2. Results and Discussion
We decided to inspect the behaviour of polyhydroxylated benzenic structures such as 5-methylbenzene-1,2,3-triol (5-methylpyrogallol) in the presence of an isolated oxidative enzyme such as HRP in the presence of H
2O
2 as oxidant. The synthesis of 5-methylpyrogallol
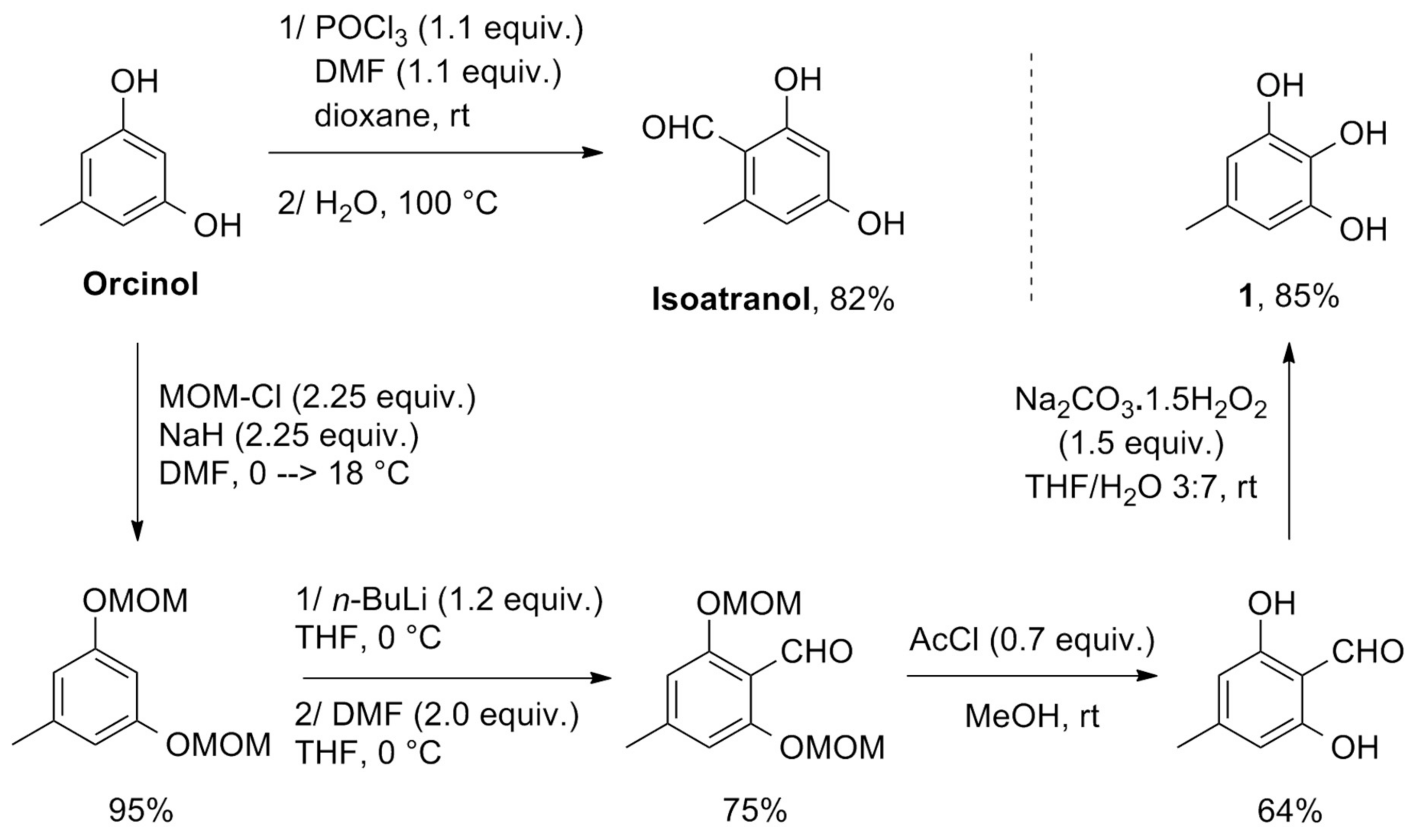
1 was achieved in four steps from commercially available orcinol by MOM-protection of both hydroxyl groups,
ortho-lithiation and formylation with DMF, deprotection and Dakin oxidation following literature procedures (
Scheme 2) [
11,
12]. It is worth noting that standard formylation procedure by POCl
3/DMF led regioselectively to isoatranol instead of
1.
