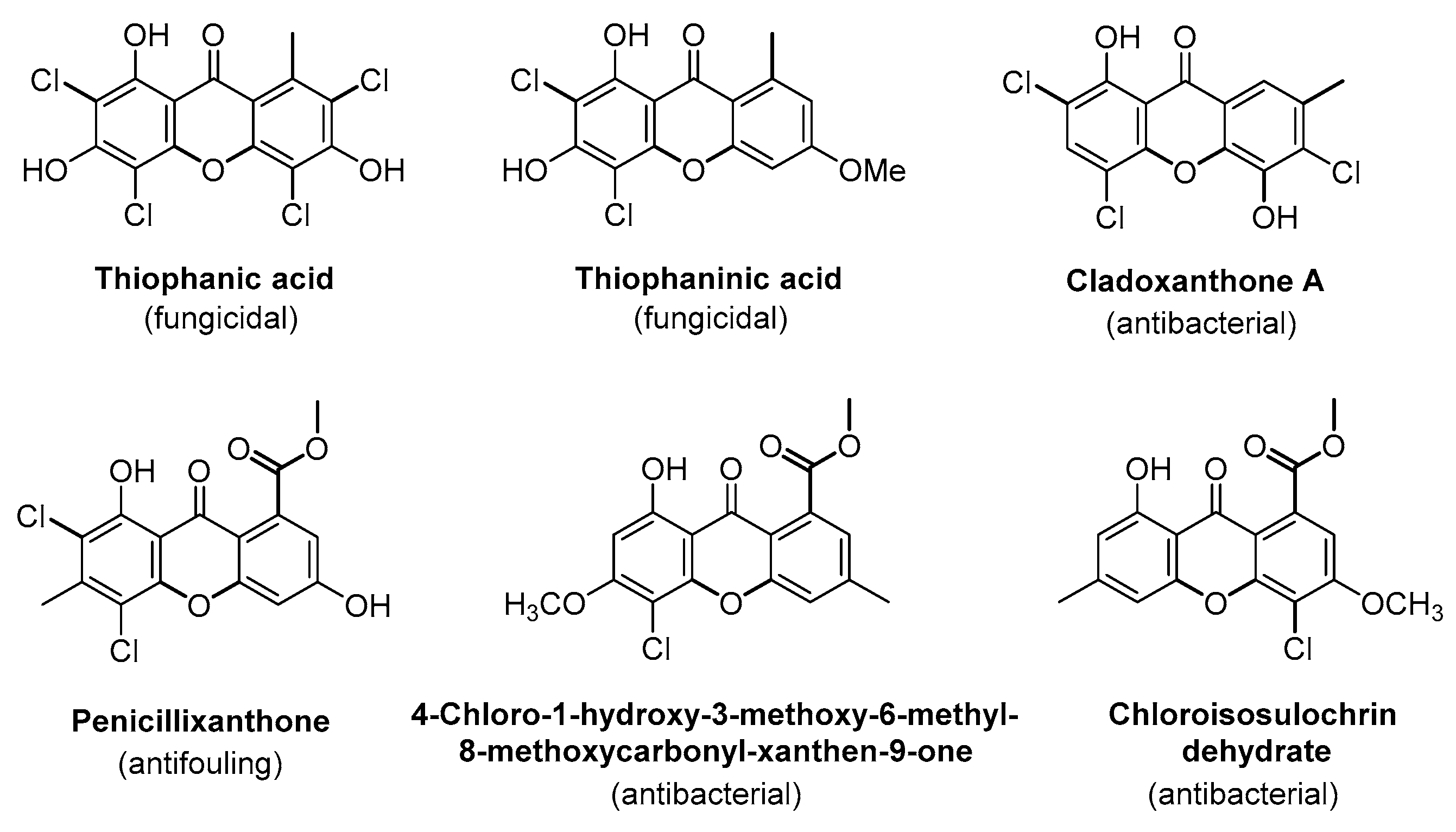
Lichen Xanthones as Models for New Antifungal Agents

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
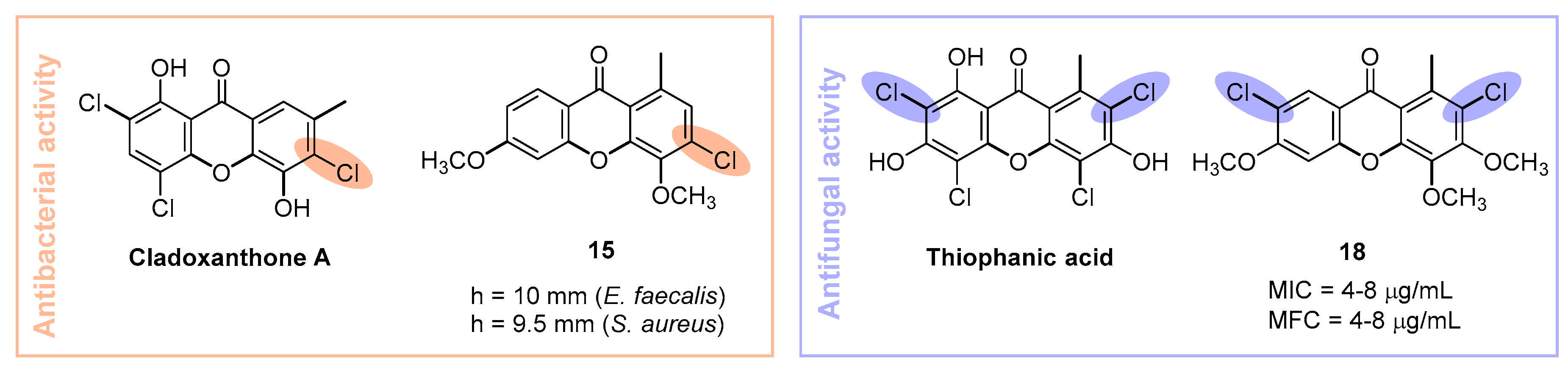
2.2. Microbiology
3. Materials and Methods
3.1. Chemistry
3.1.1. General
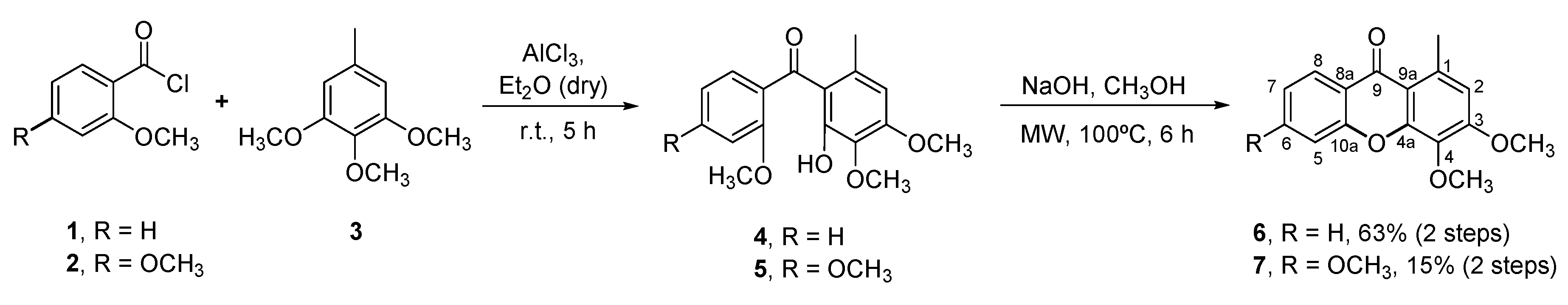
3.1.2. Synthesis of the Starting Materials 3,4-Dimethoxy-1-methyl-9H-xanthen-9-one (6) and 3,4,6-Trimethoxy-1-methyl-9H-xanthen-9-one (7)
Synthesis of the Benzophenone Intermediates 4 and 5
Synthesis of 3,4-Dimethoxy-1-methyl-9H-xanthen-9-one (6) and 3,4,6-Trimethoxy-1-methyl-9H-xanthen-9-one (7)
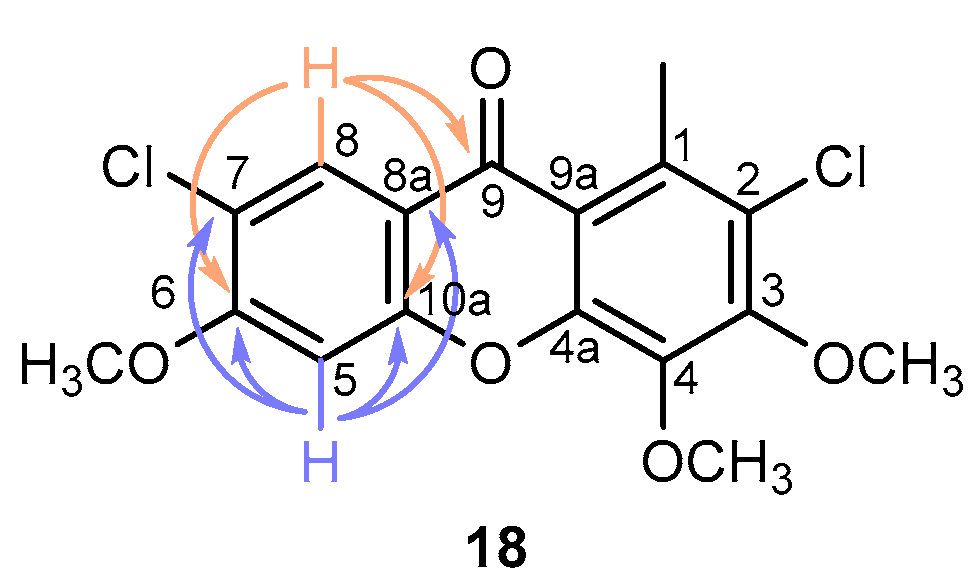
3.1.3. Chlorination Studies on 3,4-Dimethoxy-1-methyl-9H-xanthen-9-one (6)
3.1.4. Chlorination Studies on 3,4,6-Trimethoxy-1-methyl-9H-xanthen-9-one (7)
3.2. Microbiology
3.2.1. Microorganism Strains and Growth Conditions
3.2.2. Antimicrobial Susceptibility Testing
Antibacterial Activity
Antifungal Activity
3.2.3. Antibiotic Synergy Testing
Antibiotic Synergy
Antifungal Synergy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, G.; Dai, M.; Ahmed, S.; Hao, H.; Wang, X.; Yuan, Z. Antimicrobial drugs in fighting against antimicrobial resistance. Front. Microbiol. 2016, 7, 470. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone derivatives: New insights in biological activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef] [PubMed]
- Le Pogam, P.; Boustie, J. Xanthones of lichen source: A 2016 update. Molecules 2016, 21, 294. [Google Scholar] [CrossRef] [PubMed]
- Masters, K.-S.; Bräse, S. Xanthones from fungi, lichens, and bacteria: The natural products and their synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.E.; Romagni, J.G. Lichens as a potential source of pesticides. Pestic. Outlook 2001, 12, 229–232. [Google Scholar]
- Dieu, A.; Millot, M.; Champavier, Y.; Mambu, L.; Chaleix, V.; Sol, V.; Gloaguen, V. Uncommon chlorinated xanthone and other antibacterial compounds from the lichen cladonia incrassata. Planta Med. 2014, 80, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Nong, X.-H.; Zhang, X.-Y.; Xu, X.-Y.; Qi, S.-H. Antifouling compounds from the marine-derived fungus Aspergillus terreus SCSGAF0162. Nat. Prod. Commun. 2015, 10, 1033–1034. [Google Scholar] [PubMed]
