
The Low Molecular Weight Heparin Tinzaparin Attenuates Platelet Activation in Terms of Metastatic Niche Formation by Coagulation-Dependent and Independent Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Inhibition of Tumor Cell Induced Thrombin Generation
2.1.1. TF Expression by Tumor Cell Lines
2.1.2. Thrombin Generation and its Inhibition
2.2. Coagulation Independent Readouts of Platelet Activation
2.2.1. Interference with Platelet Aggregation
2.2.2. Inhibition of ATP Release from Platelets
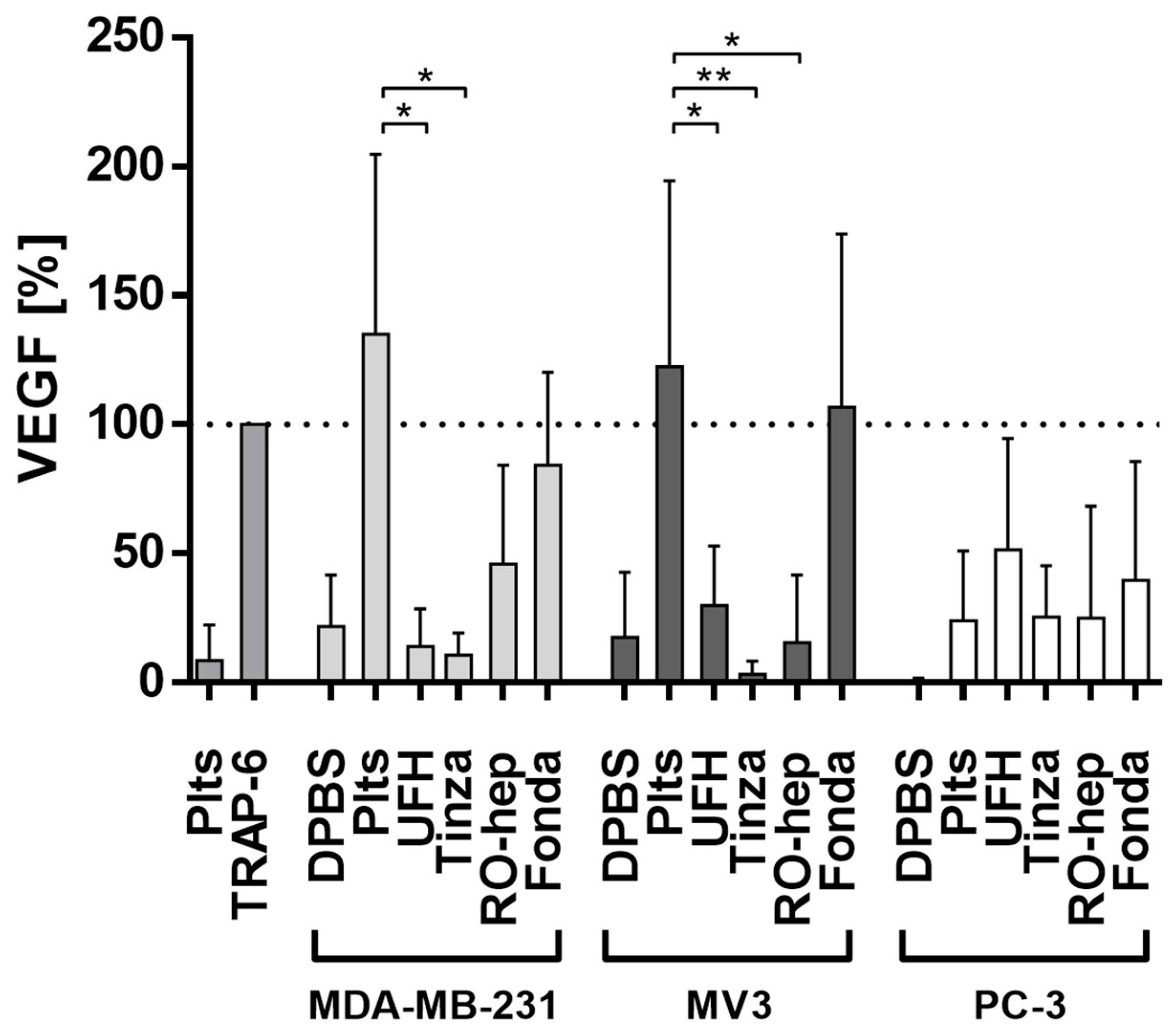
2.2.3. Inhibition of VEGF Release from Platelets and Functional Consequences
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Platelet Isolation and Activation
4.3. Flow Cytometric Detection of Tissue Factor Surface Expression
4.4. Thrombin Generation Assay
4.5. Light Transmission Aggregometry
4.6. ATP Assay
4.7. ELISA
4.8. Tube Formation Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Connors, J.M. Prophylaxis against venous thromboembolism in ambulatory patients with cancer. N. Engl. J. Med. 2014, 370, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Spek, C.A.; Versteeg, H.H.; Borensztajn, K.S. Anticoagulant therapy of cancer patients: Will patient selection increase overall survival? Thromb. Haemost. 2015, 114. [Google Scholar] [CrossRef]
- Kakkar, A.K.; Levine, M.N.; Kadziola, Z.; Lemoine, N.R.; Low, V.; Patel, H.K.; Rustin, G.; Thomas, M.; Quigley, M.; Williamson, R.C.N. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: The fragmin advanced malignancy outcome study (FAMOUS). J. Clin. Oncol. 2004, 22, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Klerk, C.P.W.; Smorenburg, S.M.; Otten, H.-M.; Lensing, A.W.A.; Prins, M.H.; Piovella, F.; Prandoni, P.; Bos, M.M.E.M.; Richel, D.J.; van Tienhoven, G.; et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J. Clin. Oncol. 2005, 23, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- Macbeth, F.; Noble, S.; Evans, J.; Ahmed, S.; Cohen, D.; Hood, K.; Knoyle, D.; Linnane, S.; Longo, M.; Moore, B.; et al. Randomized Phase III Trial of Standard Therapy Plus Low Molecular Weight Heparin in Patients With Lung Cancer: FRAGMATIC Trial. J. Clin. Oncol. 2016, 34, 488–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ek, L.; Gezelius, E.; Bergman, B.; Bendahl, P.O.; Anderson, H.; Sundberg, J.; Wallberg, M.; Falkmer, U.; Verma, S.; Belting, M.; et al. Randomized phase III trial of low-molecular-weight heparin enoxaparin in addition to standard treatment in small-cell lung cancer: the RASTEN trial. Ann. Oncol. 2018, 29, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updat. 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Zucchella, M.; Dezza, L.; Pacchiarini, L.; Meloni, F.; Tacconi, F.; Bonomi, E.; Grignani, G.; Notario, A. Human tumor cells cultured “in vitro” activate platelet function by producing ADP or thrombin. Haematologica 1989, 74, 541–545. [Google Scholar] [PubMed]
- Aitokallio-Tallberg, A.M.; Viinikka, L.U.; Ylikorkala, R.O. Increased synthesis of prostacyclin and thromboxane in human ovarian malignancy. Cancer Res. 1988, 48, 2396–2398. [Google Scholar] [PubMed]