Testing of HRP-catalysed oxidation was conducted with
1 in the presence of H
2O
2 at pH 9 varying enzyme loading and peroxide stoichiometry, along with relevant control experiments (
Table 1). In a representative reaction, the substrate
1 is dissolved in an aqueous sodium carbonate buffer solution at pH 9, containing HRP (1%
w/
w). The reactor is then wrapped with an aluminium foil and an aqueous solution of H
2O
2 is added slowly to the reaction mixture. The resulting solution is stirred at room temperature for 6 h and, after completion of the reaction, a diluted aqueous solution of HCl is added to reach a final pH of 4 and the resulting aqueous phase is finally extracted with EtOAc.
In the absence of H
2O
2 and regardless of the presence of HRP, the starting material was recovered unchanged almost quantitatively (entries 1, 2). In the presence of 2 equiv. of H
2O
2 without enzyme, only 34% of starting material was recovered upon extraction with EtOAc (entry 3). Inspection of the aqueous phase revealed the formation of a mixture of various products that could not be identified. In the presence of both H
2O
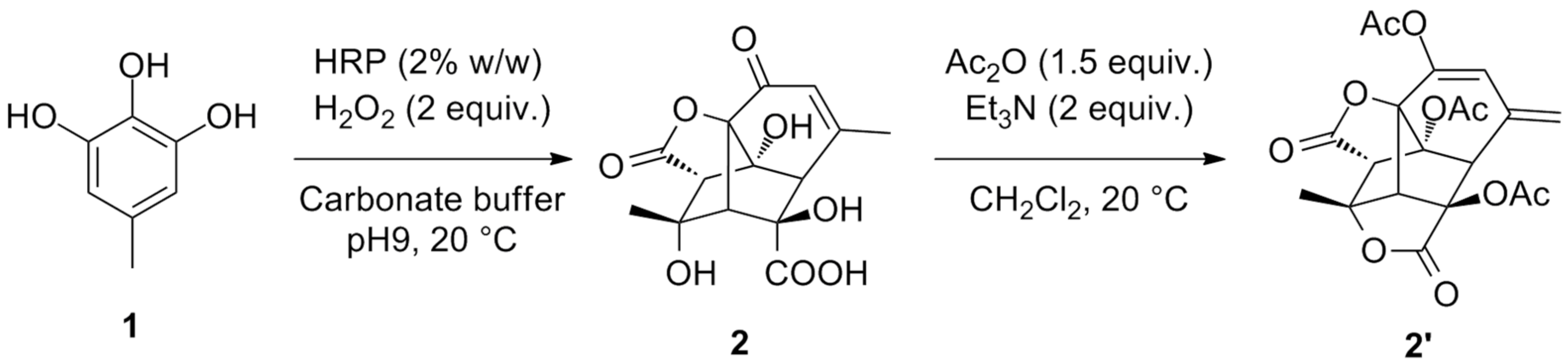
2 and HRP, the starting material was totally converted after 6 h and no product was obtained after extraction of the aqueous reaction mixture by EtOAc (entry 4). Upon evaporation of the water phase, product
2 was obtained as a white solid in 89% yield and submitted to a standard acetylation protocol leading to
2′. Variation of HRP/H
2O
2 equivalents did not show significant improvements for the conversion/yield (entries 5–10). Upon GC-MS-QTOF analysis, starting with a
m/
z of 419.0950, we identified a formula of C
20H
19O
10 for [M + H
+] (419.0978, Δ 6.6 ppm) for
2′. While the dimeric nature of the product was concluded from HRMS and
13C NMR analyses, the structure of the dimer
2′ did not match with usual oxidative products in peroxidase chemistry and we therefore embarked into a full structure elucidation of the product
2′ (
Table 2). Three acetyl groups were first evidenced by the presence of three singlets at
δH 2.05 (3H), 2.10 (3H) and 2.23 (3H), while another methyl was associated with the signal at
δH 1.67 (s, 3H, H-14) and other signals at
δH 5.16 (d,
J = 2 Hz, 1H, H-15b), 5.26 (d,
J = 2 Hz, 1H, H-15a) and
δC 120.9 (CH
2, C-15) revealed the presence of an exomethylene olefinic group. The conjugation of this first olefin with a second one was inferred from the presence of other olefinic signals at
δH 6.12 (s, 1H, H-10) and
δC 118.5 (CH, C-10) HMBC correlated with H
2-15. The dearomatisation of the first monomer
1 was then evidenced through the key H-10/C-1, H
2-15/C-8 HMBC correlations. Additional HMBC correlations from the signal at
δH 3.86 (s, 1H, H-8) like H-8/C-1 allowed closure of the ring of the first monomer with oxygen atoms connected to C-11, C-1 and C-12 due to characteristic chemical shifts. Further H-8/C-13 and H-8/C-7 correlation allowed the connection of the first monomer with the second one, C-13 corresponding to an ester function due to characteristic chemical shifts.
