NMR Structure of μ-Conotoxin GIIIC: Leucine 18 Induces Local Repacking of the N-Terminus Resulting in Reduced NaV Channel Potency

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
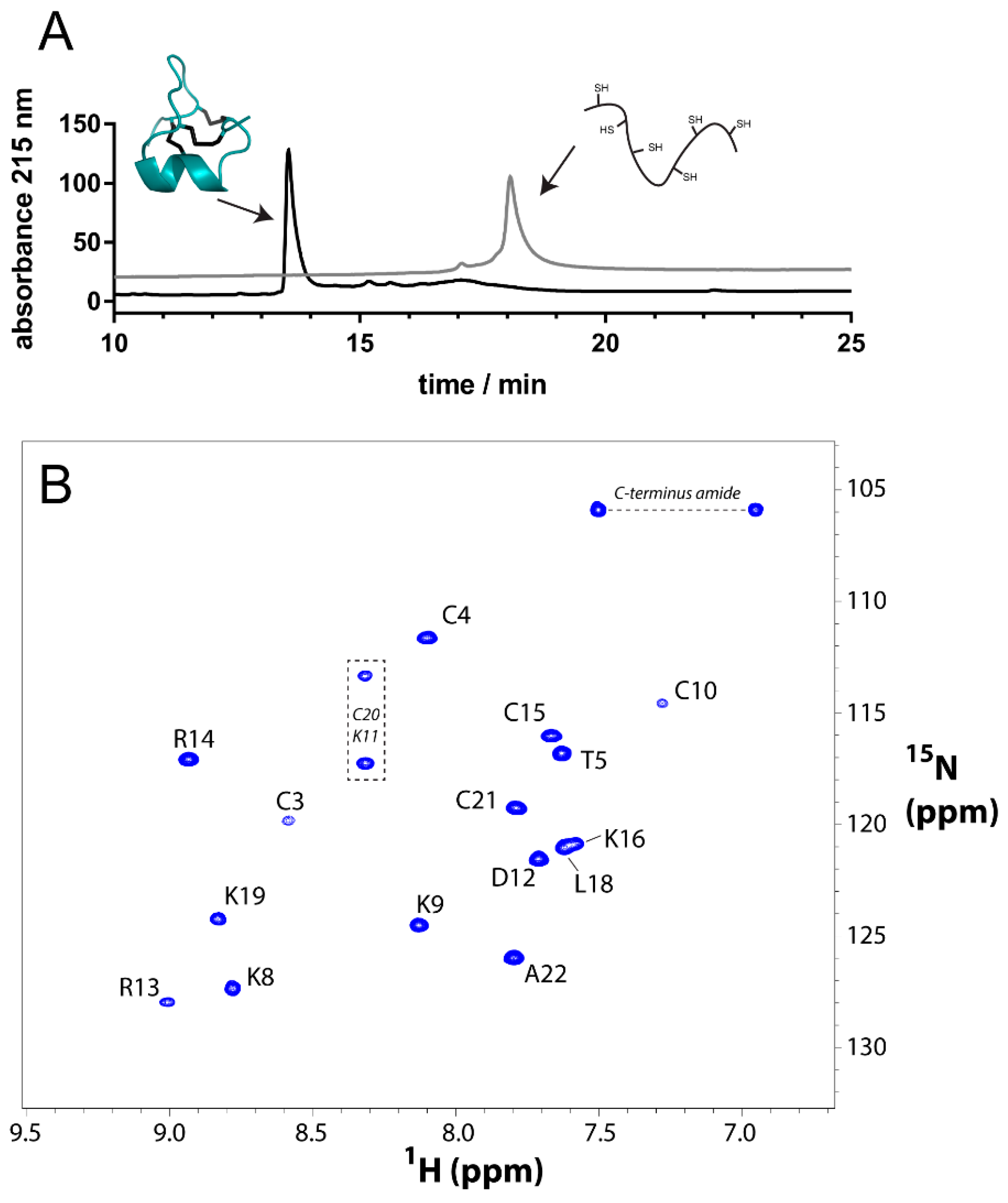
2.1. Chemical Synthesis
2.2. NMR Analysis
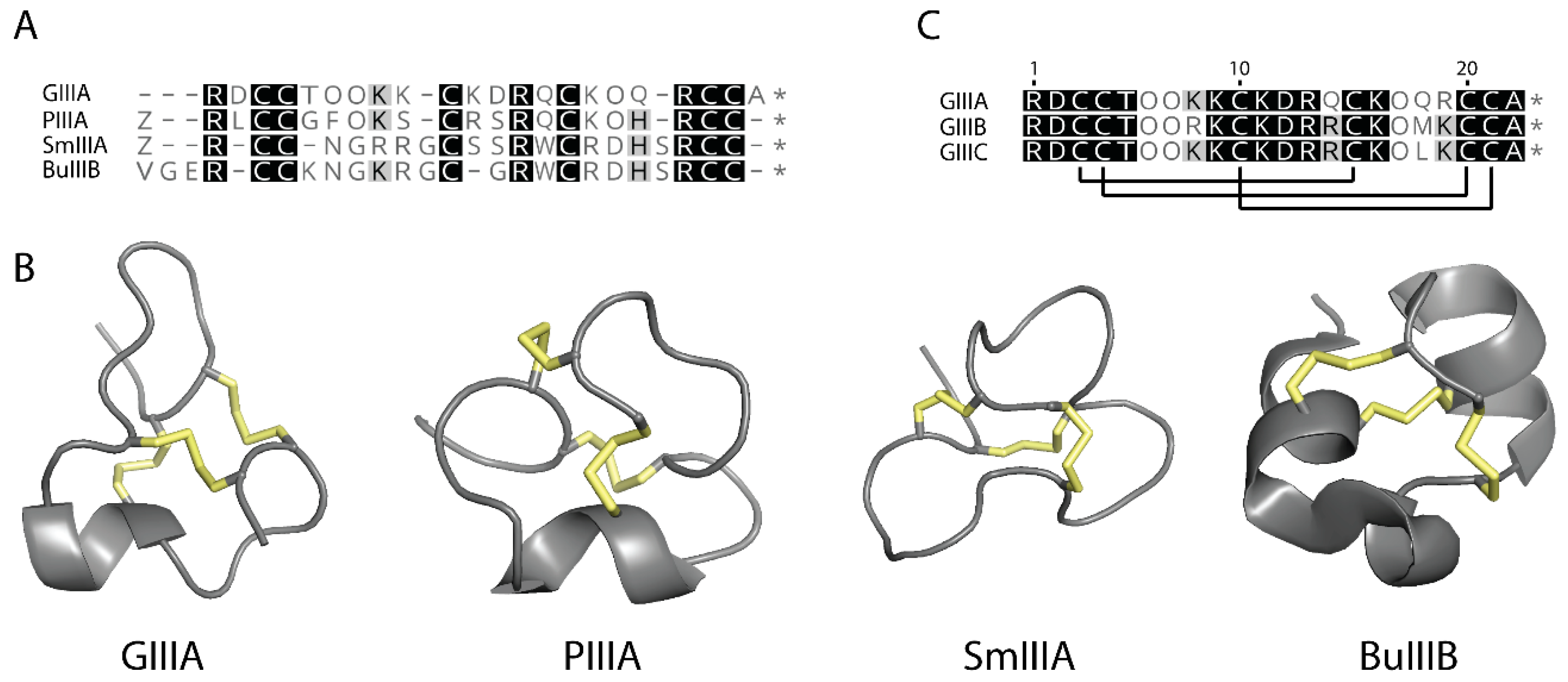
2.3. Three-Dimensional Structure of GIIIC
2.4. Pharmacological Activity of GIIIC at VGSCs
3. Discussion
3.1. Structural Comparison of GIIIC with Other μ-Conotoxins
3.2. Implications for μ-Conotoxin Interaction with Sodium Channels
4. Materials and Methods
4.1. Peptide Synthesis
4.2. NMR Spectroscopy
4.3. Structure Calculations
4.4. Pharmacology
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Catterall, W.A.; Wisedchaisri, G.; Zheng, N. The chemical basis for electrical signaling. Nat. Chem. Biol. 2017, 13, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahern, C.A.; Payandeh, J.; Bosmans, F.; Chanda, B. The hitchhiker′s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 2016, 147, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R.; Bulaj, G.; Norton, R.S. Structure and function of μ-conotoxins, peptide-based sodium channel blockers with analgesic activity. Fut. Med. Chem. 2014, 6, 1677–1698. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Yoshikami, D.; Azam, L.; Gajewiak, J.; Olivera, B.M.; Bulaj, G.; Zhang, M.M. μ-conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 10302–10307. [Google Scholar] [CrossRef] [PubMed]
- Moczydlowski, E.; Olivera, B.M.; Gray, W.R.; Strichartz, G.R. Discrimination of muscle and neuronal Na-channel subtypes by binding competition between [H-3] saxitoxin and μ-conotoxins. Proc. Natl. Acad. Sci. USA 1986, 83, 5321–5325. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S.; French, R.J.; Lipkind, G.M.; Fozzard, H.A.; Dudley, S. Predominant interactions between μ-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry 1998, 37, 4407–4419. [Google Scholar] [CrossRef] [PubMed]
- Dudley, S.C.; Todt, H.; Lipkind, G.; Fozzard, H.A. A μ-conotoxin-insensitive Na+ channel mutant: Possible localization of a binding site at the outer vestibule. Biophys. J. 1995, 69, 1657–1665. [Google Scholar] [CrossRef]
