Computational Study of Mechanism and Thermodynamics of Ni/IPr-Catalyzed Amidation of Esters


Abstract
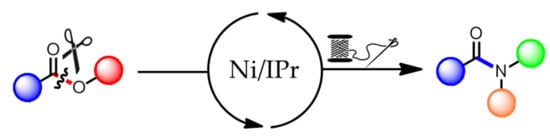
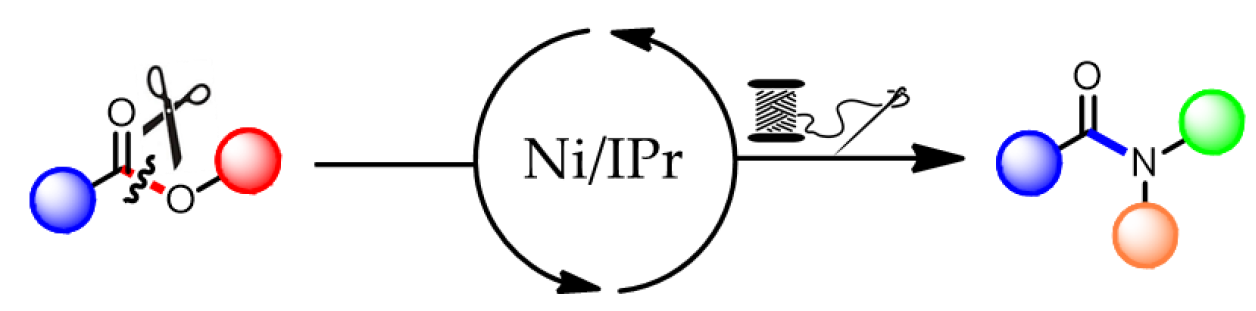
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
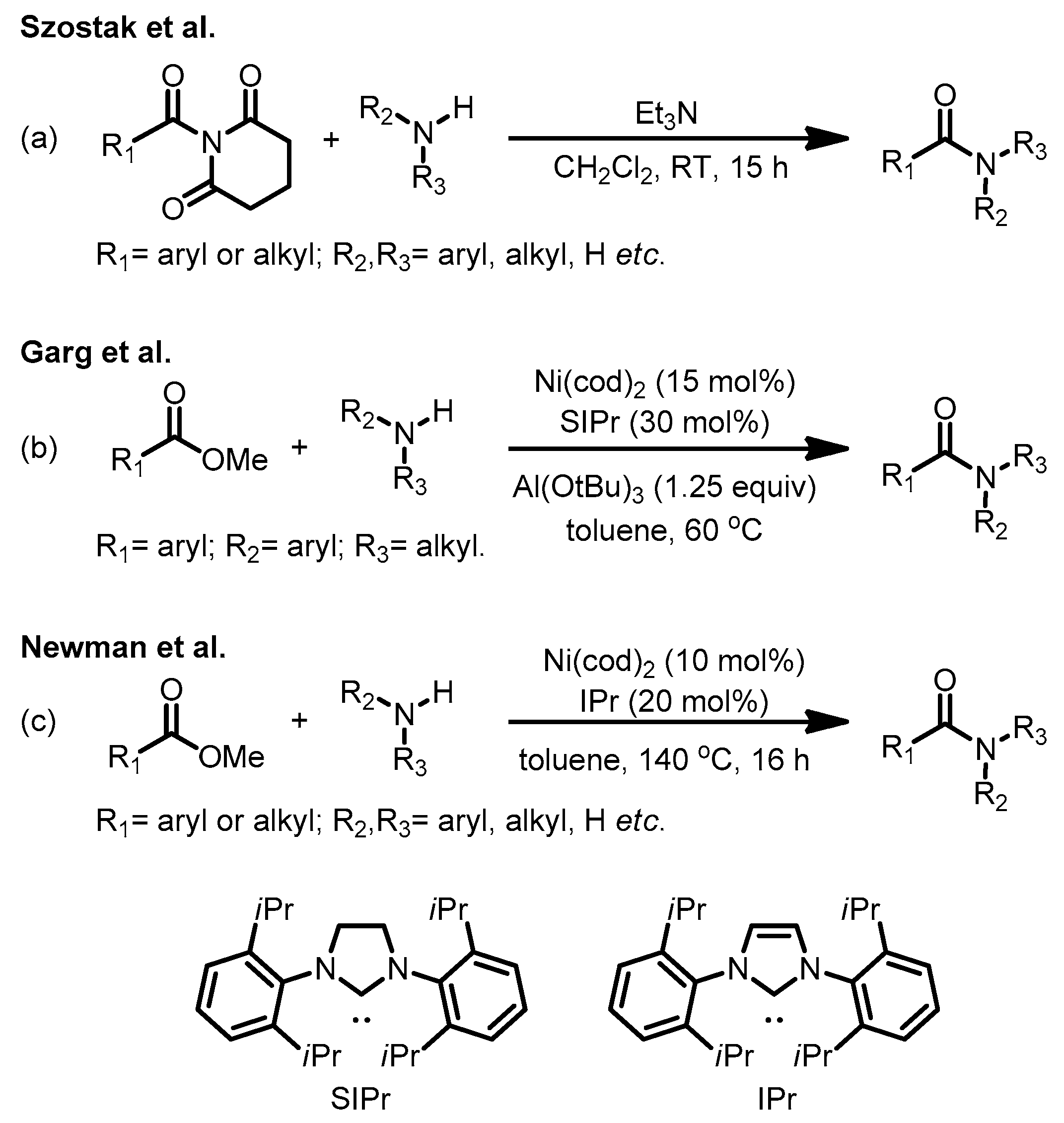
1. Introduction
2. Results and Discussion
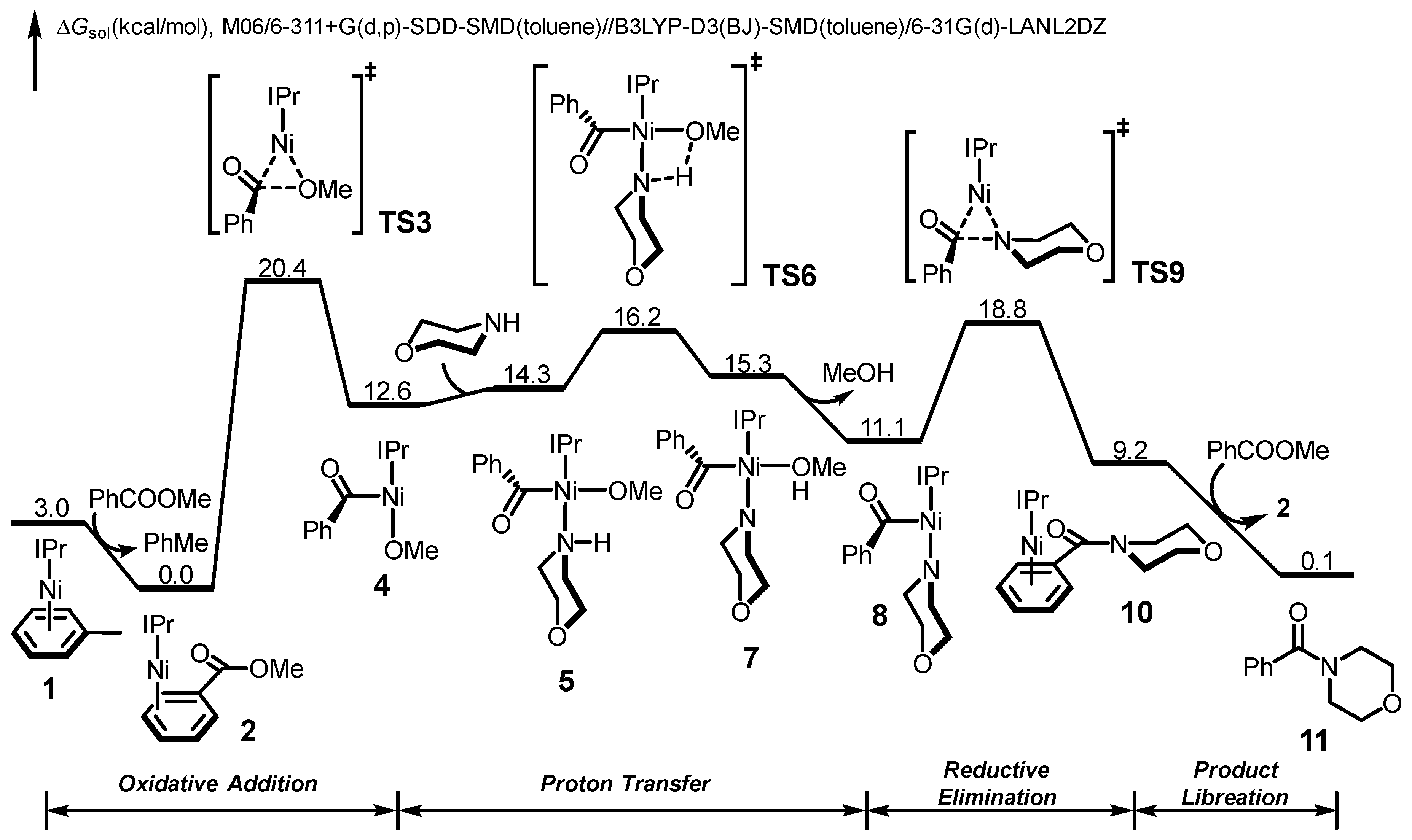
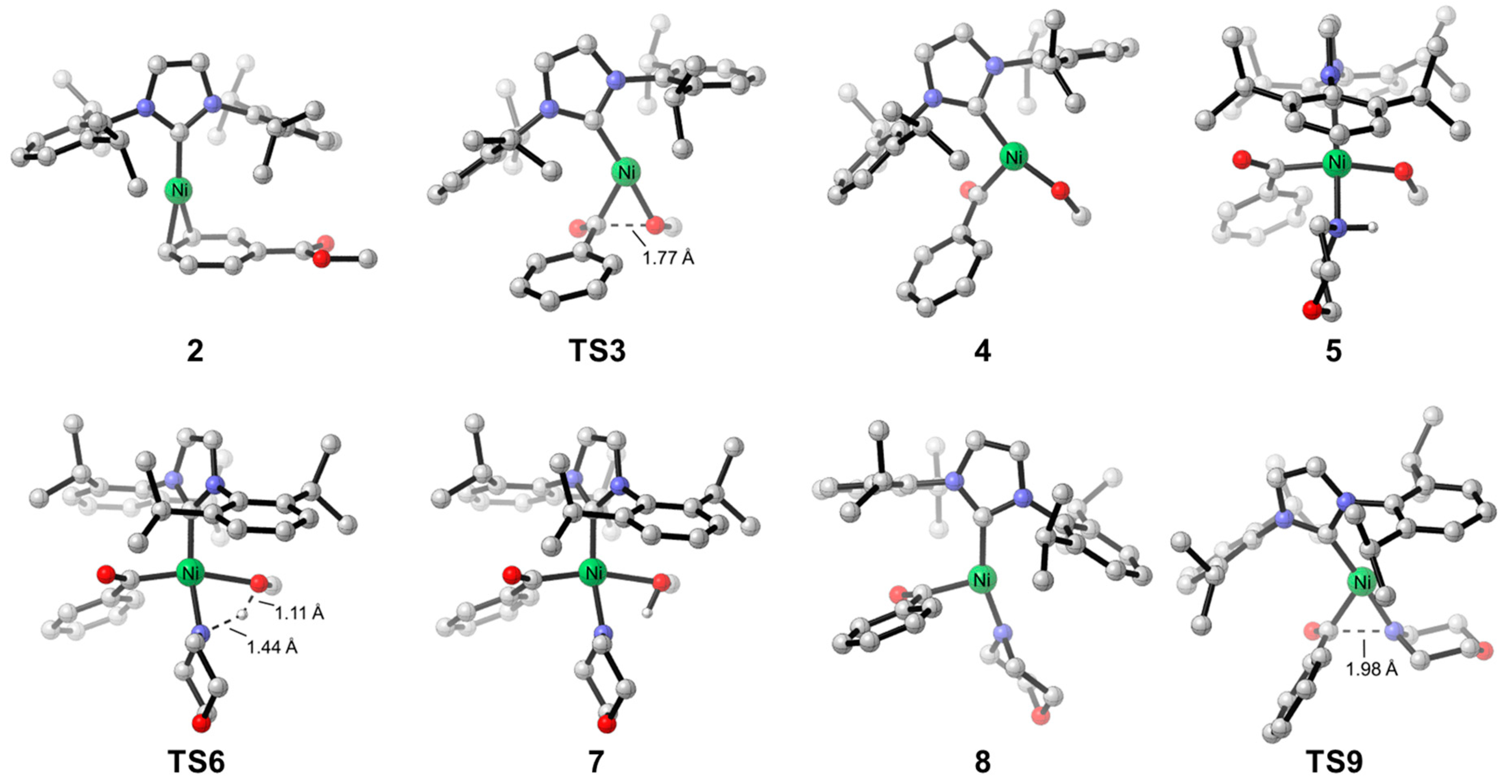
2.1. Reaction Mechanism of Aromatic Ester
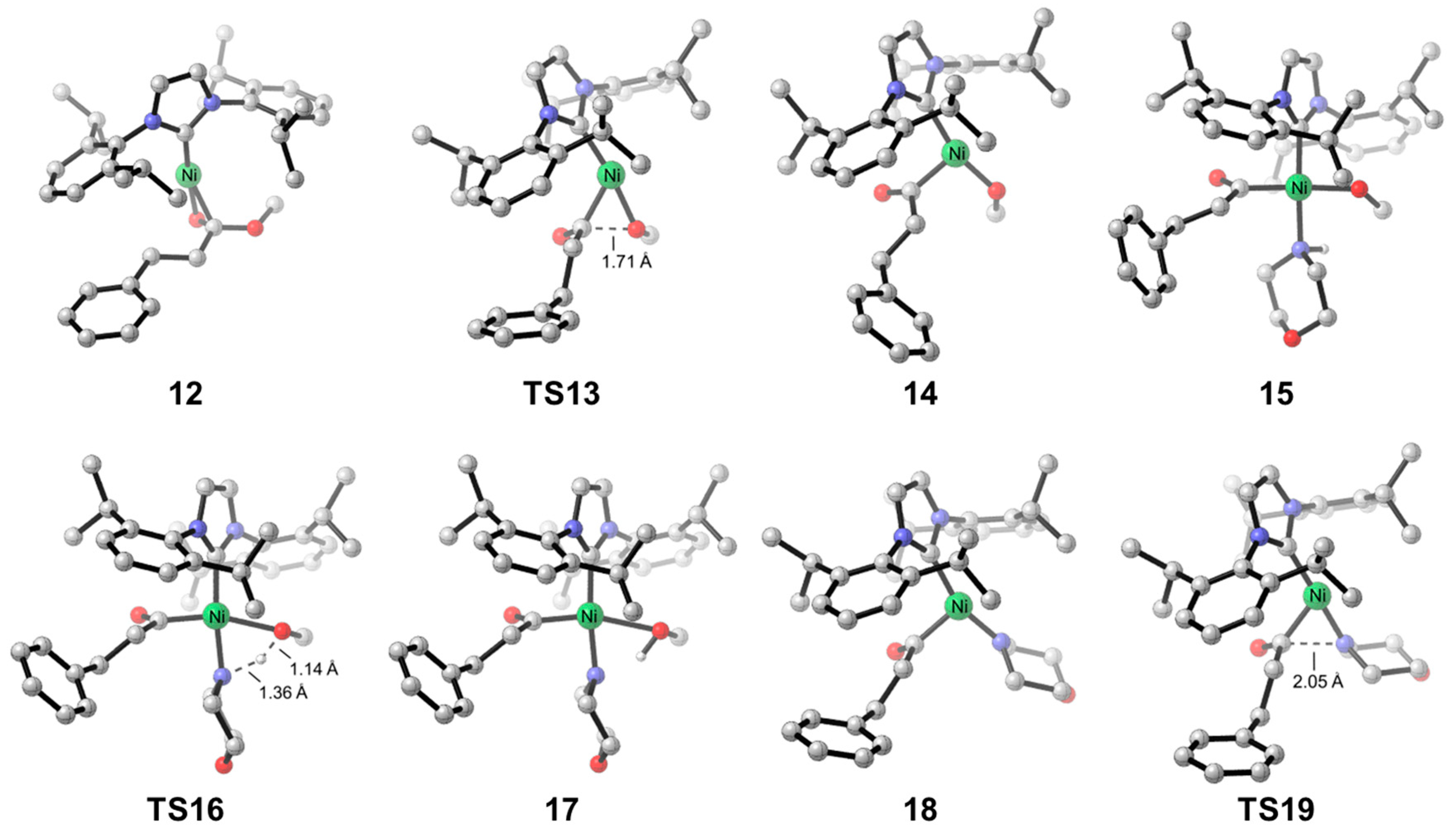
2.2. Reaction Mechanism of Aliphatic Ester
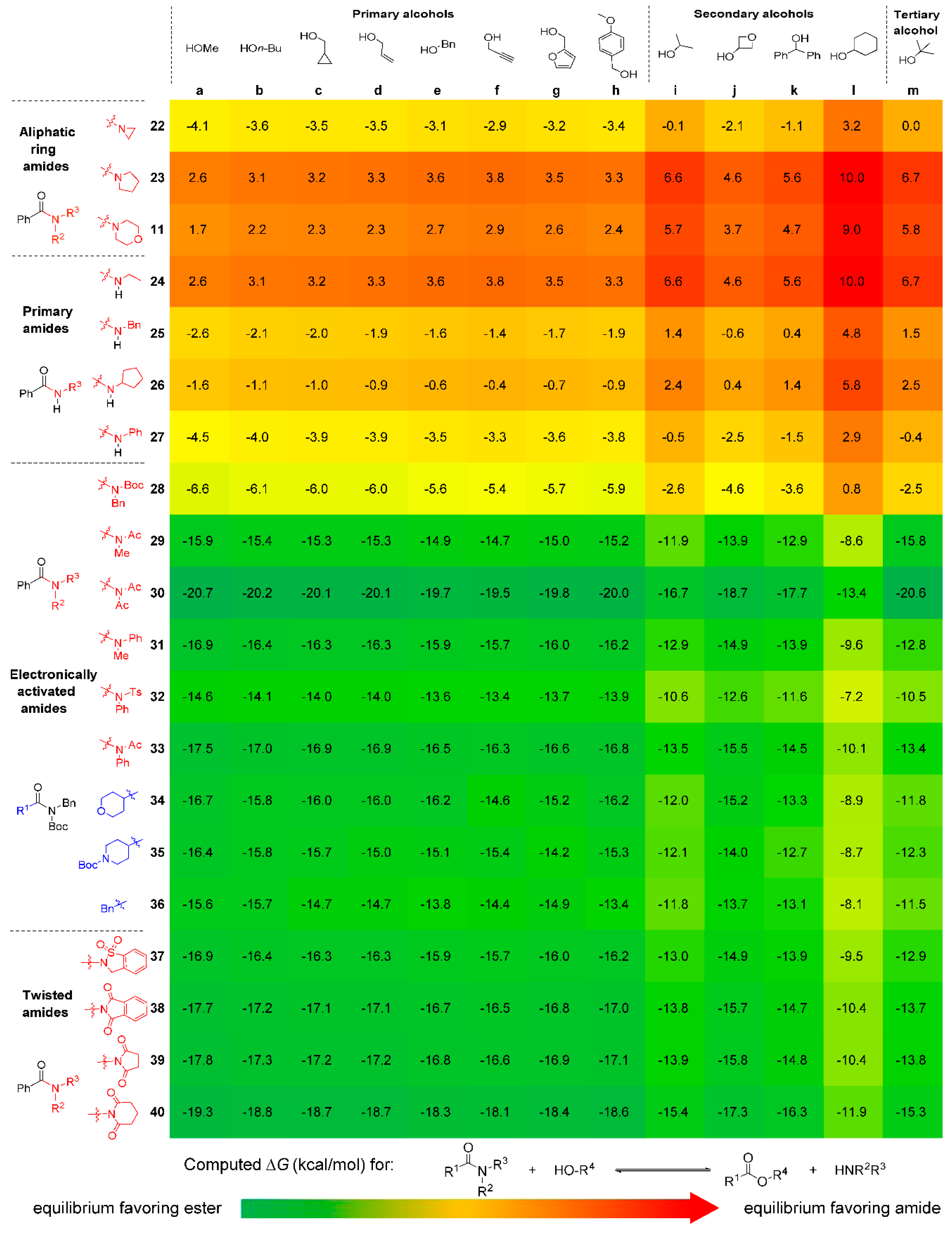
2.3. Reaction Thermodynamics
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meng, G.; Shi, S.; Szostak, M. Cross-Coupling of Amides by N–C Bond Activation. Synlett 2016, 27, 2530–2540. [Google Scholar]
- Meng, G.; Szostak, M. Palladium-Catalyzed Suzuki–Miyaura Coupling of Amides by Carbon–Nitrogen Cleavage: General Strategy for Amide N–C Bond Activation. Org. Biomol. Chem. 2016, 14, 5690–5707. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Szostak, M. Twisted Amides: From Obscurity to Broadly Useful Transition-Metal-Catalyzed Reactions by N–C Amide Bond Activation. Chem. Eur. J. 2017, 23, 7157–7173. [Google Scholar] [CrossRef] [PubMed]
- Dander, J.E.; Garg, N.K. Breaking Amides using Nickel Catalysis. ACS Catal. 2017, 7, 1413–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takise, R.; Muto, K.; Yamaguchi, J. Cross-coupling of aromatic esters and amides. Chem. Soc. Rev. 2017, 46, 5864–5888. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ji, C.-L.; Hong, X. Ni-mediated C–N activation of amides and derived catalytic transformations. Sci. China Chem. 2017, 60, 1413–1424. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Breneman, C.M. Resonance interactions in acyclic systems. 3. Formamide internal rotation revisited. Charge and energy redistribution along the C–N bond rotational pathway. J. Am. Chem. Soc. 1992, 114, 831–840. [Google Scholar] [CrossRef]
- Wiberg, K.B. The Interaction of Carbonyl Groups with Substituents. Acc. Chem. Res. 1999, 32, 922–929. [Google Scholar] [CrossRef]