H-6/C-13, C-7, C-5 and C-14 together with H-4/C-6 and C-5 HMBC correlations from the signal at
δH 3.86 (s, 1H, H-6) and 3.62 (s, 1H, H-4) finished to establish the arrangement of the second monomer therefore evidencing an oxidation at positions C-3 and C-13 followed by a cleavage into two carboxylic functions at these positions. Even if no HMBC correlation enabled location of C-3 in the chain, its presence was deduced from characteristic chemical shifts and logics into the structure of the second monomer. Connections with the first monomer were inferred from additional HMBC correlations from the methines signals. The three acetyloxy were located at positions C-11, C-12 and C-7 due to final HMBC correlations. No ambiguity was left on the final structure of
2′ using HRMS data and the molecular formula. To the best of our knowledge, this complex cage polycyclic structure is unprecedented in the literature and it shows some similarities with vinigrol [
13] epicolactone [
14] jiadifenolide [
15] or (−)-11-
O-debenzoyltashironin [
16], which require multistep synthesis to be built. Several attempts to obtain monocrystals of
2 for X-ray spectroscopy were unsuccessful.
The structure of
2 before acetylation was slightly different from the one of
2′. NMR analysis was performed in D
2O and the spectra featured some significant differences (
Table 3). No exo-methylene and of course no acetyl groups were found in this structure and only one olefinic signal was observed at a relatively low field (6.45 ppm). Consequently, two methyl groups could be identified. The three saturated and fused methines of
2′ were still present in
2. The
13C-NMR spectra featured a new signal at 197.2 ppm suggesting a conjugated carbonyl group, and two signals at 178.0 and 171.5 ppm eligible to be those of carbonyl groups of an acid and an ester function, respectively.
HPLC-MS was performed and allowed obtaining a spectrum of 2 by ESI. In negative mode, [M − H]− ion was observed at m/z 309, although weak, together with strong [2M − H]− at m/z 619 and [2M − 2H + Na]− at m/z 641. Under enantioselective SFC and upon polarimetry measurements, product 2 appeared as a racemate. However, the diastereomeric control during the overall process is impressive since only one diastereomer was formed.
Under acetylation conditions, the α,β-unsaturated carbonyl motif of
2 was thus enolised to the corresponding dienol and trapped as an acetyl dienolate. In parallel, the γ-hydroxyacid underwent lactonisation to the corresponding γ-lactone fusing a fourth cycle to the existing structure, and the two tertiary alcohol functions were acetylated (
Scheme 3).
