Heparan Sulfate Mimetics in Cancer Therapy: The Challenge to Define Structural Determinants and the Relevance of Targets for Optimal Activity



Abstract
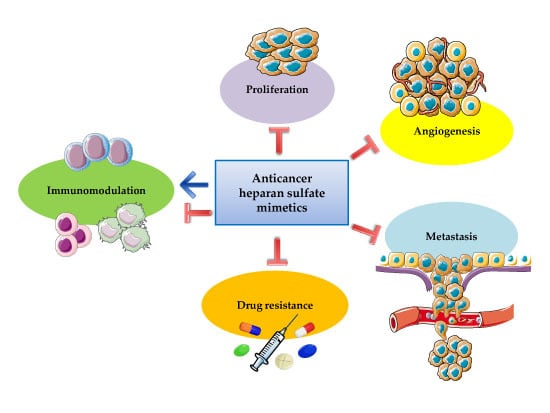
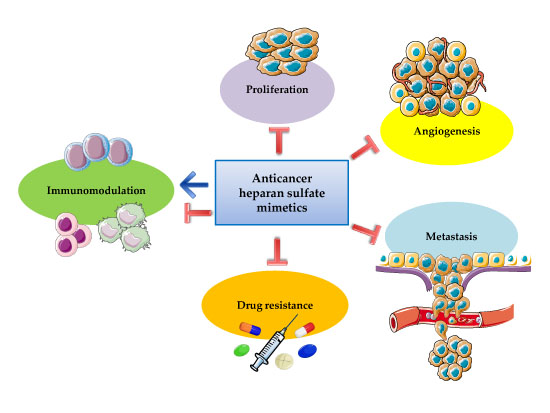
:
1. Introduction
1.1. Heparan Sulfate and Heparin in Physiology and Pharmacology
