste receptor. J. Chem. Theory Comput. 2015, 11, 4439–4449. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, N.; Pardo, L.; Fabritiis, G.D. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016, 6, 22639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provasi, D.; Bortolato, A.; Filizola, M. Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry 2009, 48, 10020–10029. [Google Scholar] [CrossRef] [PubMed]
- Horn, F.; Weare, J.; Beukers, M.W.; Horsch, S.; Bairoch, A.; Chen, W.; Edvardsen, O.; Campagne, F.; Vriend, G. GPCRDB: An information system for G protein-coupled receptors. Nucleic Acids Res. 1998, 26, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, S.; Provasi, D.; Filizola, M. The dynamic process of drug-GPCR binding at either orthosteric or allosteric sites evaluated by metadynamics. Methods Mol. Biol. (Clifton, N.J.) 2015, 1335, 277–294. [Google Scholar] [CrossRef]
- Poucher, S.M.; Keddie, J.R.; Singh, P.; Stoggall, S.M.; Caulkett, P.W.; Jones, G.; Coll, M.G. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2a selective adenosine receptor antagonist. Br. J. Pharmacol. 1995, 115, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Dore, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Segala, E.; Guo, D.; Cheng, R.K.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Pan, A.C.; Dror, R.O.; Mocking, T.; Liu, R.; Heitman, L.H.; Shaw, D.E.; IJzerman, A.P. Molecular basis of ligand dissociation from the adenosine A2A receptor. Mol. Pharmacol. 2016, 89, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.Y.; Rossetti, G.; Bauer, A.; Carloni, P. Binding of the antagonist caffeine to the human adenosine receptor hA(2A)R in nearly physiological conditions. PLoS ONE 2015, 10, e0126833. [Google Scholar] [CrossRef]
- Grouleff, J.; Irudayam, S.J.; Skeby, K.K.; Schiott, B. The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. Biochim. Biophys. Acta 2015, 1848, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Guixà-González, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Martí-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martín, M. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8, 14505. [Google Scholar] [CrossRef] [PubMed]
- Pucadyil, T.J.; Chattopadhyay, A. Cholesterol modulates ligand binding and G-protein coupling to serotonin(1A) receptors from bovine hippocampus. Biochim. Biophys. Acta 2004, 1663, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Gimpl, G.; Fahrenholz, F. Alteration of the myometrial plasma membrane cholesterol content with beta.-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 1995, 34, 13784–13793. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Taub, D. CXCR4 function requires membrane cholesterol: Implications for HIV infection. J. Immunol. 2002, 168, 4121–4126. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, M. LLC New York, NY: 2009. Available online: https://www.schrodinger.com (accessed on 8 October 2018).
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batyuk, A.; Galli, L.; Ishchenko, A.; Han, G.W.; Gati, C.; Popov, P.A.; Lee, M.Y.; Stauch, B.; White, T.A.; Barty, A.; et al. Native phasing of X-ray free-electron laser data for a G protein-coupled receptor. Sci. Adv. 2016, 2, e1600292. [Google Scholar] [CrossRef] [PubMed]
- Martin-Garcia, J.M.; Conrad, C.E.; Nelson, G.; Stander, N.; Zatsepin, N.A.; Zook, J.; Zhu, L.; Geiger, J.; Chun, E.; Kissick, D.; et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 2017, 4, 439–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikov, I.; Polovinkin, V.; Kovalev, K.; Gushchin, I.; Shevtsov, M.; Shevchenko, V.; Mishin, A.; Alekseev, A.; Rodriguez-Valera, F.; Borshchevskiy, V.; et al. Fast iodide-SAD phasing for high-throughput membrane protein structure determination. Sci. Adv. 2017, 3, e1602952. [Google Scholar] [CrossRef] [PubMed]
- Broecker, J.; Morizumi, T.; Ou, W.L.; Klingel, V.; Kuo, A.; Kissick, D.J.; Ishchenko, A.; Lee, M.Y.; Xu, S.; Makarov, O.; et al. High-throughput in situ X-ray screening of and data collection from protein crystals at room temperature and under cryogenic conditions. Nat. Protoc. 2018, 13, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.-G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 2018, 172, 68–80.e12. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.; Olieric, N.; Cheng, R.; Brunle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jiang, Q.; Glashofer, M.; Yehle, S.; Wess, J.; Jacobson, K.A. Glutamate residues in the second extracellular loop of the human A2a adenosine receptor are required for ligand recognition. Mol. Pharmacol. 1996, 49, 683–691. [Google Scholar] [PubMed]