- Zhang, M.M.; McArthur, J.R.; Azam, L.; Bulaj, G.; Olivera, B.M.; French, R.J.; Yoshikami, D. Synergistic and antagonistic interactions between tetrodotoxin and μ-conotoxin in blocking voltage-gated sodium channels. Channels 2009, 3, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nakamura, H.; Ohizumi, Y.; Kobayashi, J.; Hirata, Y. The amino-acid-sequences of homologous hydroxyproline-containing myotoxins from the marine snail Conus geographus venom. FEBS Lett. 1983, 155, 277–280. [Google Scholar] [CrossRef]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium-channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar] [PubMed]
- Leipold, E.; Ullrich, F.; Thiele, M.; Tietze, A.A.; Terlau, H.; Imhof, D.; Heinemann, S.H. Subtype-specific block of voltage-gated K+ channels by μ-conopeptides. Biochem. Biophys. Res. Commun. 2017, 482, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Aglieco, F.; Dib-Hajj, S.D. Critical molecular determinants of voltage-gated sodium channel sensitivity to μ-conotoxins GIIIA/B. Mol. Pharmacol. 2002, 61, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Prusaksochaczewski, E.; Zamponi, G.; Becksickinger, A.G.; Gordon, R.D.; French, R.J. Action of derivatives of μ-conotoxin GIIIA on sodium-channels—Single amino-acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry 1992, 31, 8229–8238. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Kohda, D.; Hatanaka, H.; Lancelin, J.M.; Ishida, Y.; Oya, M.; Nakamura, H.; Inagaki, F.; Sato, K. Structure-activity-relationships of μ-conotoxin GIIIA—structure determination of active and inactive sodium-channel blocker peptides by NMR and simulated annealing calculations. Biochemistry 1992, 31, 12577–12584. [Google Scholar] [CrossRef] [PubMed]
- Dudley, S.C.; Chang, N.; Hall, J.; Lipkind, G.; Fozzard, H.A.; French, R.J. μ-conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 2000, 116, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Li, R.A.; Ennis, I.L.; French, R.J.; Dudley, S.C.; Tomaselli, G.F.; Marban, E. Clockwise domain arrangement of the sodium channel revealed by μ-conotoxin GIIIA docking orientation. J. Biol. Chem. 2001, 276, 11072–11077. [Google Scholar] [CrossRef] [PubMed]