- Kemnitz, C.R.; Loewen, M.J. “Amide Resonance” Correlates with a Breadth of C–N Rotation Barriers. J. Am. Chem. Soc. 2007, 129, 2521–2528. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.A.; Rosser, A.A. Reliable Determination of Amidicity in Acyclic Amides and Lactams. J. Org. Chem. 2012, 77, 5492–5502. [Google Scholar] [CrossRef] [PubMed]
- Szostak, R.; Aubé, J.; Szostak, M. Determination of Structures and Energetics of Small- and Medium-Sized One-Carbon Bridged Twisted Amides using ab Initio Molecular Orbital Methods. Implications for Amidic Resonance along the C–N Rotational Pathway. J. Org. Chem. 2015, 80, 7905–7927. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Lalancette, R.; Szostak, M. Structural Characterization of N–Alkylated Twisted Amides: Consequences for Amide Bond Resonance and N–C Cleavage. Angew. Chem. Int. Ed. 2016, 55, 5062–5066. [Google Scholar] [CrossRef] [PubMed]
- Szostak, R.; Meng, G.; Szostak, M. Resonance Destabilization in N-Acylanilines (Anilides): Electronically-Activated Planar Amides of Relevance in N–C(O) Cross-Coupling. J. Org. Chem. 2017, 82, 6373–6378. [Google Scholar] [CrossRef] [PubMed]
- Szostak, R.; Szostak, M. N-Acyl-Glutarimides: Resonance and Proton Affinities of Rotationally-Inverted Twisted Amides Relevant to N–C(O) Cross-Coupling. Org. Lett. 2018, 20, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E.; Piontek, A.; Dziuk, B.; Szostak, R.; Szostak, M. Barriers to Rotation in ortho-Substituted Tertiary Aromatic Amides: Effect of Chloro-Substitution on Resonance and Distortion. J. Org. Chem. 2018, 83, 3159–3163. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Reversible Twisting of Primary Amides via Ground State N–C(O) Destabilization: Highly Twisted Rotationally Inverted Acyclic Amides. J. Am. Chem. Soc. 2018, 140, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Hie, L.; Nathel, N.F.F.; Shah, T.K.; Baker, E.L.; Hong, X.; Yang, Y.