- Dang, Q.; Liu, J.; Li, J.; Sun, Y. Podoplanin: A novel regulator of tumor invasion and metastasis. Med. Oncol. 2014, 31, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, N. Platelets in cancer metastasis: To help the “villain” to do evil. Int. J. Cancer 2016, 138, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.-G.; Kopp, H.-G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Kopp, H.-G.; Salih, H.R. Modulation of natural killer cell anti-tumor reactivity by platelets. J. Innate Immun. 2011, 3, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, L.; Schön, M.P. Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Läubli, H.; Borsig, L. Selectins promote tumor metastasis. Semin. Cancer Biol. 2010, 20, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B.Z.; Kahn, M.L. Platelets and tumor cells: a new form of border control. Cancer Cell 2013, 24, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Honn, K.V.; Grossi, I.M.; Diglio, C.A.; Wojtukiewicz, M.; Taylor, J.D. Enhanced tumor cell adhesion to the subendothelial matrix resulting from 12(S)-HETE-induced endothelial cell retraction. FASEB J. 1989, 3, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Möhle, R.; Green, D.; Moore, M.A.; Nachman, R.L.; Rafii, S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. USA 1997, 94, 663–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, R.J.; Boehme, B.; Podda, M.; Henschler, R.; Jager, E.; Tandi, C.; Boehncke, W.-H.; Zollner, T.M.; Kaufmann, R.; Gille, J. Endothelial P-selectin as a target of heparin action in experimental melanoma lung metastasis. Cancer Res. 2004, 64, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.L.; Choi, S.H.; Varki, A. Differential metastasis inhibition by clinically relevant levels of heparins—Correlation with selectin inhibition, not antithrombotic activity. Clin. Cancer Res. 2005, 11, 7003–7011. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.L.; Varki, A.; Borsig, L. Heparin attenuates metastasis mainly due to inhibition of P- and L-selectin, but non-anticoagulant heparins can have additional effects. Thromb. Res. 2007, 120, S107–S111. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Y.; Gao, Y.; Shen, J.; Zheng, S.; Wei, M.; Zeng, X. Modified heparins inhibit integrin αIIbβ3 mediated adhesion of melanoma cells to platelets in vitro and in vivo. Int. J. Cancer. 2009, 125, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Harenberg, J. New developments in anticoagulants: Past, present and future. Thromb. Haemost. 2017, 117, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casu, B.; Vlodavsky, I.; Sanderson, R.D. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol. Haemost. Thromb. 2008, 36, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. Vascular permeability—The essentials. Ups. J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.Y.; Kamphuisen, P.W.; Meyer, G.; Bauersachs, R.; Janas, M.S.; Jarner, M.F.; Khorana, A.A.; CATCH Investigators. Tinzaparin vs Warfarin for Treatment of Acute Venous Thromboembolism in Patients with Active Cancer: A Randomized Clinical Trial. JAMA 2015, 314, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Kalaska, B.; Kaminski, K.; Miklosz, J.; Yusa, S.-I.; Sokolowska, E.; Blazejczyk, A.; Wietrzyk, J.; Kasacka, I.; Szczubialka, K.; Pawlak, D.; et al. Heparin-binding copolymer reverses effects of unfractionated heparin, enoxaparin, and fondaparinux in rats and mice. Transl. Res. 2016, 177, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Kalathottukaren, M.T.; Abbina, S.; Yu, K.; Shenoi, R.A.; Creagh, A.L.; Haynes, C.; Kizhakkedathu, J.N. A Polymer Therapeutic Having Universal Heparin Reversal Activity: Molecular Design and Functional Mechanism. Biomacromolecules 2017, 18, 3343–3358. [Google Scholar] [CrossRef] [PubMed]