- Safo, P.; Rosenbaum, T.; Shcherbatko, A.; Choi, D.Y.; Han, E.; Toledo-Aral, J.J.; Olivera, B.M.; Brehm, P.; Mandel, G. Distinction among neuronal subtypes of voltage-activated sodium channels by μ-conotoxin PIIIA. J. Neurosci. 2000, 20, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.J.; Watson, M.; Adams, D.J.; Hammarstrom, A.K.; Gage, P.W.; Hill, J.M.; Craik, D.J.; Thomas, L.; Adams, D.; Alewood, P.F.; et al. Solution structure of μ-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J. Biol. Chem. 2002, 277, 27247–27255. [Google Scholar] [CrossRef] [PubMed]
- Lancelin, J.M.; Kohda, D.; Tate, S.; Yanagawa, Y.; Abe, T.; Satake, M.; Inagaki, F. Tertiary structure of conotoxin GIIIA in aqueous-solution. Biochemistry 1991, 30, 6908–6916. [Google Scholar] [CrossRef] [PubMed]
- Keizer, D.W.; West, P.J.; Lee, E.F.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structural basis for tetrodotoxin-resistant sodium channel binding by μ-conotoxin SmIIIA. J. Biol. Chem. 2003, 278, 46805–46813. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.H.; Zhang, M.M.; Gupta, K.; Gajewiak, J.; Gulyas, J.; Balaram, P.; Rivier, J.E.; Olivera, B.M.; Yoshikami, D.; Bulaj, G.; et al. Mammalian neuronal sodium channel blocker μ-conotoxin BuIIIB has a structured N-terminus that influences potency. ACS Chem. Biol. 2013, 8, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Alewood, P.F.; Craik, D.J. Three-dimensional solution structure of μ-conotoxin GIIIB, a specific blocker of skeletal muscle sodium channels. Biochemistry 1996, 35, 8824–8835. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Conformational preferences of amino-acids in globular proteins. Biochemistry 1978, 17, 4277–4284. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Loughnan, M.; Thomas, L.; Adams, D.A.; Drinkwater, R.; Adams, D.J.; Alewood, P.F. Isolation and structure-activity of μ-conotoxin TIIIA, a potent inhibitor of tetrodotoxin-sensitive voltage-gated sodium channels. Mol. Pharmacol. 2007, 71, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley Interscience: New York, NY, USA, 1986. [Google Scholar]
- Hill, J.M.; Alewood, P.F.; Craik, D.J. Solution structure of the sodium channel antagonist conotoxin GS: A new molecular caliper for probing sodium channel geometry. Structure 1997, 5, 571–583. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. Molprobity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, L.A.; Gassner, N.C.; Snow, S.D.; Eldridge, A.M.; Baase, W.A.; Drew, D.L.; Matthews, B.W. Context-dependent protein stabilization by methionine-to-leucine substitution shown in T4 lysozyme. Protein Sci. 1998, 7, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ishida, Y.; Wakamatsu, K.; Kato, R.; Honda, H.; Ohizumi, Y.; Nakamura, H.; Ohya, M.; Lancelin, J.M.; Kohda, D.; et al. Active-site of μ-conotoxin GIIIA, a peptide blocker of muscle sodium-channels. J. Biol. Chem. 1991, 266, 16989–16991. [Google Scholar] [PubMed]