-F.; Liu, P.; Houk, K.N.; Garg, N.K. Conversion of amides to esters by the nickel-catalysed activation of amide C–N bonds. Nature 2015, 524, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weires, N.A.; Baker, E.L.; Garg, N.K. Nickel-Catalysed Suzuki-Miyaura Coupling of Amides. Nat. Chem. 2016, 8, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Simmons, B.J.; Weires, N.A.; Dander, J.E.; Garg, N.K. Nickel-Catalyzed Alkylation of Amide Derivatives. ACS Catal. 2016, 6, 3176–3179. [Google Scholar] [CrossRef]
- Baker, E.L.; Yamano, M.M.; Zhou, Y.; Anthony, S.M.; Garg, N.K. A Two-Step Approach to Achieve Secondary Amide Transamidation Enabled by Nickel Catalysis. Nat. Commun. 2016, 7, 11554. [Google Scholar] [CrossRef] [PubMed]
- Dander, J.E.; Weires, N.A.; Garg, N.K. Benchtop Delivery of Ni(cod)2 using Paraffin Capsules. Org. Lett. 2016, 18, 3934–3936. [Google Scholar] [CrossRef] [PubMed]
- Hie, L.; Baker, E.L.; Anthony, S.M.; Desrosier, J.; Senanayake, C.; Garg, N.K. Nickel-Catalyzed Esterification of Aliphatic Amides. Angew. Chem. Int. Ed. 2016, 55, 15129–15132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, J.M.; Moreno, J.; Racine, S.; Du, S.; Garg, N.K. Mizoroki–Heck Cyclizations of Amide Derivatives for the Introduction of Quaternary Centers. Angew. Chem. Int. Ed. 2017, 56, 6567–6571. [Google Scholar] [CrossRef] [PubMed]
- Weires, N.A.; Caspi, D.D.; Garg, N.K. Kinetic Modeling of the Nickel-Catalyzed Esterification of Amides. ACS Catal. 2017, 7, 4381–4385. [Google Scholar] [CrossRef] [PubMed]
- Dander, J.E.; Baker, E.L.; Garg, N.K. Nickel-Catalyzed Transamidation of Aliphatic Amide Derivatives. Chem. Sci. 2017, 8, 6433–6438. [Google Scholar] [CrossRef] [PubMed]
- Boit, T.B.; Weires, N.A.; Kim, J.; Garg, N.K. Nickel-Catalyzed Suzuki–Miyaura Coupling of Aliphatic Amides. ACS Catal. 2018, 8, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Sterically-Controlled Pd-Catalyzed Chemoselective Ketone Synthesis via N–C Cleavage in Twisted Amides. Org. Lett. 2015, 17, 4364–4367. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. General Olefin Synthesis by the Palladium-Catalyzed Heck Reaction of Amides: Sterically-Controlled Chemoselective N–C Activation. Angew. Chem. Int. Ed. 2015, 54, 14518–14522. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Szostak, M. Rhodium-Catalyzed C–H Bond Functionalization with Amides by Double C–H/C–N Bond Activation. Org. Lett. 2016, 18, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Meng, G.; Szostak, M. Synthesis of Biaryls via Nickel Catalyzed Suzuki–Miyaura Coupling of Amides by Carbon–Nitrogen Cleavage. Angew. Chem. Int. Ed. 2016, 55, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Meng, G.; Liu, R.; Szostak, M. Sterically-Controlled Intermolecular Friedel-Crafts Acylation with Twisted Amides via Selective N–C Cleavage under Mild Conditions. Chem. Commun. 2016, 52, 6841–6844. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Szostak, M. Efficient Synthesis of Diaryl Ketones by Nickel-Catalyzed Negishi Cross-Coupling of Amides via Carbon–Nitrogen Bond Cleavage at Room Temperature Accelerated by Solvent Effect. Chem. Eur. J. 2016, 22, 10420–10424. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Meng, G.; Szostak, M. General Method for the Suzuki–Miyaura Cross-Coupling of Amides Using Commercially Available, Air- and Moisture-Stable Palladium/NHC (NHC = N-Heterocyclic Carbene) Complexes. ACS Catal. 2017, 7, 1960–1965. [Google Scholar] [CrossRef]
- Shi, S.; Szostak, M. Decarbonylative Cyanation of Amides by Palladium Catalysis. Org. Lett. 2017, 19, 3095–3098. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Szostak, M. Nickel-Catalyzed Negishi Cross-Coupling of N-Acylsuccinimides: Stable, Amide-Based, Twist-Controlled Acyl-Transfer Reagents via N–C Activation. Synthesis 2017, 49, 3602–3608. [Google Scholar]
- Lei, P.; Meng, G.; Ling, Y.; An, J.; Szostak, M. Pd-PEPPSI: Pd-NHC Precatalyst for Suzuki−Miyaura Cross-Coupling Reactions of Amides. J. Org. Chem. 2017, 82, 6638–6646. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Szostak, M. Decarbonylative Phosphorylation of Amides by Palladium and Nickel Catalysis: The Hirao Cross-Coupling of Amide Derivatives. Angew. Chem. Int. Ed. 2017, 56, 12718–12722. [Google Scholar] [CrossRef] [PubMed]