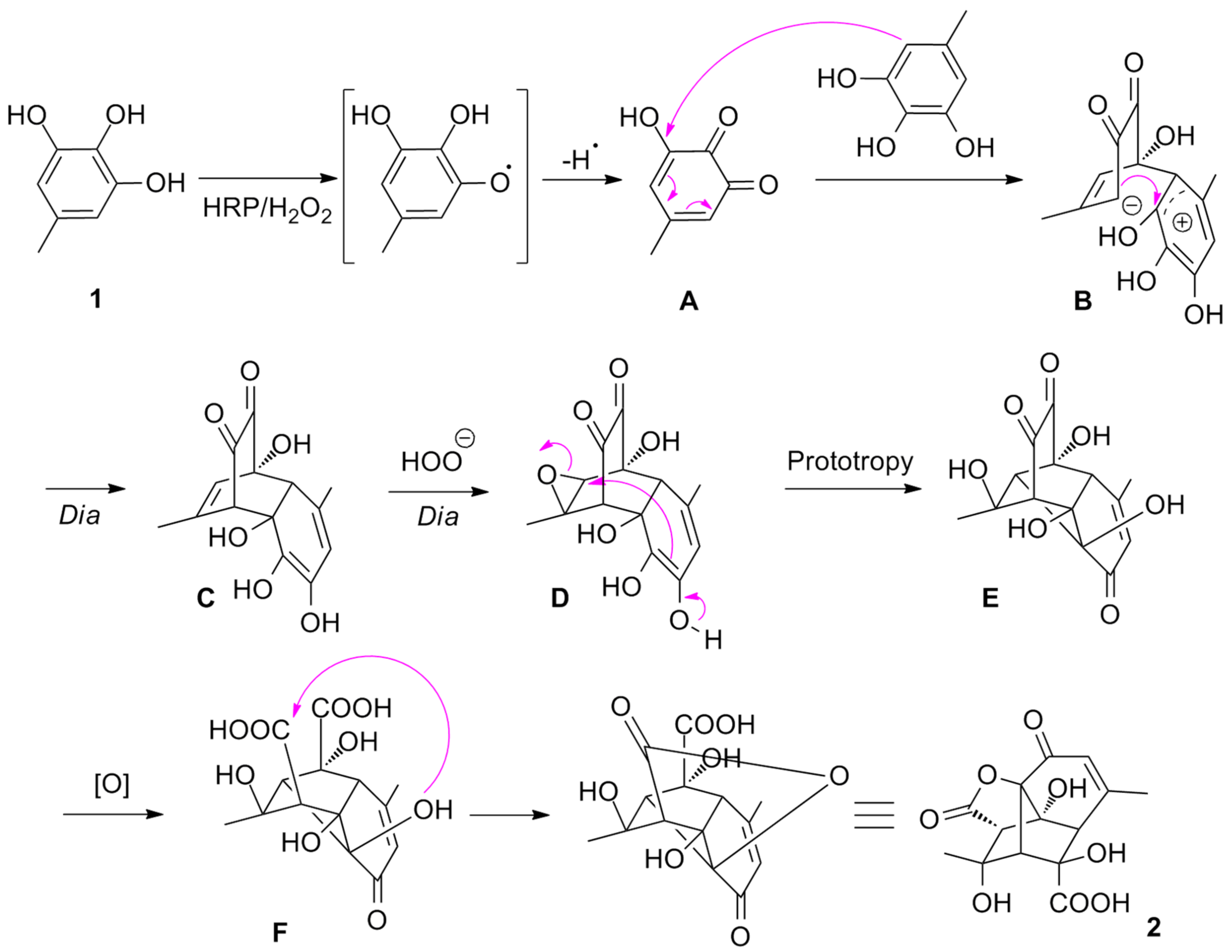
From a mechanistic point of view, we propose a series of oxidation/cyclisation to convert
1 into
2 (
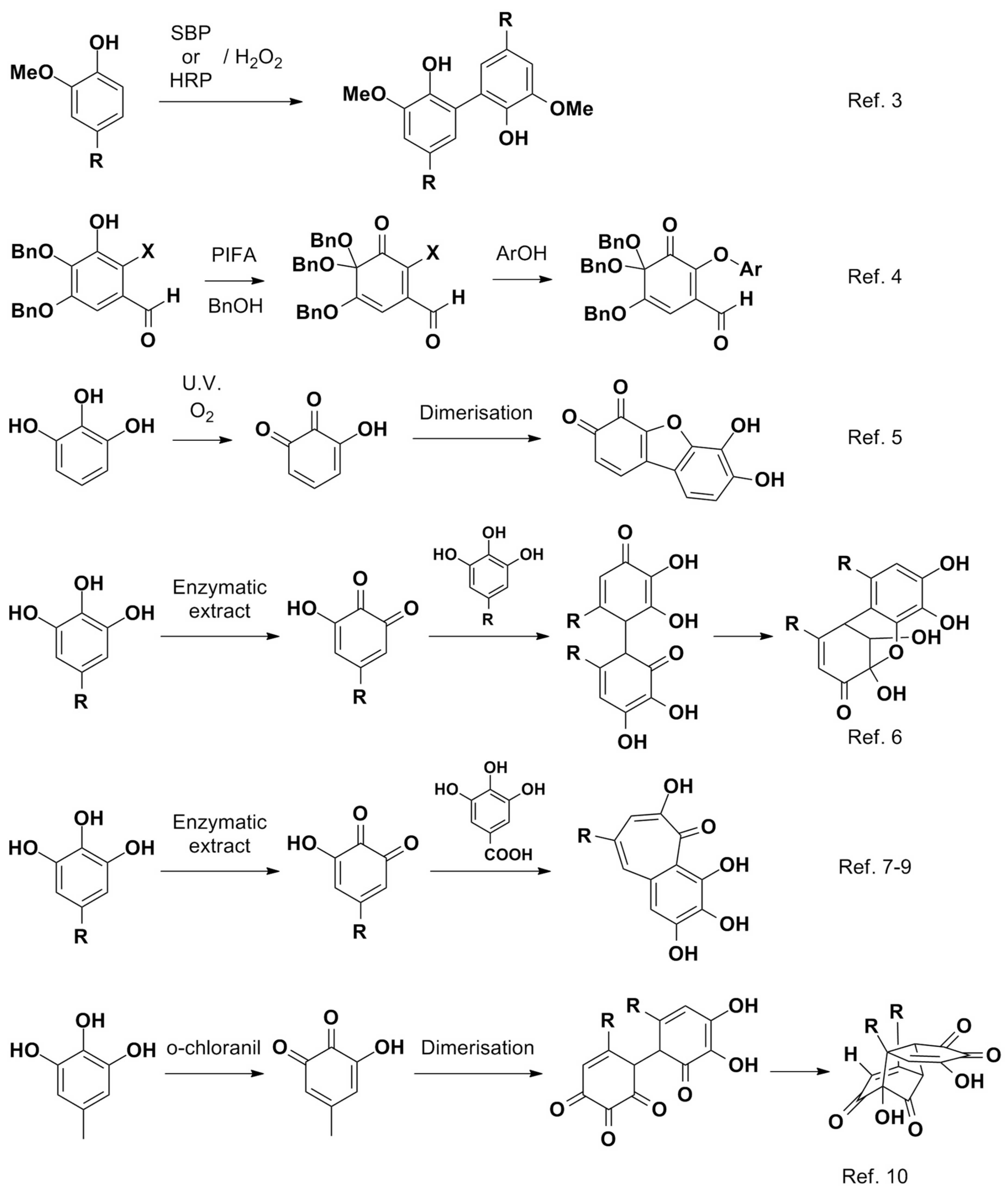
Scheme 4). Inspired by the reactivity of gallate derivatives in oxidative conditions [
10], we hypothesised that substrate
1 could be slowly oxidised into the corresponding
o-quinone
A by HRP/H
2O
2, which could quickly react with unconverted
1 to yield intermediate
B upon a vinylogous Michael addition followed ring closure by attack of the resulting enolate towards the arenium intermediate to form
C in a distereoselective fashion. Epoxidation of the trisubstituted double bond by H
2O
2 present in excess in basic medium could then lead to
D diastereoselectively. A subsequent regio- and stereoselective nucleophilic attack of an enol on the epoxide would then deliver
E after prototropy. Upon oxidative cleavage of the α-diketone motif to
F and subsequent lactonisation, product
2 would then be obtained, the oxidative cleavage possibly taking place earlier in the sequence.