- Chahine, M.; Chen, L.Q.; Fotouhi, N.; Walsky, R.; Fry, D.; Santarelli, V.; Horn, R.; Kallen, R.G. Characterizing the μ-conotoxin binding site on voltage-sensitive sodium channels with toxin analogs and channel mutations. Receptor Channel 1995, 3, 161–174. [Google Scholar]
- Braunschweiler, L.; Ernst, R.R. Coherence transfer by isotropic mixing—Application to proton correlation spectroscopy. J. Magn. Reson. 1983, 53, 521–528. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Jeener, J.; Meier, B.H.; Bachmann, P.; Ernst, R.R. Investigation of exchange processes by 2-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Palmer, A.G.; Cavanagh, J.; Wright, P.E.; Rance, M. Sensitivity improvement in proton-detected 2-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 1991, 93, 151–170. [Google Scholar] [CrossRef]
- Griesinger, C.; Sorensen, O.W.; Ernst, R.R. Practical aspects of the eCOSY technique—Measurement of scalar spin spin coupling-constants in peptides. J. Magn. Reson. 1987, 75, 474–492. [Google Scholar]
- Hwang, T.L.; Shaka, A.J. Water suppression that works—Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, P.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar]
- Nederveen, A.J.; Doreleijers, J.F.; Vranken, W.; Miller, Z.; Spronk, C.A.E.M.; Nabuurs, S.B.; Guntert, P.; Livny, M.; Markley, J.L.; Nilges, M.; et al. Recoord: A recalculated coordinate database of 500+ proteins from the pdb using restraints from the Biomagresbank. Proteins 2005, 59, 662–672. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of GIIIA and GIIIC are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Restraints | |
|---|---|
| total no. distance restraints | 179 |
| intraresidue | 71 |
| sequential | 59 |
| medium range, i − j < 5 | 35 |
| long range, i − j ≥ 5 | 14 |
| hydrogen bond restraints | 6 |
| dihedral angle restraints | |
| phi | 17 |
| psi chi1 | 11 5 |
| Deviations from idealized geometry | |
| bond lengths (Å) | 0.010 ± 0.001 |
| bond angles (deg) | 1.294 ± 0.046 |