1.2. Pleiotropic Effects of Heparin and Heparan Sulfate Mimetics in Cancer Therapy
2. Heparan Sulfate and Cancer
2.1. Inflammation
2.2. Proliferation, Angiogenesis, and Metastasis
2.3. Therapeutic Resistance
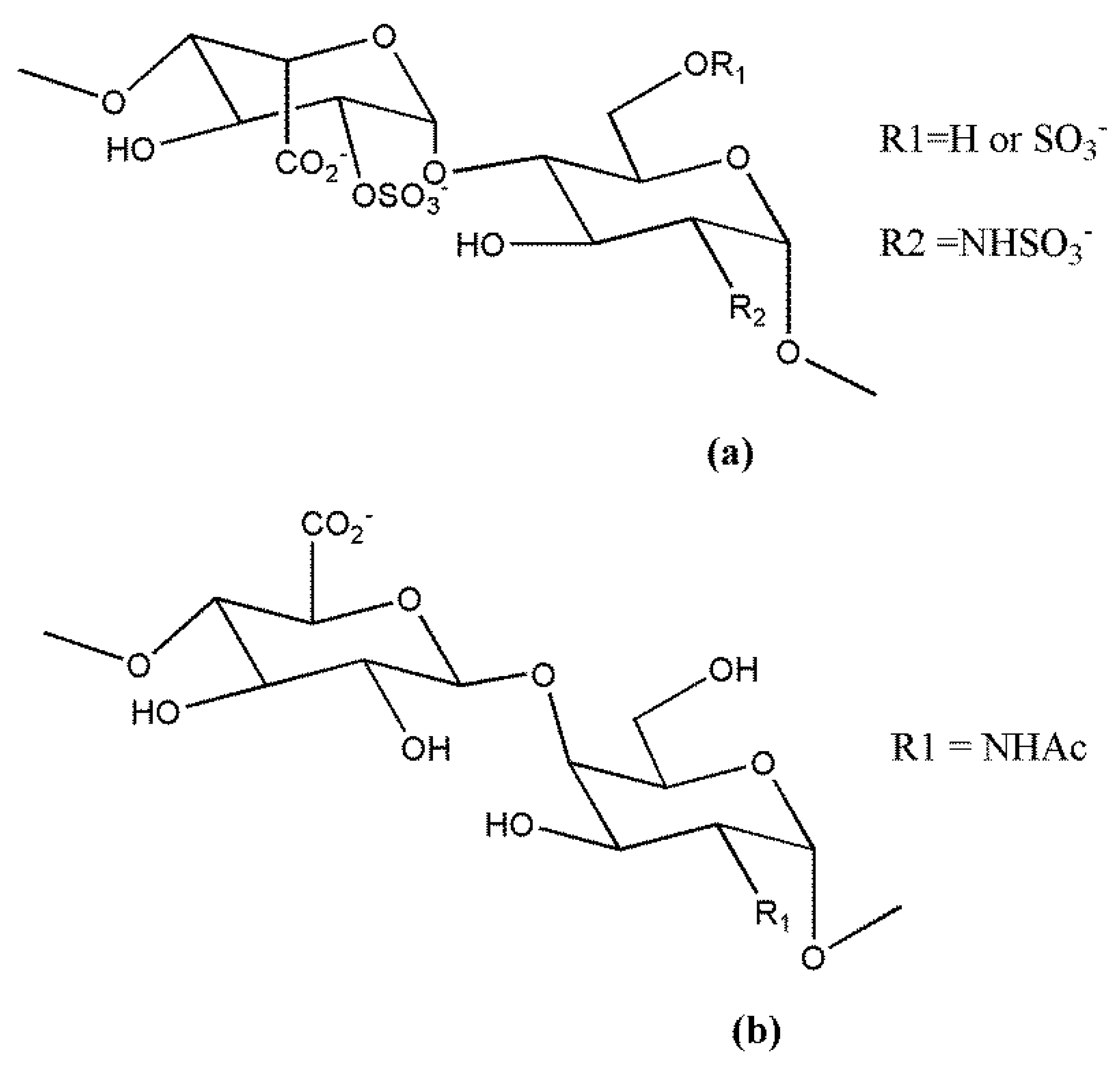