- Sun, B.; Bachhawat, P.; Chu, M.L.; Wood, M.; Ceska, T.; Sands, Z.A.; Mercier, J.; Lebon, F.; Kobilka, T.S.; Kobilka, B.K. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.-P.; Lane, J.R.; Lin, J.Y.; Katritch, V.; IJzerman, A.P.; Stevens, R.C. Identification and characterization of amino acid residues essential for human A2A adenosine receptor: ZM241385 binding and subtype selectivity. J. Biol. Chem. 2010, 285, 13032–13044. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Dreier, J.L.; Mandala, S.; Proia, R.L. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J. Biol. Chem. 2004, 279, 15396–15401. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Lee, C.T.; Durrant, J.D.; Malmstrom, R.D.; Feher, V.A.; Amaro, R.E. Emerging computational methods for the rational discovery of allosteric drugs. Chem. Rev. 2016, 116, 6370–6390. [Google Scholar] [CrossRef] [PubMed]
- Baker, F.N.; Porollo, A. CoeViz: A web-based tool for coevolution analysis of protein residues. BMC Bioinform. 2016, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Burger, L.; van Nimwegen, E. Disentangling direct from indirect co-evolution of residues in protein alignments. PLoS Comput. Biol. 2010, 6, e1000633. [Google Scholar] [CrossRef] [PubMed]
- De Juan, D.; Pazos, F.; Valencia, A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 2013, 14, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Yang, M.; Han, K. Antagonist binding and induced conformational dynamics of GPCR A2A adenosine receptor. Proteins 2013, 81, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lyman, E. Agonist dynamics and conformational selection during microsecond simulations of the A(2A) adenosine receptor. Biophys. J. 2012, 102, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta Biomembr. 2004, 1666, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Fenalti, G.; Brown, M.A.; Katritch, V.; Abagyan, R.; Cherezov, V.; Stevens, R.C. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc. 2010, 132, 11443–11445. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, A.N.; Chevalier, A.; Cousins, G.D.; Ayettey, E.; McCusker, E.C.; Wenk, C.; Robinson, A.S. Conserved disulfide bond is not essential for the adenosine A2A receptor: Extracellular cysteines influence receptor distribution within the cell and ligand-binding recognition. Biochim. Biophys. Acta 2015, 1848, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, A. GPCRs: Lipid-dependent membrane receptors that act as drug targets. Adv. Biol. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Dijkman, P.M.; Watts, A. Lipid modulation of early G protein-coupled receptor signalling events. Biochim. Biophys. Acta 2015, 1848, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A Lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W. Oxidation reactions in Parkinson’s disease. Neurology 1990, 40, 37–39. [Google Scholar]
- Ikeda, K.; Kurokawa, M.; Aoyama, S.; Kuwana, Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J. Neurochem. 2002, 80, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.M.; Wilkins, A.D.; Rodriguez, G.J.; Wensel, T.G.; Lichtarge, O. Intramolecular allosteric communication in dopamine D2 receptor revealed by evolutionary amino acid covariation. Proc. Natl. Acad. Sci. USA 2016, 113, 3539–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, M.; Burgueno, J.; Marcellino, D.; Cabello, N.; Canela, E.I.; Mallol, J.; Agnati, L.; Ferre, S.; Bouvier, M.; Fuxe, K.; et al. Homodimerization of adenosine A2A receptors: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Neurochem. 2004, 88, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Martinez-Pinilla, E.; Lanciego, J.L.; Navarro, G. Basic pharmacological and structural evidence for class A G-protein-coupled receptor heteromerization. Front. Pharmacol. 2016, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, F.; Felline, A. Dimerization and ligand binding affect the structure network of A2A adenosine receptor. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, Y.; Komuro, Y.; Kobayashi, C.; Jung, J.; Mori, T.; Sugita, Y. Dimensionality of collective variables for describing conformational changes of a multi-domain protein. J. Phys. Chem. Lett. 2016, 7, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Jafurulla, M.; Chattopadhyay, A. Sphingolipids in the function of G protein-coupled receptors. Eur. J. Pharmacol. 2015, 763, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Massink, A.; Gutierrez-de-Teran, H.; Lenselink, E.B.; Ortiz Zacarias, N.V.; Xia, L.; Heitman, L.H.; Katritch, V.; Stevens, R.C.; AP, I.J. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol. Pharmacol. 2015, 87, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W. Role of cholesterol in synapse formation and function. Biochim. Biophys. Acta 2003, 1610, 271–280. [Google Scholar] [CrossRef]