- Osumi, Y.; Liu, C.; Szostak, M. N-Acylsuccinimides: Twist-Controlled, Acyl-Transfer Reagents in Suzuki–Miyaura Cross-Coupling by N–C Amide Bond Activation. Org. Biomol. Chem. 2017, 15, 8867–8871. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Achtenhagen, M.; Liu, R.; Szostak, M. Transamidation of N-Acyl-Glutarimides with Amines. Org. Biomol. Chem. 2018, 16, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhao, Y.; Liu, J.; Zhang, Y.; Shi, Z. Nickel-Catalyzed Decarbonylative Borylation of Amides: Evidence for Acyl C-N Bond Activation. Angew. Chem. Int. Ed. 2016, 55, 8718–8722. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, M.; Pu, X.; Shi, Z. Nickel-Catalyzed Retro-Hydroamidocarbonylation of Aliphatic Amides to Olefins. Nat. Commun. 2017, 8, 14993. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zou, G. Acylative Suzuki coupling of amides: Acyl-nitrogen activation via synergy of independently modifiable activating groups. Chem. Commun. 2015, 51, 5089–5092. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Guo, L.; Lee, S.; Liu, X.; Rueping, M. Selective Reductive Removal of Ester and Amide Groups from Arenes and Heteroarenes through Nickel–Catalyzed C–O and C–N Bond Activation. Angew. Chem. Int. Ed. 2017, 56, 3972–3976. [Google Scholar] [CrossRef] [PubMed]
- Srimontree, W.; Chatupheeraphat, A.; Liao, H.; Rueping, M. Amide to Alkyne Interconversion via a Nickel/Copper-Catalyzed Deamidative Cross-Coupling of Aryl and Alkenyl Amides. Org. Lett. 2017, 19, 3091–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatupheeraphat, A.; Liao, H.; Lee, S.; Rueping, M. Nickel-Catalyzed C–CN Bond Formation via Decarbonylative Cyanation of Esters, Amides, and Intramolecular Recombination Fragment Coupling of Acyl Cyanides. Org. Lett. 2017, 19, 4255–4258. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yue, H.; Jia, J.; Guo, L.; Rueping, M. Synthesis of Amidines from Amides using a Nickel Catalyzed Decarbonylative Amination via CO Extrusion Intramolecular Recombination Fragment Coupling. Chem. Eur. J. 2017, 23, 11771–11775. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Guo, L.; Yue, H.; Liao, H.; Rueping, M. Nickel-Catalyzed Decarbonylative Silylation, Borylation, and Amination of Arylamides via a Deamidative Reaction Pathway. Synlett 2017, 28, 2594–2598. [Google Scholar]
- Lee, S.; Liao, H.; Chatupheeraphat, A.; Rueping, M. Nickel-Catalyzed C–S Bond Formation via Decarbonylative Thioetherification of Esters, Amides and Intramolecular Recombination Fragment Coupling of Thioesters. Chem. Eur. J. 2018, 24, 3608–3612. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hsiao, C.; Guo, L.; Rueping, M. Cross-Coupling of Amides with Alkylboranes via Nickel-Catalyzed C–N Bond Cleavage. Org. Lett. 2018, 20, 2976–2979. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Sasmal, S.; Seth, K.; Lahiri, G.K.; Maiti, D. Nickel-Catalyzed Deamidative Step-Down Reduction of Amides to Aromatic Hydrocarbons. ACS Catal. 2017, 7, 433–437. [Google Scholar] [CrossRef]
- Walker, J.A.; Vickerman, K.L.; Humke, J.N.; Stanley, L.M. Ni-Catalyzed Alkene Carboacylation via Amide C–N Bond Activation. J. Am. Chem. Soc. 2017, 30, 10228–10231. [Google Scholar] [CrossRef] [PubMed]
- Amani, J.; Alam, R.; Badir, S.; Molander, G.A. Synergistic Visible-Light Photoredox/Nickel-Catalyzed Synthesis of Aliphatic Ketones via N–C Cleavage of Imides. Org. Lett. 2017, 19, 2426–2429. [Google Scholar] [CrossRef] [PubMed]
- Hie, L.; Nathel, N.F.F.; Hong, X.; Yang, Y.-F.; Houk, K.N.; Garg, N.K. Nickel-Catalyzed Activation of Acyl C–O Bonds of Methyl Esters. Angew. Chem. Int. Ed. 2016, 55, 2810–2814. [Google Scholar] [CrossRef] [PubMed]
- Halima, T.B.; Masson-Makdissi, J.; Newman, S.G. Nickel-Catalyzed Amide Bond Formation from Methyl Esters. Angew. Chem. Int. Ed. 2018, 57, 12925–12929. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, S.; Fu, Y.; Guo, Q.; Liu, L. Mechanism of Ni-Catalyzed Selective C–O Bond Activation in Cross-Coupling of Aryl Esters. J. Am. Chem. Soc. 2009, 131, 8815–8823. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-Z.; Fu, Y. Mechanistic Origin of Cross-Coupling Selectivity in Ni-Catalysed Tishchenko Reactions. Chem. Eur. J. 2012, 18, 16765–16773. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Liang, Y.; Houk, K.N. Mechanisms and Origins of Switchable Chemoselectivity of Ni-Catalyzed C(aryl)–O and C(acyl)–O Activation of Aryl Esters with Phosphine Ligands. J. Am. Chem. Soc. 2014, 136, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yu, H.-Z.; Fu, Y. Mechanistic Study of Chemoselectivity in Ni-Catalyzed Coupling Reactions between Azoles and Aryl Carboxylates. J. Am. Chem. Soc. 2014, 136, 8252–8260. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Muto, K.; Yamaguchi, J.; Zhao, C.; Itami, K.; Musaev, D.G. Key Mechanistic Features of Ni-Catalyzed C–H/C–O Biaryl Coupling of Azoles and Naphthalen-2-yl Pivalates. J. Am. Chem. Soc. 2014, 136, 14834–14844. [Google Scholar] [CrossRef] [PubMed]
- Muto, K.; Yamaguchi, J.; Musaev, D.G.; Itami, K. Decarbonylative organoboron cross-coupling of esters by nickel catalysis. Nat. Commun. 2015, 6, 7508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, L. Recent advances in mechanistic studies on Ni catalyzed cross-coupling reactions. Chin. J. Catal. 2015, 36, 3–14. [Google Scholar] [CrossRef]
- Liu, L.; Chen, P.; Sun, Y.; Wu, Y.; Chen, S.; Zhu, J.; Zhao, Y. Mechanism of Nickel-Catalyzed Selective C–N Bond Activation in Suzuki–Miyaura Cross-Coupling of Amides: A Theoretical Investigation. J. Org. Chem. 2016, 81, 11686–11696. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Y.; Yu, H.-Z.; Fu, Y. Mechanism of Nickel-Catalyzed Suzuki–Miyaura Coupling of Amides. Chem. Asian J. 2017, 12, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.-L.; Hong, X. Factors Controlling the Reactivity and Chemoselectivity of Resonance Destabilized Amides in Ni-Catalyzed Decarbonylative and Nondecarbonylative Suzuki–Miyaura Coupling. J. Am. Chem. Soc. 2017, 139, 15522–15529. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-Y.; Liu, T.; Sun, X.; Xu, Z.; Fan, X.; Zhu, L.; Bi, S.