The Pro-Tumoral Activity of Heparan Sulfate 3-O-Sulfotransferase 3B (HS3ST3B) in Breast Cancer MDA-MB-231 Cells Is Dependent on the Expression of Neuropilin-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
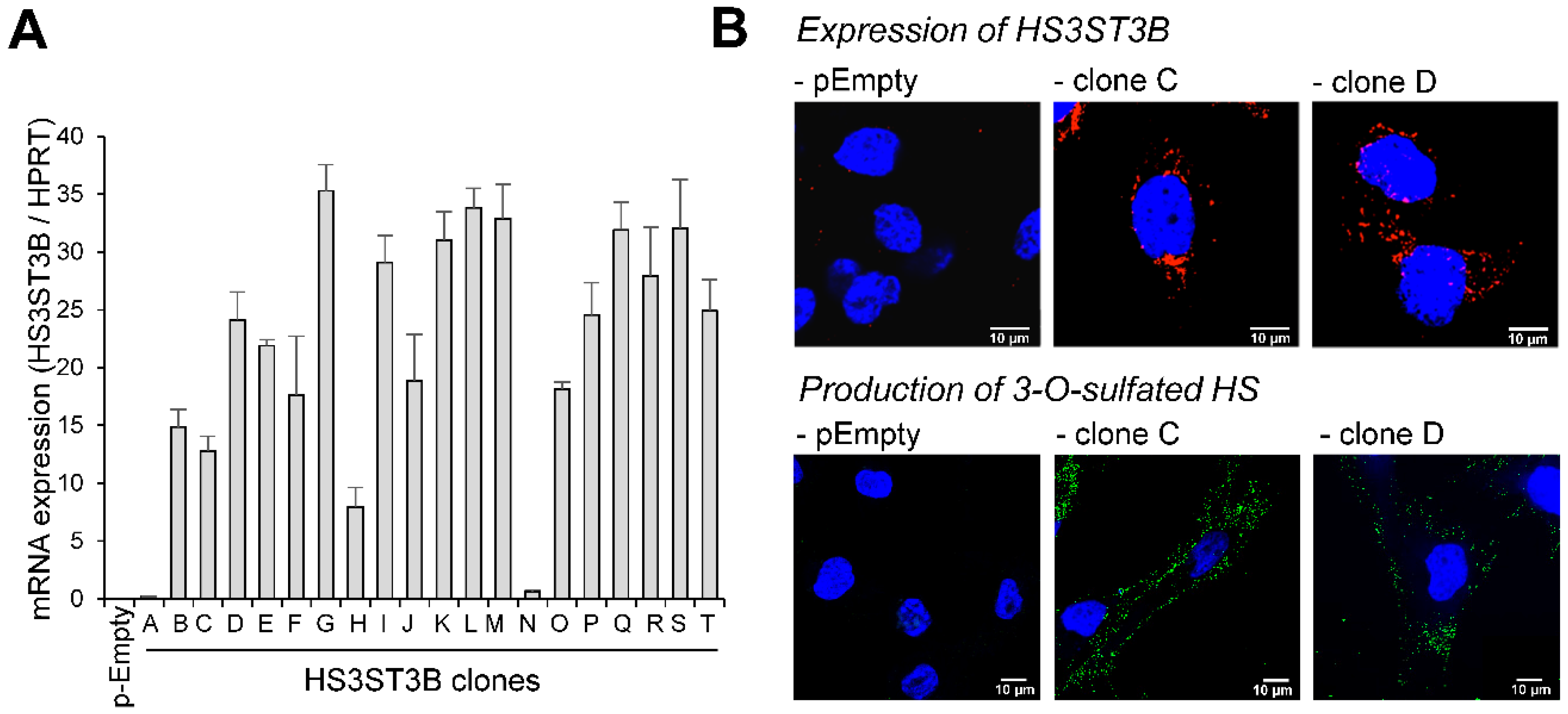
2.1. Expression of HS3ST3B by Stable Transfection in MDA-MB-231 Cells
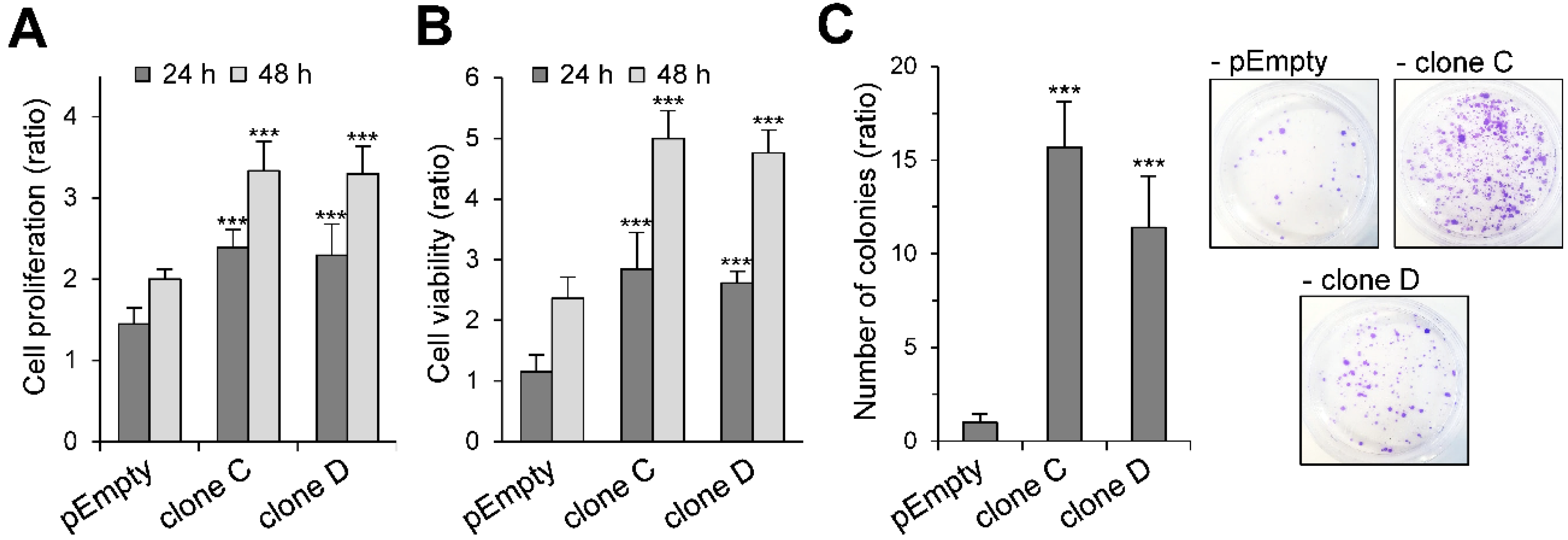
2.2. Effect of the Stable Expression of HS3ST3B on the Proliferation and Viability of MBA-MB-231 Cells
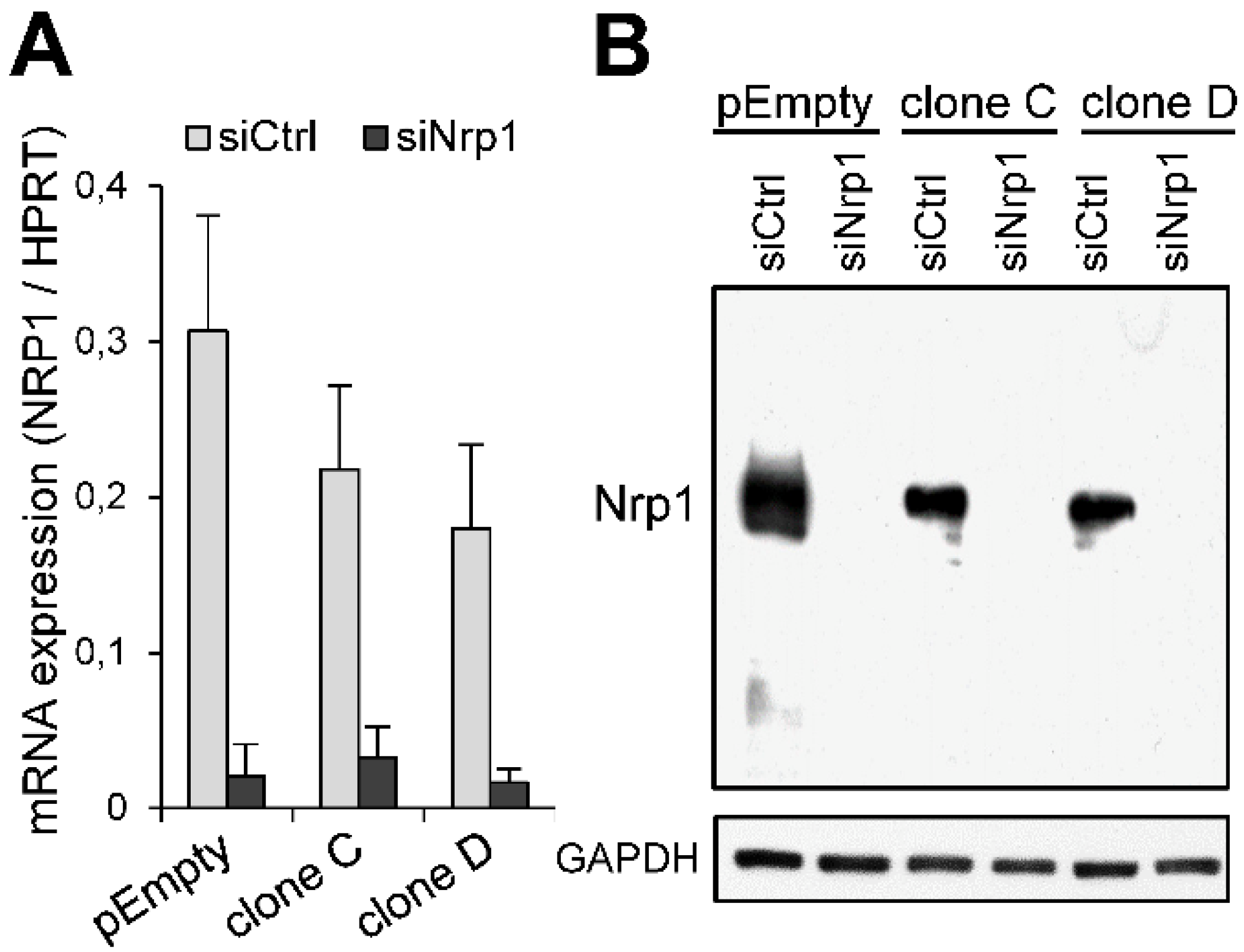
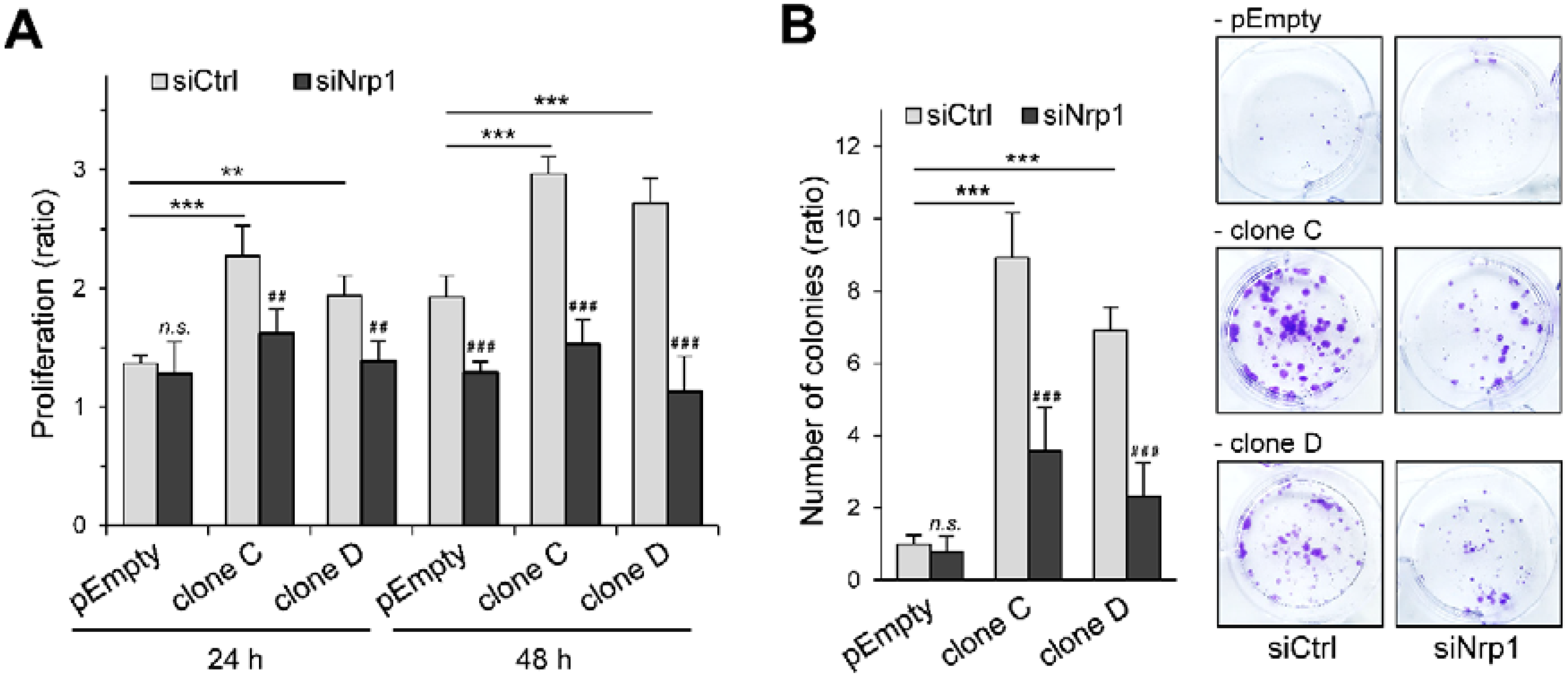
2.3. Participation of Nrp1 to the Enhancing Effect of HS3ST3B on MDA-MB-231 Cell Growth
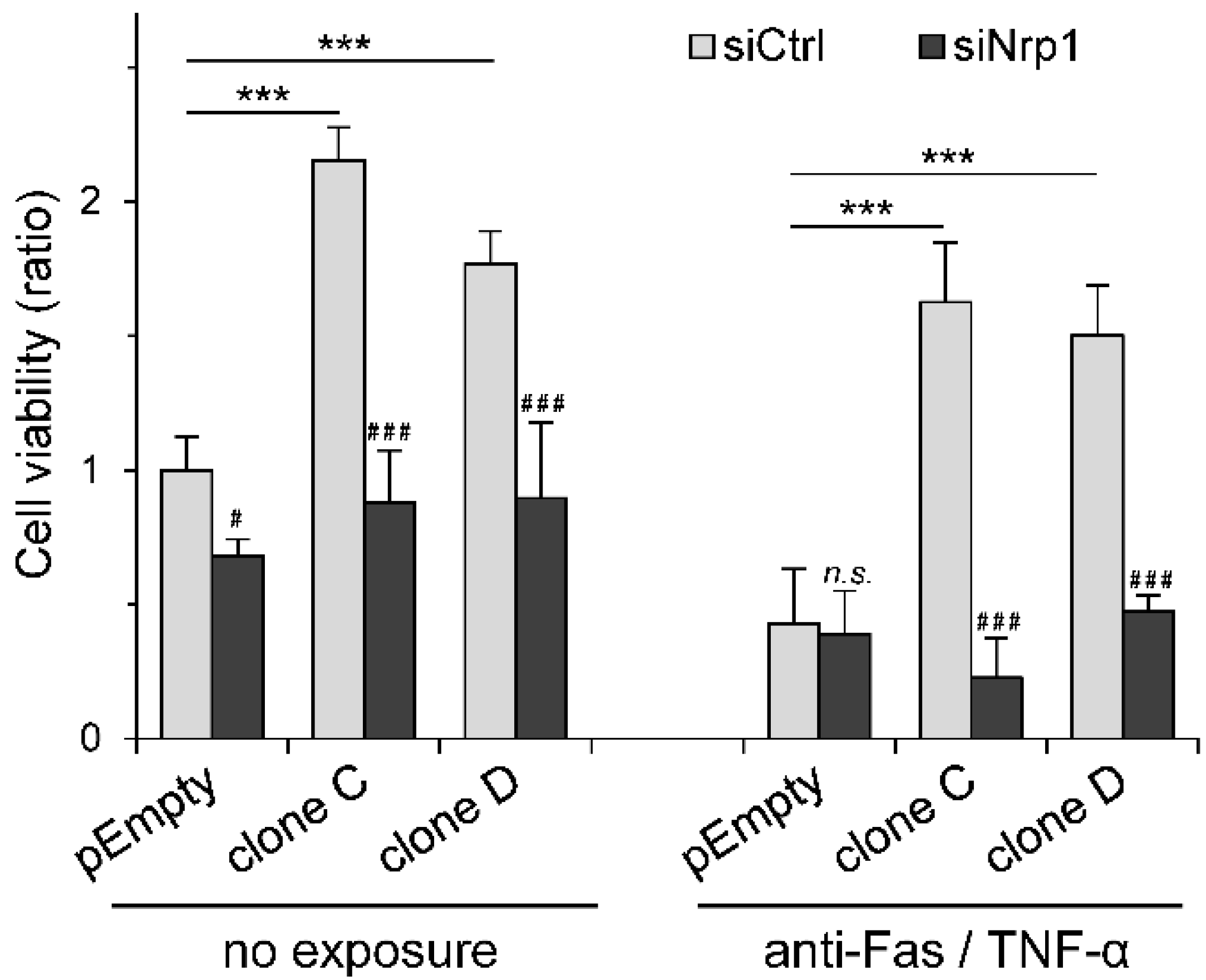
2.4. Participation of Nrp1 to HS3ST3B-Mediated Protection against Cell Death
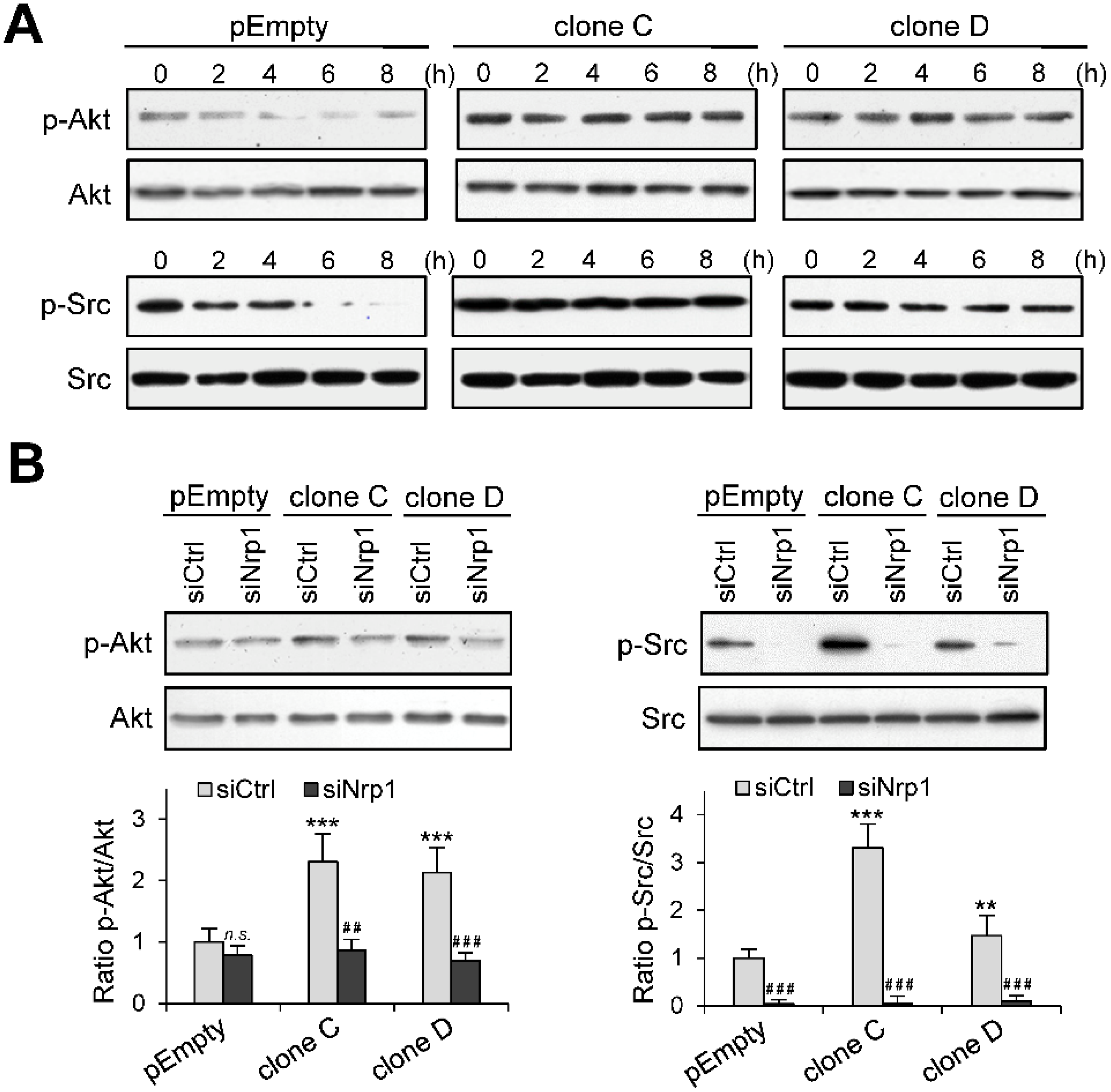
2.5. Role of Nrp1 in the Activation of Akt and Src in HS3ST3B Expressing Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Transfection
4.3. Measurement of Cell Proliferation and Viability
4.4. Colony Formation Assay
4.5. RNA Interference
4.6. RNA Isolation and Real-Time RT-PCR
4.7. Immunofluorescence Staining and Analysis
4.8. SDS-PAGE and Western Blot
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Esko, J.D.; Selleck, S.B. Order out of chaos: Assembly of ligand binding sites in heparan sulphate. Annu. Rev. Biochem. 2002, 71, 435–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Glycosaminoglycan (GAG) biosynthesis and GAG-binding proteins. Prog. Mol. Biol. Transl. Sci. 2010, 93, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Powell, A.; Guimond, S. Heparan sulfate: Decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001, 11, 75–82. [Google Scholar] [CrossRef]