3. Non-Anticoagulant Heparin Derivatives and Oligosaccharidic HS Mimetics
3.1. Non-Anticoagulant Heparin Derivatives
3.2. Oligosaccharidic HS Mimetics
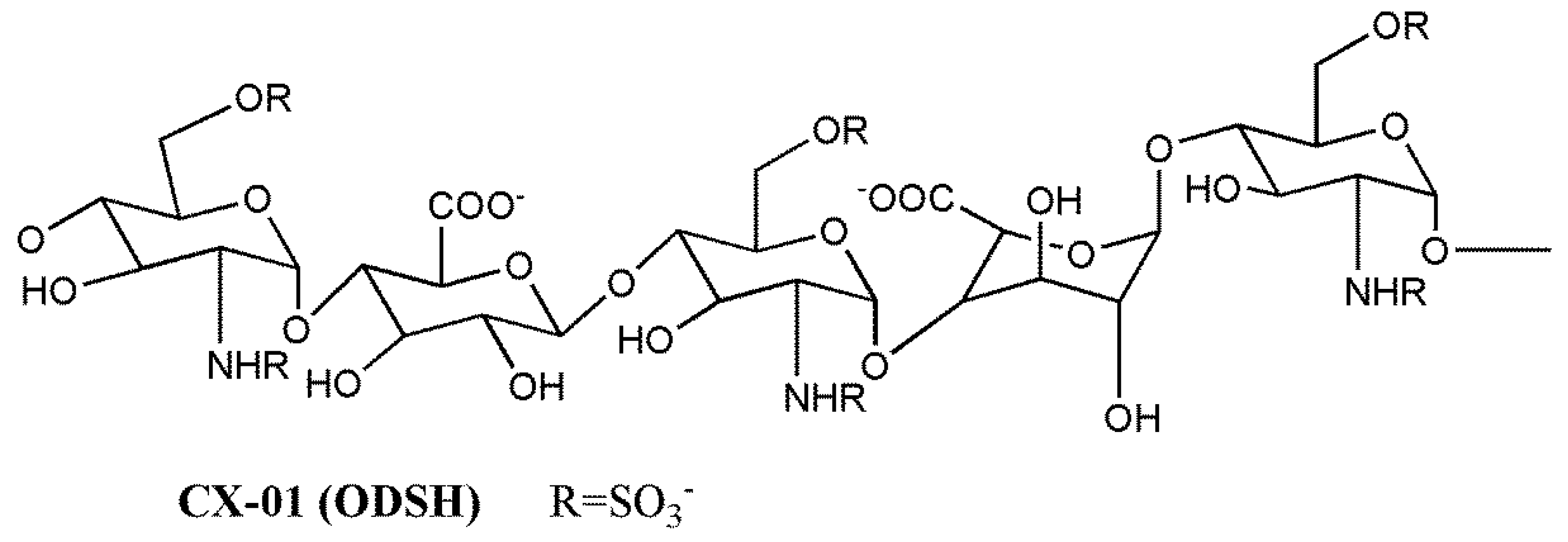
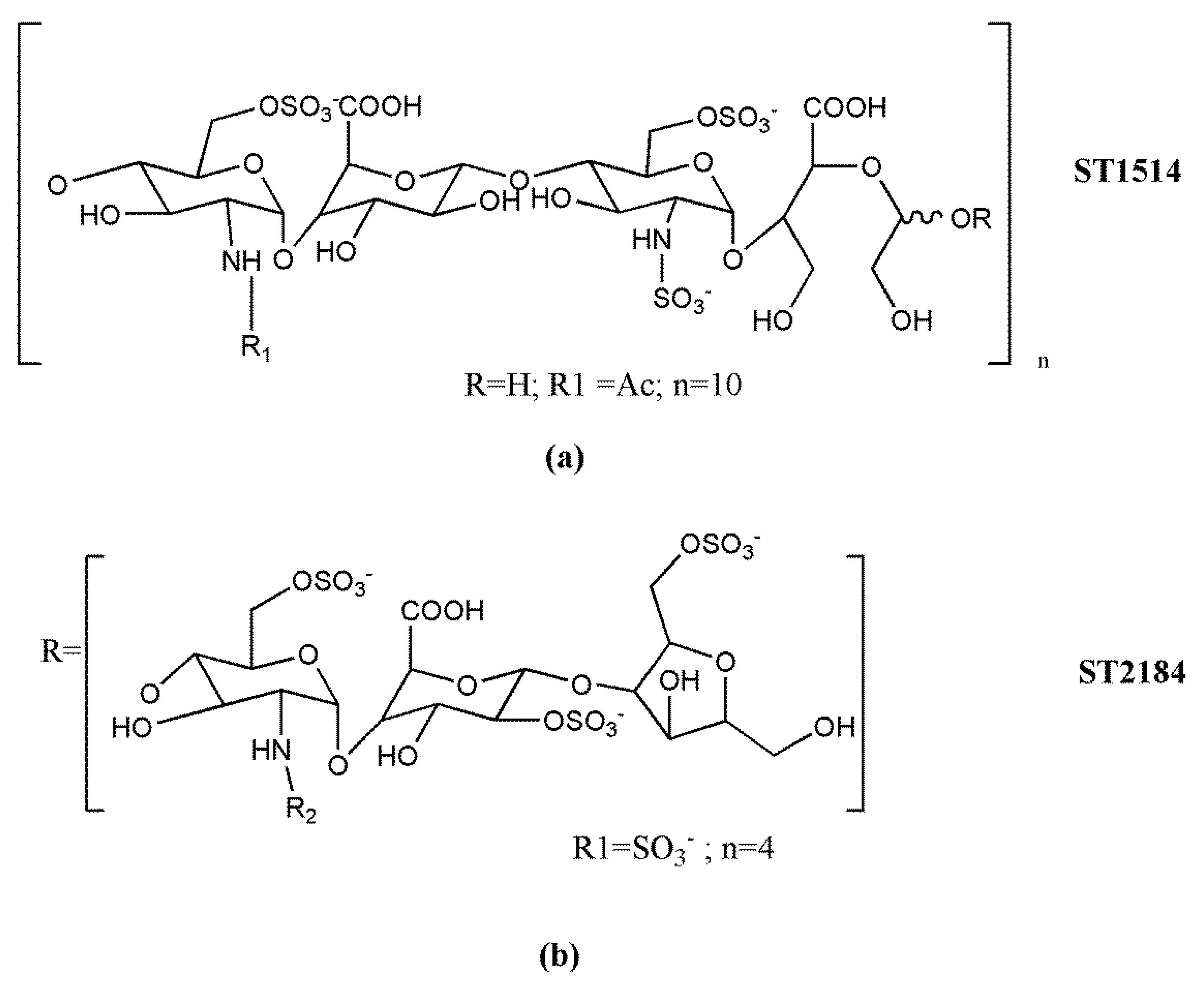
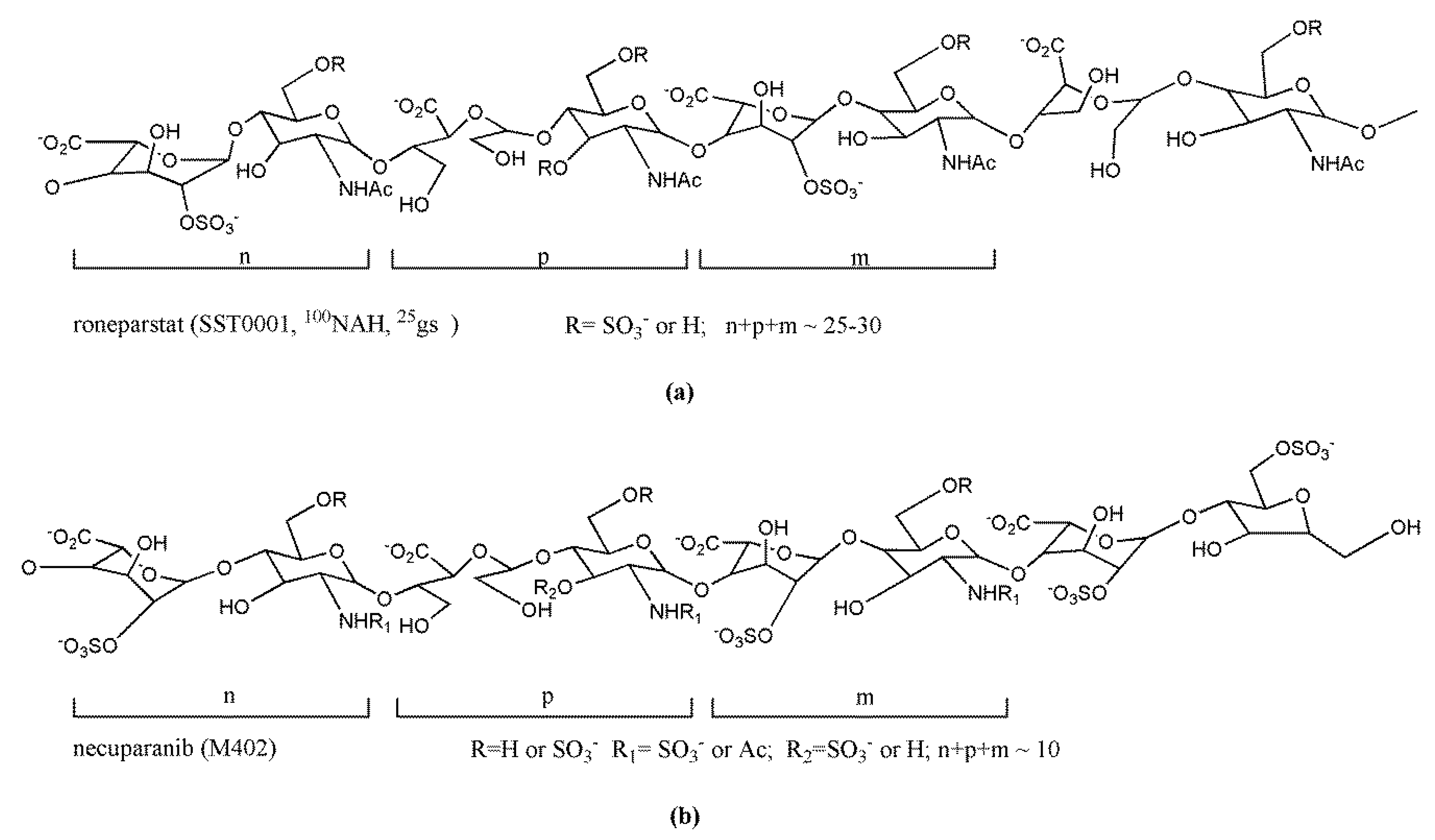
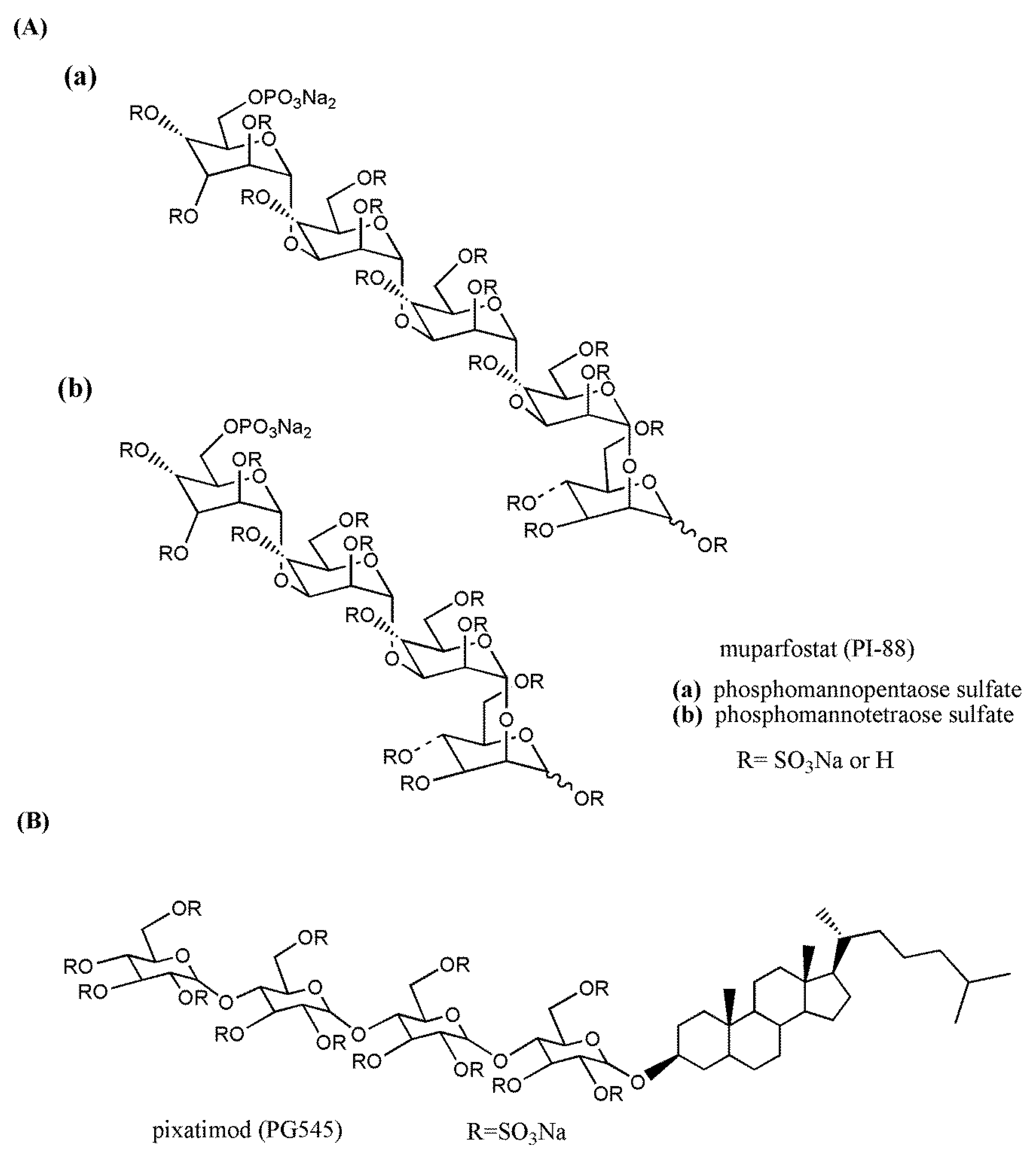