3. Materials and Methods
1H NMR and 13C NMR spectra were recorded on a Bruker (Billerica, MA, USA) AC (200 and 400 MHz). 1H NMR spectra are reported as follows: Chemical shifts in ppm (δ) relative to the chemical shift of TMS at 0 ppm, integration, multiplicities (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and br = broad), and coupling constants (Hz). 13C NMR spectra chemical shifts are reported in ppm (δ) relative to CDCl3 at 77.16 ppm. Identity was assessed by comparison with data of authentic samples or literature data for 1 and its synthetic intermediates.
Column chromatography was carried out on silica gel (spherical 15–30 µm, neutral, 63–200 µm, Geduran Si 60, Merck KGaA, Darmstadt, Germany).
GC-TCD analyses were carried out using a Shimadzu (Kyoto, Japan) QP2010 plus gas chromatograph, under the following operation conditions: Vector gas, He; injector temperature, 250 °C; detector temperature, 210 °C at 60 mA; split ratio, 1/20; total flow, 22.5 mL min−1; Phenomenex (Torrance, CA, USA) Zebron ZB5MS column, polydimethylsiloxane (10 m, inside diameter 0.10 mm, film thickness 0.10 μm); temperature program, 80–200 °C at 10 °C min−1 and 200 °C for 8 min.
GC/MS analyses were performed by using a Shimadzu QP2010 gas chromatograph (conditions: carrier gas, He; injector and detector temperatures, 250 °C; injected volume, 0.5 μL; split ratio, 1/100; pressure, 180 kPa; SLB-5 ms capillary column (thickness: 0.25 mm, length: 30 m, inside diameter: 0.25 mm); temperature program, 60–315 °C at 10 °C min−1, and 10 min at 315 °C) coupled to a mass selective detector. Mass spectra were obtained by electron ionisation at 70 eV, m/z 35–400, source temperature 250 °C; only the most abundant ions are given.
High resolution mass spectrometry (HRMS) was performed at ERINI platform (Grasse, France) using a Waters UPLC coupled with a Waters (Milford, MA, USA) Xevo G2 QTOF spectrometer.
Enantioselective SFC was performed on a Jasco (Tokyo, Japan) Extrema apparatus equipped with Daicel (Osaka, Japan) ChiralPak IA column coupled with a dual wavelength 190 to 600 nm UV-4070/75 detector. Pressure: 150 bars; flow: 4 mL/min; MeOH: 15%. Wavelength: 245 nm.
Polarimetry was performed on an Anton Paar (Graz, Austria) MCP150 polarimeter at 20 °C in MeOH/H2O 10:6 v/v. Wavelength: 589 nm.
Materials. Dimethyl formamide (DMF), tetrahydrofuran (THF), methanol (MeOH), Ethanol (EtOH), cyclohexane (CHX) were purchased from Sigma-Aldrich (Saint-Louis, MO, USA) and dried and/or distilled according to conventional procedures. Orcinol, POCl3, MOMCl, NaH, n-BuLi, AcCl, Na2CO3, NaHCO3, H2O2 (30% w/w in water) and HRP were purchased from Sigma-Aldrich and used as received.
1,3-di(methoxymethoxy)-5-methylbenzene: Orcinol (0.8 g; 6.5 mmol) was dissolved in freshly dried and distilled DMF (40 mL). To this solution was added NaH (0.58 g as a 60% dispersion in mineral oil, 14.5 mmol). The mixture was stirred under a nitrogen atmosphere at 0 °C in a round-bottomed flask equipped with a refrigerant and a bubbler allowing to monitor H2 evolution. After 15 min, MOM-Cl (1.10 mL; 14.5 mmol) was added and the mixture stirred during pendant 18 h. Water was then carefully added (30 mL) and the resulting mixture extracted with Et2O (5 × 30 mL). Organic layers were then pooled and washed with a 2 M aqueous NaOH solution (3 × 20 mL) and brine (20 mL). After drying over MgSO4, filtration and solvent removal, a yellow oil was obtained, which was submitted to column chromatography over silica gel (petroleum ether/EtOAc 9:1) to yield the MOM-protected orcinol as a colourless liquid (1.13 g, 95%). Rf 0.7 (petroleum ether/EtOAc 8:2). 1H NMR (CDCl3, 200 MHz): δ ppm 6.6–6.5 (m, 3H, ArH), 5.15 (s, 4H), 3.48 (s, 6H) 2.32 (s, 3H). 13C NMR: (CDCl3, 50 MHz) δ ppm 158.2 (C), 140.3 (C), 110.4 (CH), 102.1 (CH), 94.4 (CH), 55.9 (OCH3), 21.7 (CH3); MS (EI) m/z: 212(8), 182(1), 152(3), 136(2), 123(1), 108(2), 91(1), 77(2), 45(100).