- Thacker, B.E.; Xu, D.; Lawrence, R.; Esko, J.D. Heparan sulfate 3-O-sulfation: A rare modification in search of a function. Matrix Biol. 2014, 35, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shworak, N.W.; Fritze, L.M.; Edelberg, J.M.; Rosenberg, R.D. Purification of heparan sulfate d-glucosaminyl 3-O-sulfotransferase. J. Biol. Chem. 1996, 271, 27072–27082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Lawrence, R.; Schwartz, J.J.; Bai, X.M.; Wei, G.; Esko, J.D.; Rosenberg, R.D. The effect of precursor structures on the action of glucosaminyl 3-O-sulfotransferase-1 and the biosynthesis of anticoagulant heparan sulfate. J. Biol. Chem. 2001, 276, 28806–28813. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Yoshida, K.; Gotoh, M.; Sugioka, S.; Kikuchi, N.; Kwon, Y.D.; Tawada, A.; Maeyama, K.; Inaba, N.; Hiruma, T.; et al. Characterization of a heparan sulfate 3-O-sulfotransferase-5, an enzyme synthesizing a tetrasulfated disaccharide. J. Biol. Chem. 2003, 278, 26780–26787. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Chen, J.; Tiwari, V.; Ju, W.; Li, J.P.; Malmstrom, A.; Shukla, D.; Liu, J. Heparan sulfate 3-O-sulfotransferase isoform 5 generates both an antithrombin-binding site and an entry receptor for herpes simplex virus, type 1. J. Biol. Chem. 2002, 277, 37912–37919. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shriver, Z.; Blaiklock, P.; Yoshida, K.; Sasisekharan, R.; Rosenberg, R.D. Heparan sulfate d-glucosaminyl 3-O-sulfotransferase-3A sulfates N-unsubstituted glucosamine residues. J. Biol. Chem. 1999, 274, 38155–38162. [Google Scholar] [CrossRef] [PubMed]
- O'Donnell, C.D.; Tiwari, V.; Oh, M.J.; Shukla, D. A role for heparan sulfate 3-O sulfotransferase isoform 2 in herpes simplex virus type 1 entry and spread. Virology 2006, 346, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.M.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef]