- He, K.Y.; Zhang, C.; Duan, Y.R.; Huang, G.L.; Yang, C.Y.; Lu, X.R.; Zheng, C.J.; Chen, G.Y. New chlorinated xanthone and anthraquinone produced by a mangrove-derived fungus Penicillium citrinum HL-5126. J. Antibiot. 2017, 70, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Jayalakshmi, V.; Neelakan, S.; Seshadri, T.R. A synthesis of thiophanic acid. Curr. Sci. 1968, 37, 196–197. [Google Scholar]
- Arshad, M.; Devlin, J.P.; Ollis, W.D. Synthesis of sordidone and thiophanic acid, two chlorine-containing lichen metabolites. J. Chem. Soc. C 1971, 1324–1326. [Google Scholar] [CrossRef]
- Neelakantan, S.; Thillaichidambaram, N. New synthesis of thiophanic acid. Curr. Sci. 1973, 42, 21–22. [Google Scholar]
- Elix, J.; Crook, C.; Hui, J.; Zhu, Z. Synthesis of new lichen xanthones. Aust. J. Chem. 1992, 45, 845–855. [Google Scholar] [CrossRef]
- Elix, J.A.; Gaul, K.L.; Jiang, H. The structure and synthesis of some minor xanthones from the lichen Rinodina thiomela. Aust. J. Chem. 1993, 46, 95–110. [Google Scholar] [CrossRef]
- Elix, J.A.; Portelli, V.J. A synthesis of the lichen xanthone thiomelin. Aust. J. Chem. 1990, 43, 1773–1778. [Google Scholar] [CrossRef]
- Elix, J.A.; Jiang, H.; Portelli, V.J. Structure and synthesis of the lichen xanthone isoarthothelin (2,5,7-trichloronorlichexanthone). Aust. J. Chem. 1990, 43, 1291–1295. [Google Scholar] [CrossRef]
- Mahajan, T.; Kumar, L.; Dwivedi, K.; Agarwal, D.D. Efficient and facile chlorination of industrially-important aromatic compounds using NaCl/p-TsOH/NCS in aqueous media. Ind. Eng. Chem. Res. 2012, 51, 3881–3886. [Google Scholar] [CrossRef]
- Bernini, R.; Pasqualetti, M.; Provenzano, G.; Tempesta, S. Ecofriendly synthesis of halogenated flavonoids and evaluation of their antifungal activity. New J. Chem. 2015, 39, 2980–2987. [Google Scholar] [CrossRef]
- Soidinsalo, O.; Wähälä, K. Aromatic chlorination with thionyl chloride. Applications in the synthesis of chlorinated isoflavones. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 2761–2767. [Google Scholar] [CrossRef]
- Quillinan, A.J.; Scheinmann, F. Studies in the xanthone series. Part xii. A general synthesis of polyoxygenated xanthones from benzophenone precursors. J. Chem. Soc. Perkin Trans. 1973, 1, 1329–1337. [Google Scholar] [CrossRef]