| impropers (deg) | 1.38 ± 0.18 |
| NOE (Å) | 0.014 ± 0.002 |
| cDih (deg) | 0.126 ± 0.132 |
| Mean energies (kcal/mol) | |
| overall | −664 ± 42 |
| bonds | 9.3 ± 1.0 |
| angles | 34.0 ± 2.7 |
| improper | 10.7 ± 2.1 |
| van Der Waals | −56.6 ± 5.7 |
| NOE | 0.04 ± 0.01 |
| cDih | 0.13 ± 0.18 |
| electrostatic | −760 ± 43 |
| Violations | |
| NOE violations exceeding 0.2 Å | 0 |
| dihedral violations exceeding 2.0 Å | 0 |
| Rms deviation from mean structure, Å | |
| backbone atoms | 1.44 ± 0.37 |
| all heavy atoms | 2.51 ± 0.48 |
| Stereochemical quality b | |
| residues in most favoured Ramachandran region, % | 96.6 ± 4.1 |
| Ramachandran outliers, % | 0.2 ± 1.1 |
| unfavourable sidechain rotamers, % | 0.0 ± 0.0 |
| clashscore, all atoms | 6.5 ± 3.0 |
| overall MolProbity score | 1.5 ± 0.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harvey, P.J.; Kurniawan, N.D.; Finol-Urdaneta, R.K.; McArthur, J.R.; Van Lysebetten, D.; Dash, T.S.; Hill, J.M.; Adams, D.J.; Durek, T.; Craik, D.J. NMR Structure of μ-Conotoxin GIIIC: Leucine 18 Induces Local Repacking of the N-Terminus Resulting in Reduced NaV Channel Potency. Molecules 2018, 23, 2715. https://doi.org/10.3390/molecules23102715
Harvey PJ, Kurniawan ND, Finol-Urdaneta RK, McArthur JR, Van Lysebetten D, Dash TS, Hill JM, Adams DJ, Durek T, Craik DJ. NMR Structure of μ-Conotoxin GIIIC: Leucine 18 Induces Local Repacking of the N-Terminus Resulting in Reduced NaV Channel Potency. Molecules. 2018; 23(10):2715. https://doi.org/10.3390/molecules23102715
Chicago/Turabian StyleHarvey, Peta J., Nyoman D. Kurniawan, Rocio K. Finol-Urdaneta, Jeffrey R. McArthur, Dorien Van Lysebetten, Thomas S. Dash, Justine M. Hill, David J. Adams, Thomas Durek, and David J. Craik. 2018. "NMR Structure of μ-Conotoxin GIIIC: Leucine 18 Induces Local Repacking of the N-Terminus Resulting in Reduced NaV Channel Potency" Molecules 23, no. 10: 2715. https://doi.org/10.3390/molecules23102715
APA StyleHarvey, P. J., Kurniawan, N. D., Finol-Urdaneta, R. K., McArthur, J. R., Van Lysebetten, D., Dash, T. S., Hill, J. M., Adams, D. J., Durek, T., & Craik, D. J. (2018). NMR Structure of μ-Conotoxin GIIIC: Leucine 18 Induces Local Repacking of the N-Terminus Resulting in Reduced NaV Channel Potency. Molecules, 23(10), 2715. https://doi.org/10.3390/molecules23102715