- Shworak, N.W.; Liu, J.A.; Petros, L.M.; Zhang, L.J.; Kobayashi, M.; Copeland, N.G.; Jenkins, N.A.; Rosenberg, R.D. Multiple isoforms of heparan sulfate d-glucosaminyl 3-O-sulfotransferase—Isolation, characterization, and expression of human cDNAs and identification of distinct genomic loci. J. Biol. Chem. 1999, 274, 5170–5184. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; O'Donnell, C.D.; Oh, M.J.; Valyi-Nagy, T.; Shukla, D. A role for 3-O sulfotransferase isoform-4 in assisting HSV-1 entry and spread. Biochem. Biophys. Res. Commun. 2005, 338, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Tiwari, V.; Xia, G.; Clement, C.; Shukla, D.; Liu, J. Characterization of heparan sulphate 3-O-sulphotransferase isoform 6 and its role in assisting the entry of herpes simplex virus type 1. Biochem. J. 2005, 385, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.; Ouzzine, M.; Talhaoui, I.; Sharp, S.; Prydz, K.; Coughtrie, M.W.; Fournel-Gigleux, S. Epigenetics: Methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of H-EMCSS chondrosarcoma cells. FASEB J. 2010, 24, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.A.; Kim, Y.; Hong, S.H.; Lee, J.; Cho, Y.G.; Han, J.Y.; Kim, Y.H.; Han, J.; Shim, Y.M.; Lee, Y.S.; et al. Epigenetic inactivation of heparan sulfate (glucosamine) 3-O-sulfotransferase 2 in lung cancer and its role in tumorigenesis. PLoS ONE 2013, 8, e79634. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Gauche, C.; Coughtrie, M.W.; Bui, C.; Gulberti, S.; Merhi-Soussi, F.; Ramalanjaona, N.; Bertin-Jung, I.; Diot, A.; Dumas, D.; et al. The heparan sulfate sulfotransferase 3-OST3A (HS3ST3A) is a novel tumor regulator and a prognostic marker in breast cancer. Oncogene 2016, 35, 