-W. Computational study of the mechanism of amide bond formation via CS2-releasing 1,3-acyl transfer. Org. Biomol. Chem. 2018, 16, 5808–5815. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zou, L.; Bai, R.; Lan, Y. Ni(I)–Ni(III) vs. Ni(II)–Ni(IV): Mechanistic study of Ni-catalyzed alkylation of benzamides with alkyl halides. Org. Chem. Front. 2018, 5, 615–622. [Google Scholar] [CrossRef]
- Saper, N.I.; Hartwig, J.F. Mechanistic Investigations of the Hydrogenolysis of Diaryl Ethers Catalyzed by Nickel Complexes of N-Heterocyclic Carbene Ligands. J. Am. Chem. Soc. 2017, 139, 17667–17676. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Head-Gordon, M.; Pople, J.A.; Frisch, M. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Häussermann, U.; Dolg, M.; Stoll, H.; Preuss, H.; Schwerdtfeger, P.; Pitzer, R.M. Accuracy of energy-adjusted quasirelativistic ab initio pseudopotentials. Mol. Phys. 1993, 78, 1211–1224. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- CYLview, 1.0b; CYLview 2.0; Universitéde Sherbrooke: Sherbrooke, QC, Canada, 2009.
Sample Availability: Samples of the compounds are not available from the authors. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, C.-L.; Xie, P.-P.; Hong, X. Computational Study of Mechanism and Thermodynamics of Ni/IPr-Catalyzed Amidation of Esters. Molecules 2018, 23, 2681. https://doi.org/10.3390/molecules23102681
Ji C-L, Xie P-P, Hong X. Computational Study of Mechanism and Thermodynamics of Ni/IPr-Catalyzed Amidation of Esters. Molecules. 2018; 23(10):2681. https://doi.org/10.3390/molecules23102681
Chicago/Turabian StyleJi, Chong-Lei, Pei-Pei Xie, and Xin Hong. 2018. "Computational Study of Mechanism and Thermodynamics of Ni/IPr-Catalyzed Amidation of Esters" Molecules 23, no. 10: 2681. https://doi.org/10.3390/molecules23102681
APA StyleJi, C. -L., Xie, P. -P., & Hong, X. (2018). Computational Study of Mechanism and Thermodynamics of Ni/IPr-Catalyzed Amidation of Esters. Molecules, 23(10), 2681. https://doi.org/10.3390/molecules23102681