- Serebryany, E.; Zhu, G.A.; Yan, E.C. Artificial membrane-like environments for in vitro studies of purified G-protein coupled receptors. Biochim. Biophys. Acta 2012, 1818, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, T.E.; Hoffman, J.F.; Fanestil, D.D. Membrane Physiology; Springer: Boston, MA, USA, 1980. [Google Scholar]
- DeLano, W.L. The PyMol Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Hummer, G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.P.; Lyubartsev, A.P. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.P.; Lyubartsev, A.P. An extension and further validation of an all-atomistic force field for biological membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hünenberger, P. Thermostat algorithms for molecular dynamics simulations. Adv. Comput. Simul. 2005, 105–149. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single-crystals—A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald—An N.Log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Bochicchio, A.; Rossetti, G.; Tabarrini, O.; Kraubeta, S.; Carloni, P. Molecular view of ligands specificity for CAG repeats in anti-Huntington therapy. J. Chem. Theory Comput. 2015, 11, 4911–4922. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Rossetti, G.; Arnesano, F.; Ippoliti, E.; Natile, G.; Carloni, P. Molecular recognition of platinated DNA from chromosomal HMGB1. J. Chem. Theory Comput. 2014, 10, 3578–3584. [Google Scholar] [CrossRef] [PubMed]
- Kranjc, A.; Bongarzone, S.; Rossetti, G.; Biarnes, X.; Cavalli, A.; Bolognesi, M.L.; Roberti, M.; Legname, G.; Carloni, P. Docking ligands on protein surfaces: The case study of prion protein. J. Chem. Theory Comput. 2009, 5, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Paramo, T.; East, A.; Garzon, D.; Ulmschneider, M.B.; Bond, P.J. Efficient characterization of protein cavities within molecular simulation trajectories: Trj_cavity. J. Chem. Theory Comput. 2014, 10, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Porollo, A.A.; Adamczak, R.; Meller, J. POLYVIEW: A flexible visualization tool for structural and functional annotations of proteins. Bioinformatics 2004, 20, 2460–2462. [Google Scholar] [CrossRef] [PubMed]
- Klotz, K.-N.; Hessling, J.; Hegler, J.; Owman, C.; Kull, B.; Fredholm, B.; Lohse, M. Comparative pharmacology of human adenosine receptor subtypes–characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1997, 357, 1–9. [Google Scholar] [CrossRef]
- Neuhoff, V.; Philipp, K.; Zimmer, H.G.; Mesecke, S. A Simple, Versatile, Sensitive and Volume-Independent Method for Quantitative Protein Determination which is Independent of Other External Influences. Biol. Chem. 1979, 360, 1657–1670. [Google Scholar] [CrossRef]
- Stevens, R.C.; Cherezov, V.; Katritch, V.; Abagyan, R.; Kuhn, P.; Rosen, H.; Wuthrich, K. The GPCR network: A large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov. 2013, 12, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Pietrucci, F.; Marinelli, F.; Carloni, P.; Laio, A. Substrate binding mechanism of HIV-1 protease from explicit-solvent atomistic simulations. J. Am. Chem. Soc. 2009, 131, 11811–11818. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, R.; Giorgetti, A.; Bauer, A.; Neumaier, B.; Rossetti, G.; Carloni, P. Role of Extracellular Loops and Membrane Lipids for Ligand Recognition in the Neuronal Adenosine Receptor Type 2A: An Enhanced Sampling Simulation Study. Molecules 2018, 23, 2616. https://doi.org/10.3390/molecules23102616
Cao R, Giorgetti A, Bauer A, Neumaier B, Rossetti G, Carloni P. Role of Extracellular Loops and Membrane Lipids for Ligand Recognition in the Neuronal Adenosine Receptor Type 2A: An Enhanced Sampling Simulation Study. Molecules. 2018; 23(10):2616. https://doi.org/10.3390/molecules23102616
Chicago/Turabian StyleCao, Ruyin, Alejandro Giorgetti, Andreas Bauer, Bernd Neumaier, Giulia Rossetti, and Paolo Carloni. 2018. "Role of Extracellular Loops and Membrane Lipids for Ligand Recognition in the Neuronal Adenosine Receptor Type 2A: An Enhanced Sampling Simulation Study" Molecules 23, no. 10: 2616. https://doi.org/10.3390/molecules23102616
APA StyleCao, R., Giorgetti, A., Bauer, A., Neumaier, B., Rossetti, G., & Carloni, P. (2018). Role of Extracellular Loops and Membrane Lipids for Ligand Recognition in the Neuronal Adenosine Receptor Type 2A: An Enhanced Sampling Simulation Study. Molecules, 23(10), 2616. https://doi.org/10.3390/molecules23102616