5043–5055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, K.; Asada, K.; Fukutomi, T.; Okochi, E.; Yagi, Y.; Hasegawa, T.; Asahara, T.; Sugimura, T.; Ushijima, T. Methylation-associated silencing of heparan sulfate D-glucosaminyl 3-O-sulfotransferase-2 (3-OST-2) in human breast, colon, lung and pancreatic cancers. Oncogene 2003, 22, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Vijaya-Kumar, A.; Salem-Gassar, E.; Spillmann, D.; Stock, C.; Sen, Y.P.; Zhang, T.; Van Kuppevelt, T.H.; Hülsewig, C.; Koszlowski, E.O.; Pavao, M.S.; et al. HS3ST2 modulates breast cancer cell invasiveness via MAP kinase- and Tcf4 (Tcf7l2)-dependent regulation of protease and cadherin expression. Int. J. Cancer 2014, 135, 2579–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Li, Q.; Jiang, Z.Z.; Guo, C.W.; Li, P. Heparan sulfate d-glucosaminyl 3-O-sulfotransferase-3B1, a novel epithelial-mesenchymal transition inducer in pancreatic cancer. Cancer Biol. Ther. 2011, 12, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Song, K.; Zhou, L.; Xie, Z.; Zhou, P.; Zhao, Y.; Han, Y.; Xu, X.; Li, P. Heparan sulfate d-glucosaminyl 3-O-sulfotransferase-3B1 (HS3ST3B1) promotes angiogenesis and proliferation by induction of VEGF in acute myeloid leukemia cells. J. Cell Biochem. 2015, 116, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Biroccio, A.; Cherfils-Vicini, J.; Augereau, A.; Pinte, S.; Bauwens, S.; Ye, J.; Simonet, T.; Horard, B.; Jamet, K.; Cervera, L.; et al. TRF2 inhibits a cell-extrinsic pathway through which natural killer cells eliminate cancer cells. Nat. Cell Biol. 2013, 15, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Hellec, C.; Delos, M.; Carpentier, M.; Denys, A.; Allain, F. The heparan sulfate 3-O-sulfotransferases (HS3ST) 2, 3B and 4 enhance proliferation and survival in breast cancer MDA-MB-231 cells. PLoS ONE 2018, 13, e0194676. [Google Scholar] [CrossRef] [PubMed]