1,3-di(methoxymethoxy)-4-methylbenzaldehyde: 1,3-di(methoxymethoxy)-5-methylbenzene (0.95 g; 4.5 mmol) was dissolved in freshly distilled THF (50 mL) and stirred under a nitrogen atmosphere at 0 °C while n-butyllithium was added dropwise (3.4 mL as a 1.6 M solution in hexane, 5.4 mmol). The mixture was stirred during 1.5 h while let warm to room temperature and the reaction was quenched with DMF (0.7 mL, 9 mmol). The resulting mixture was washed with water (50 mL) and extracted with Et2O (4 × 30 mL). Organic layers were pooled and washed with water (40 mL) and brine (40 mL), dried over MgSO4, filtrated and concentrated in vacuo. The resulting yellow oil was submitted to column chromatography over silica gel (petroleum ether/EtOAc 95:5) to yield a yellow solid (0.81 g, 75%). 1H NMR (CDCl3, 200 MHz): δ ppm 10.40 (s, 1H, CHO), 6.58 (s, 2H, ArH), 5.17 (s, 4H), 3.42 (s, 6H) 2.26 (s, 3H); 13C NMR: (CDCl3, 50 MHz) δ ppm 188.7 (CHO), 159.4 (C), 147.3 (C), 113.7 (CH), 109.3 (CH), 94.6 (CH), 56.3 (OCH3), 22.5 (CH3); MS m/z: 240(2), 209(2), 195(3), 179(4), 178(10), 165(3), 164(4), 136(2), 77(2), 46(3), 45(100).
Atranol: 1,3-di(methoxymethoxy)-4-methylbenzaldehyde (0.315 g, 1.3 mmol) was dissolved in 30 mL of MeOH at room temperature under a nitrogen atmosphere. Acetyl chloride was then added dropwise (60 µl, 0.9 mmol). The mixture was stirred for 20 h and concentrated in vacuo. An aqueous HCl solution was then added (30 mL at 0.1 M) and the resulting mixture extracted with EtOAc (3 × 150 mL). Organic layers were pooled and dried over MgSO4, filtrated and concentrated in vacuo. The resulting light-yellow oil was submitted to column chromatography over silica gel (petroleum ether/EtOAc 8:2), and atranol 1 was obtained as a light yellow oil (0.164 g, 90%). 1H NMR (Acetone-d6, 200 MHz): δ ppm 10.71 (s, 2H, OH), 10.26 (s, 1H, CHO), 6.25 (s, 2H, ArH), 2.23 (s, 3H). 13C NMR (Acetone-d6, 50 MHz): δ ppm 194.2 (CHO), 163.1 (C), 151.6 (C), 109.3 (C), 108.4 (CH), 22.4 (CH3). MS m/z: 152(84), 151(100), 134 (6), 123(4), 106 (16), 95(9), 77(14), 69 (6), 67(11), 55(12).
5-Methylpyrogallol1: Atranol (0.152 g, 1 mmol) and sodium percarbonate (0.236 g, 1.5 mmol) were dissolved in THF/water 3:7 mixture (5 mL). The reaction mixture is then stirred at room temperature for 2 h. After completion of the reaction, aqueous 0.1 M HCl solution was added (5 mL) and the mixture extracted with EtOAc (2 × 10 mL). Organic layers were pooled and dried over MgSO4, filtrated and concentrated in vacuo. An orange solid was obtained (0.119 g, 85%). 1H NMR (Acetone-d6, 200 MHz): δ ppm 7.64 (s, 2H, OH), 7.02 (s, 1H, OH), 6.20 (s, 2H, ArH), 2.10 (s, 3H). 13C NMR (Acetone-d6, 50 MHz): δ ppm 146.3 (C), 131.0 (C), 129.2 (C), 108.5 (CH), 20.9 (CH3). MS m/z: 140(100), 139(34), 134 (6), 123(11), 122(19), 121(9), 94 (35), 77(4), 66(37), 65(21), 55(6), 53(17).