- Välimäki, S.; Khakalo, A.; Ora, A.; Johansson, L.-S.; Rojas, O.J.; Kostiainen, M.A. Effect of PEG-PDMAEMA Block Copolymer Architecture on Polyelectrolyte Complex Formation with Heparin. Biomacromolecules 2016, 17, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, G.; Becattini, C.; Bauersachs, R.; Brenner, B.; Campanini, M.; Cohen, A.; Connors, J.M.; Fontanella, A.; Gussoni, G.; Huisman, M.V.; et al. Apixaban versus Dalteparin for the Treatment of Acute Venous Thromboembolism in Patients with Cancer: The Caravaggio Study. Thromb. Haemost. 2018. [Google Scholar] [CrossRef]
- Xing, J.; Yin, X.; Chen, D. Rivaroxaban versus enoxaparin for the prevention of recurrent venous thromboembolism in patients with cancer: A meta-analysis. Medicine (Baltimore) 2018, 97, e11384. [Google Scholar] [CrossRef] [PubMed]
- Raskob, G.E.; van Es, N.; Verhamme, P.; Carrier, M.; Di Nisio, M.; Garcia, D.; Grosso, M.A.; Kakkar, A.K.; Kovacs, M.J.; Mercuri, M.F.; et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N. Engl. J. Med. 2018, 378, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Ornstein, D.L.; Zacharski, L.R. The use of heparin for treating human malignancies. Haemostasis 1999, 29, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Simonis, D.; Fritzsche, J.; Alban, S.; Bendas, G. Kinetic analysis of heparin and glucan sulfates binding to P-selectin and its impact on the general understanding of selectin inhibition. Biochemistry 2007, 46, 6156–6164. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, N.; Naggi, A.; Torri, G.; Ishai-Michaeli, R.; Casu, B.; Vlodavsky, I.; Borsig, L. P-selectin- and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. FASEB J. 2007, 21, 3562–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel, M.; Fish, W.R.; Toma, N.; Luo, S.; Bird, K.; Mori, K.; Kusumoto, S.; Blystone, S.D.; Suda, Y. Heparin modulates integrin function in human platelets. J. Vasc. Surg. 2001, 33, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, A.; Casu, B.; Cassinelli, G.; Guerrini, M.; Torri, G.; Naggi, A. Structural features of glycol-split low-molecular-weight heparins and their heparin lyase-generated fragments. Anal. Bioanal. Chem. 2014, 406, 249–265. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of this study were commercially available, the non-anticoagulant heparin derivative RO-heparin has been prepared by Dr. Naggi et al. [41]. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gockel, L.M.; Ponert, J.M.; Schwarz, S.; Schlesinger, M.; Bendas, G. The Low Molecular Weight Heparin Tinzaparin Attenuates Platelet Activation in Terms of Metastatic Niche Formation by Coagulation-Dependent and Independent Pathways. Molecules 2018, 23, 2753. https://doi.org/10.3390/molecules23112753
Gockel LM, Ponert JM, Schwarz S, Schlesinger M, Bendas G. The Low Molecular Weight Heparin Tinzaparin Attenuates Platelet Activation in Terms of Metastatic Niche Formation by Coagulation-Dependent and Independent Pathways. Molecules. 2018; 23(11):2753. https://doi.org/10.3390/molecules23112753
Chicago/Turabian StyleGockel, Lukas Maria, Jan Moritz Ponert, Svenja Schwarz, Martin Schlesinger, and Gerd Bendas. 2018. "The Low Molecular Weight Heparin Tinzaparin Attenuates Platelet Activation in Terms of Metastatic Niche Formation by Coagulation-Dependent and Independent Pathways" Molecules 23, no. 11: 2753. https://doi.org/10.3390/molecules23112753
APA StyleGockel, L. M., Ponert, J. M., Schwarz, S., Schlesinger, M., & Bendas, G. (2018). The Low Molecular Weight Heparin Tinzaparin Attenuates Platelet Activation in Terms of Metastatic Niche Formation by Coagulation-Dependent and Independent Pathways. Molecules, 23(11), 2753. https://doi.org/10.3390/molecules23112753