- Thacker, B.E.; Seamen, E.; Lawrence, R.; Parker, M.W.; Xu, Y.; Liu, J.; Vander Kooi, C.W.; Esko, J.D. Expanding the 3-O-sulfate proteome–Enhanced binding of neuropilin-1 to 3-O-sulfated heparan sulfate modulates its activity. ACS Chem. Biol. 2016, 11, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, B.M.; Tan, P.K.; Niederleithner, H.; Ferrara, N.; Petzelbauer, P.; Sibilia, M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 2010, 140, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J.; Glinka, Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget 2012, 3, 921–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, C.; Ruhrberg, C. Neuropilin signalling in vessels, neurons and tumours. Semin. Cell. Dev. Biol. 2013, 24, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Uniewicz, K.A.; Fernig, D.G. Neuropilins: A versatile partner of extracellular molecules that regulate development and disease. Front. Biosci. 2008, 13, 4339–4360. [Google Scholar] [CrossRef] [PubMed]
- West, D.C.; Rees, C.G.; Duchesne, L.; Patey, S.J.; Terry, C.J.; Turnbull, J.E.; Delehedde, M.; Heegaard, C.W.; Allain, F.; Vanpouille, C.; et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J. Biol. Chem. 2005, 280, 13457–13464. [Google Scholar] [CrossRef] [PubMed]
- Delos, M.; Foulquier, F.; Hellec, C.; Vicogne, D.; Fifre, A.; Carpentier, M.; Papy-Garcia, D.; Allain, F.; Denys, A. Heparan sulfate 3-O-sulfotransferase 2 (HS3ST2) displays an unexpected subcellular localization in the plasma membrane. Biochim. Biophys. Acta 2018, 1862, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Debatin, K.M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsiades, C.S.; Mitsiades, N.; Koutsilieris, M. The