- Fernandes, C.; Masawang, K.; Tiritan, M.E.; Sousa, E.; de Lima, V.; Afonso, C.; Bousbaa, H.; Sudprasert, W.; Pedro, M.; Pinto, M.M. New chiral derivatives of xanthones: Synthesis and investigation of enantioselectivity as inhibitors of growth of human tumor cell lines. Bioorg. Med. Chem. 2014, 22, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Scheyer, T.; Romano, C.S.; Vojnov, A.A. Antioxidant and antimicrobial activities of rosemary extracts linked to their polyphenol composition. Free Radic. Res. 2006, 40, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.M.; Markham, J.L. A new method for determining the minimum inhibitory concentration of essential oils. J. Appl. Microbiol. 1998, 84, 538–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, A. Anticandidal low molecular compounds from higher plants with special reference to compounds from essential oils. Med. Res. Rev. 2006, 26, 223–268. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.R.; Aires-de-Sousa, M.; Conceição, T.; Antunes, F.; da Costa, P.M.; de Lencastre, H. High prevalence of emrsa-15 in portuguese public buses: A worrisome finding. PLoS ONE 2011, 6, e17630. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, Â.; Vaz-Pires, P.; Martins da Costa, P. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. In river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.; Bessa, L.; Buttachon, S.; Costa, P.; Buaruang, J.; Dethoup, T.; Silva, A.; Kijjoa, A. Antibacterial and antibiofilm activities of tryptoquivalines and meroditerpenes isolated from the marine-derived fungi neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis. Mar. Drugs 2014, 12, 822–839. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Palmeira, A.; Gomes, A.S.; Vasconcelos, V.; Sousa, E.; Pinto, M.; Martins da Costa, P. Synergistic effects between thioxanthones and oxacillin against methicillin-resistant Staphylococcus aureus. Microb. Drug Resist. 2015, 21, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 10th ed.; Document M07-A10; CLSI: Wayne, PA, USA, 2015. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 3rd ed.; Document M27-A3; CLSI: Wayne, PA, USA, 2008. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi, 2nd ed.; Document M38-A2; CLSI: Wayne, PA, USA, 2008. [Google Scholar]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 6–20 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Comp. | R1 | R2 | R3 | Method | Yield (%) |