Dimer2: 5-methylpyrogallol (200 mg, 1.43 mmol) was dissolved in a carbonate aqueous buffer at pH9 (20 mM, 65 mL), containing HRP (124 U/mg, 4 mg). The flask was covered with an aluminium foil and H2O2 (30% aqueous solution) was added at the controlled flow of 0.1 mL/h to reach a final amount of 2 equiv. The reaction mixture was stirred at room temperature for 6 h followed by the addition of an aqueous HCl solution (40 mL, 0.1 M) further extracted with EtOAc (3 × 70 mL). Evaporation of aqueous phase a white powder of dimer 2 and small amounts of salt (0.193 g, 88%). 1H NMR (D2O, 400 MHz): δ ppm 6.37 (t, 1H), 3.97 (d, 1H), 3.37 (s, H), 2.68 (s, 2H), 2.62 (s, H), 2.14 (s, 3H), 1.79 (s, 3H). 13C RMN: (D2O, 100 MHz) δ ppm 197.2 (C), 178.0 (C), 171.5 (C), 164.1 (C), 127.1 (CH), 88.7 (C), 87.1 (C), 85.7 (C), 77.7 (C), 60.1 (CH), 54.4 (CH), 52.9 (CH), 25.7 (CH3), 23.6 (CH3). MS (ESI): [M − H]− ion was observed at m/z 309, [2M − H]+ at m/z 619, [2M − 2H + Na]+ at m/z 641. A (20 °C, 589 nm) = −0.002° ± 0.000.
Acetylated dimer2′: To a solution of dimer 2 (0.100 g, 0.32 mmol) in freshly distilled dichloromethane (2 mL) were added Et3N (0.28 mL, 2.1 mmol) and Ac2O (0.2 mL, 2.1 mmol). The mixture was stirred overnight at room temperature under a nitrogen atmosphere. After concentration in vacuo, water was added (10 mL) and the resulting solution extracted with EtOAc (2 × 10 mL). The organic layers were pooled, dried over MgSO4, filtrated and concentrated in vacuo. A colourless oil was obtained (0.12 g, 90%). 1H NMR (CDCl3, 400 MHz): δ ppm 6.12 (s, 1H), 5.26 (s, 1H), 5.16 (s, 1H), 3.88 (s, 1H), 3.86 (s, 1H), 3.62 (s, 1H), 2.23 (s, 3H), 2.10 (s, 3H), 2.05 (s, 3H), 1.67 (s, 3H). 13C NMR: (CDCl3, 100 MHz) δ ppm 170.9 (CO), 169.8 (CO), 169.7 (CO), 169.5 (CO), 169.1 (CO), 140.6 (C), 134.3 (C), 120.9 (C), 88.6(C), 87.3(C), 80.9 (C), 77.7 (C), 58.6 (CH), 55.5 (CH), 50.6 (CH), 23.2 (CH3), 20.9 (CH3), 20.7 (CH3), 20.7 (CH3), 20,5 (CH3). MS m/z: 418(1), 376(20), 334(7), 290(2), 273(3), 266(1), 246(2), 231(3), 214(6), 182(23), 175(3), 162(6), 141(28), 140(100), 91(1), 77(2), 69(2), 43(100). HRMS: 419.0950, calculated for [M.H]+ C20H19O10 419.0978, ∆ = −6.7 ppm. 377.0859, calculated for [M(-CH3CO + H).H]+ C18H17O9 377.0873, ∆ = −3.7 ppm. 335.0754, calculated for [M(-2CH3CO + 2H).H]+ C16H15O8 335.0767, ∆ = −3.9 ppm.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