Akt pathway: Molecular targets for anti-cancer drug development. Curr. Cancer Drug Targets 2004, 4, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Bailón, M.P.; Calcabrini, A.; Gómez-Domínguez, D.; Morte, B.; Martín-Forero, E.; Gómez-López, G.; Molinari, A.; Wagner, K.U.; Martín-Pérez, J. Src kinases catalytic activity regulates proliferation, migration and invasiveness of MDA-MB-231 breast cancer cells. Cell Signal. 2012, 6, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Fuh, G.; Garcia, K.C.; de Vos, A.M. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J. Biol. Chem. 2000, 275, 26690–26695. [Google Scholar] [CrossRef] [PubMed]
- Teran, M.; Nugent, M.A. Synergistic binding of vascular endothelial growth factor-A and its receptors to heparin selectively modulates complex affinity. J. Biol. Chem. 2015, 290, 16451–16462. [Google Scholar] [CrossRef] [PubMed]
- Bachelder, R.E.; Crago, A.; Chung, J.; Wendt, M.A.; Shaw, L.M.; Robinson, G.; Mercurio, A.M. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 2001, 61, 5736–5740. [Google Scholar] [PubMed]
- Miao, H.Q.; Lee, P.; Lin, H.; Soker, S.; Klagsbrun, M. Neuropilin-1 expression by tumor cells promotes tumor angiogenesis and progression. FASEB J. 2000, 14, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Rizzolio, S.; Rabinowicz, N.; Rainero, E.; Lanzetti, L.; Serini, G.; Norman, J.; Neufeld, G.; Tamagnone, L. Neuropilin-1-dependent regulation of EGF-receptor signaling. Cancer Res. 2012, 72, 5801–5811. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Mac Gabhann, F. VEGF-A121a binding to neuropilins—A concept revisited. Cell Adhes. Migr. 2018, 12, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Yoshida, K.; Shibata, Y.; Kimata, K. Tetrasulfated disaccharide unit in heparan sulfate: Enzymatic formation and tissue distribution. J. Biol. Chem. 2008, 283, 31237–31245. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hsieh, P.H.; Xu, Y.; Thieker, D.; Chai, E.J.; Xie, S.; Cooley, B.; Woods, R.J.; Chi, L.; Liu, J. Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate. J. Am. Chem. Soc. 2017, 139, 5249–5256. [Google Scholar] [CrossRef] [PubMed]
- Krenn, E.C.; Wille, I.; Gesslbauer, B.; Poteser, M.; van Kuppevelt, T.H.; Kungl, A.J. Glycanogenomics: A qPCR-approach to investigate biological glycan function. Biochem. Biophys. Res. Commun. 2008, 375, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Denys, A.; Delos, M.; Sikora, A.S.; Carpentier, M.; Julien, S.; Pestel, J.; Allain, F. Macrophage polarization alters the expression and sulfation pattern of glycosaminoglycans. Glycobiology 2015, 25, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.S.; Delos, M.; Martinez, P.; Carpentier, M.; Allain, F.; Denys, A. Regulation of the expression of heparan sulfate 3-O-sulfotransferase 3B (HS3ST3B) by inflammatory stimuli in human monocytes. J. Cell. Biochem. 2016, 117, 1529–1542. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.S.; Hellec, C.; Carpentier, M.; Martinez, P.; Delos, M.; Denys, A.; Allain, F. Tumour-necrosis factor-α induces heparan sulfate 6-O-endosulfatase 1 (Sulf-1) expression in fibroblasts. Int. J. Biochem. Cell. Biol. 2016, 80, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, H.; Wang, Y.; Shi, M. Heparan sulfate d-glucosamine 3-O-sulfotransferase 3B1 is a novel regulator of transforming growth factor-beta-mediated epithelial-to-mesenchymal transition and regulated by miR-218 in non-small cell lung cancer. J. Can. Res. Ther. 2018, 14, 24–29. [Google Scholar] [CrossRef]