|---|---|---|---|---|---|
| 8 | CH2Cl | H | OCH3 | A B | 5 6 |
| 9 | CH3 | H | OH | A B | 2 4 |
| 10 | CH3 | H | Cl | A B | 12 12 |
| 11 | CH3 | Cl | OCH3 | C | 26 |

| Comp. | R1 | R2 | R3 | R4 | R5 | R6 | Method | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 12 | CH3 | Cl | OCH3 | H | OCH3 | H | A D E | 7 12 14 |
| 13 | CH3 | Cl | Cl | H | OCH3 | H | A B C | Traces 12 8 |
| 14 | CH2Cl | H | OCH3 | H | OCH3 | H | A B D | Traces 8 13 |
| 15 | CH3 | H | Cl | H | OCH3 | H | B C | 5 10 |
| 16 | CH3 | H | Cl | H | Cl | H | C | 6 |
| 17 | CH3 | Cl | OCH3 | Cl | OCH3 | H | E | traces |
| 18 | CH3 | Cl | OCH3 | H | OCH3 | Cl | E | traces |
| 19 | CH3 | H | OCH3 | Cl | OCH3 | H | E | 5 |
| 20 | CH3 | H | OCH3 | H | OCH3 | Cl | E | 1 |
| Comp. | E. coli ATCC 25922 | E. coli SA/2 | P. aeruginosa ATCC 27853 | E. faecalis ATCC 29212 | E. faecalis B3/101 (VRE) | S. aureus ATCC 29213 | S. aureus 66/1 (MRSA) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Halo | MIC | MBC | Halo | MIC | MBC | Halo | MIC | MBC | Halo | MIC | MBC | Halo | MIC | MBC | Halo | MIC | MBC | Halo | MIC | MBC | |
| 6 | 0 | >16 | ND | 0 | ND | ND | 0 | >16 | ND | 0 | >16 | ND | 0 | ND | ND | 0 | >16 | ND | 0 | ND | ND |
| 7 | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | ND | ND |
| 8 | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | ND | ND |
| 9 | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | ND | ND |
| 10 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 11 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 12 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 13 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 14 | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | >64 | ND | 0 | ND | ND | 0 | >64 | ND | 0 | ND | ND |
| 15 | 0 | >16 | ND | 0 | ND | ND | 0 | >16 | ND | 10 | >16 | ND | 0 | ND | ND | 9.5 | >16 | ND | 0 | ND | ND |
| 16 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 17 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 18 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 19 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| 20 | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | >32 | ND | 0 | ND | ND | 0 | >32 | ND | 0 | ND | ND |