- Aung, N.Y.; Ohe, R.; Meng, H.; Kabasawa, T.; Yang, S.; Kato, T.; Yamakawa, M. Specific neuropilins expression in alveolar macrophages among tissue-specific macrophages. PLoS ONE 2016, 11, e0147358. [Google Scholar] [CrossRef] [PubMed]
- Delos, M.; Hellec, C.; Foulquier, F.; Carpentier, M.; Allain, F.; Denys, A. Participation of 3-O-sulfated heparan sulfates in the protection of macrophages by herpes simplex virus-1 glycoprotein D and cyclophilin B against apoptosis. FEBS Open Bio 2016, 7, 133–148. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hellec, C.; Diawara, M.; Carpentier, M.; Denys, A.; Allain, F. The Pro-Tumoral Activity of Heparan Sulfate 3-O-Sulfotransferase 3B (HS3ST3B) in Breast Cancer MDA-MB-231 Cells Is Dependent on the Expression of Neuropilin-1. Molecules 2018, 23, 2718. https://doi.org/10.3390/molecules23102718
Hellec C, Diawara M, Carpentier M, Denys A, Allain F. The Pro-Tumoral Activity of Heparan Sulfate 3-O-Sulfotransferase 3B (HS3ST3B) in Breast Cancer MDA-MB-231 Cells Is Dependent on the Expression of Neuropilin-1. Molecules. 2018; 23(10):2718. https://doi.org/10.3390/molecules23102718
Chicago/Turabian StyleHellec, Charles, Mariama Diawara, Mathieu Carpentier, Agnès Denys, and Fabrice Allain. 2018. "The Pro-Tumoral Activity of Heparan Sulfate 3-O-Sulfotransferase 3B (HS3ST3B) in Breast Cancer MDA-MB-231 Cells Is Dependent on the Expression of Neuropilin-1" Molecules 23, no. 10: 2718. https://doi.org/10.3390/molecules23102718
APA StyleHellec, C., Diawara, M., Carpentier, M., Denys, A., & Allain, F. (2018). The Pro-Tumoral Activity of Heparan Sulfate 3-O-Sulfotransferase 3B (HS3ST3B) in Breast Cancer MDA-MB-231 Cells Is Dependent on the Expression of Neuropilin-1. Molecules, 23(10), 2718. https://doi.org/10.3390/molecules23102718