| Comp. | C. albicans ATCC 10231 | A. fumigatus ATCC 46645 | T. rubrum FF5 | M. canis FF1 | E. floccosum FF9 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MIC | MFC | MIC | MFC | MIC | MFC | MIC | MFC | MIC | MFC | |
| 6 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 7 | >128 | >128 | >128 | >128 | >128 | >128 | ND | ND | ND | ND |
| 8 | >128 | >128 | >128 | >128 | ≥128 | >128 | >128 | >128 | 128 | 128 |
| 9 | >128 | >128 | >128 | >128 | ≥128 | >128 | ≥128 | >128 | ≥128 | >128 |
| 10 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 11 | >32 | > 32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 12 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 13 | >128 | >128 | >128 | >128 | >128 | >128 | ND | ND | ND | ND |
| 14 | >128 | >128 | >128 | >128 | >128 | >128 | ND | ND | ND | ND |
| 15 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 16 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 17 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 182 | >128 | >128 | >128 | >128 | 8 | 8 | 8 | 8 | 4 | 4 |
| 19 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
| 20 | >32 | >32 | >32 | >32 | >32 | >32 | ND | ND | ND | ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Resende, D.I.S.P.; Pereira-Terra, P.; Inácio, Â.S.; Costa, P.M.d.; Pinto, E.; Sousa, E.; Pinto, M.M.M. Lichen Xanthones as Models for New Antifungal Agents. Molecules 2018, 23, 2617. https://doi.org/10.3390/molecules23102617
Resende DISP, Pereira-Terra P, Inácio ÂS, Costa PMd, Pinto E, Sousa E, Pinto MMM. Lichen Xanthones as Models for New Antifungal Agents. Molecules. 2018; 23(10):2617. https://doi.org/10.3390/molecules23102617
Chicago/Turabian StyleResende, Diana I. S. P., Patrícia Pereira-Terra, Ângela S. Inácio, Paulo Martins da Costa, Eugénia Pinto, Emília Sousa, and Madalena M. M. Pinto. 2018. "Lichen Xanthones as Models for New Antifungal Agents" Molecules 23, no. 10: 2617. https://doi.org/10.3390/molecules23102617
APA StyleResende, D. I. S. P., Pereira-Terra, P., Inácio, Â. S., Costa, P. M. d., Pinto, E., Sousa, E., & Pinto, M. M. M. (2018). Lichen Xanthones as Models for New Antifungal Agents. Molecules, 23(10), 2617. https://doi.org/10.3390/molecules23102617