3.3. Clinical Candidate HS Mimetics
4. New Molecules and Perspectives
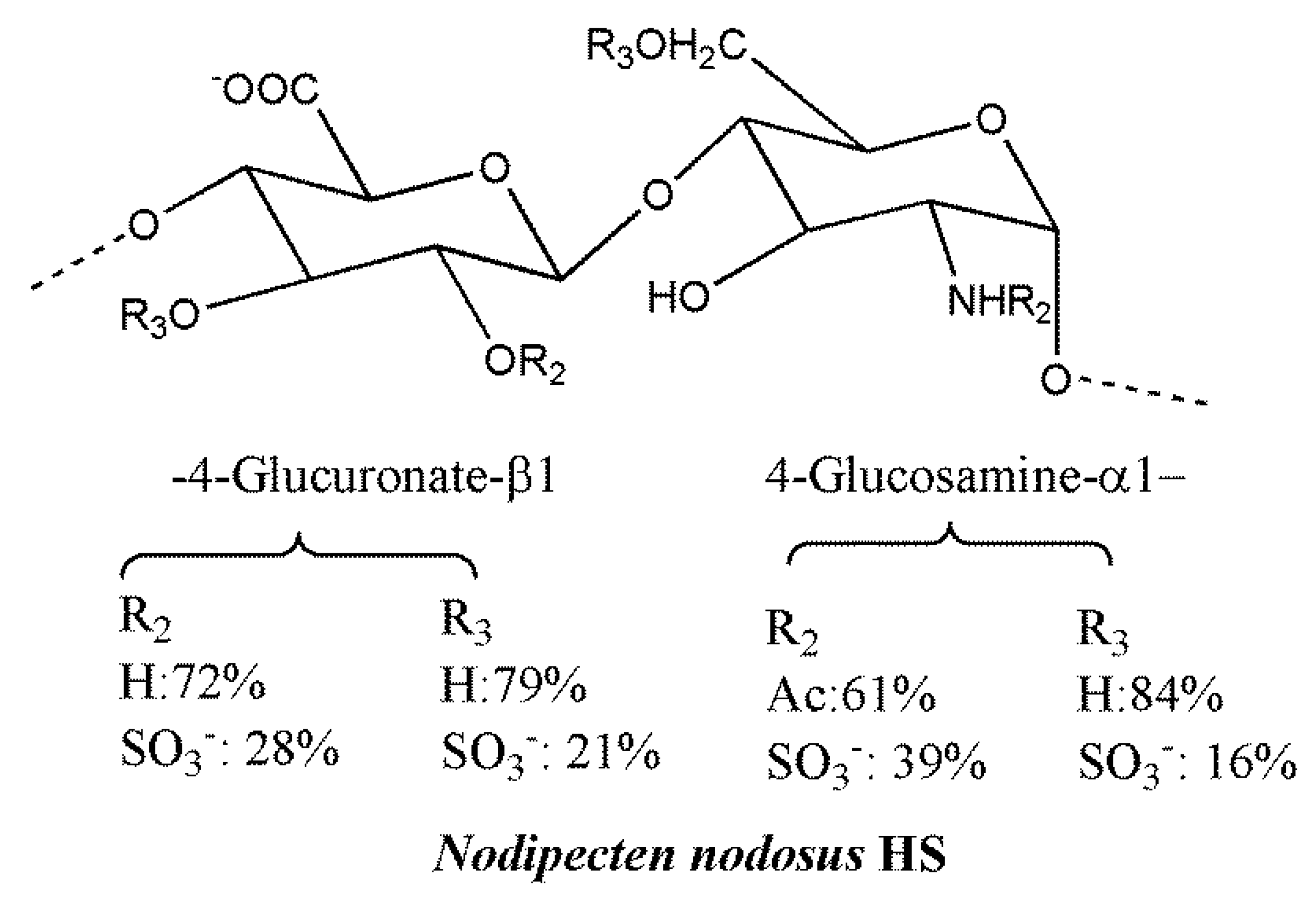
4.1. HS and Heparin-Like GAGs from Marine and Terrestrial Invertebrates
4.2. Bacterial HS and Semisynthetic Heparin-Like GAGs
4.3. Synthetic Oligosaccharidic HS Mimetics
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Rudd, T.R.; Skidmore, M.A.; Guerrini, M.; Hricovini, M.; Powell, A.K.; Siligardi, G.; Yates, E.A. The conformation and structure of GAGs: Recent progress and perspectives. Curr. Opin. Struct. Biol. 2010, 20, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Shriver, Z.; Capila, I.; Venkataraman, G.; Sasisekharan, R. Heparin and heparan sulfate: Analyzing structure and microheterogeneity. In Handbook Experimental Pharmacology; Heparin-A century of Progress, Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207, pp. 159–176. [Google Scholar]
- Gallangher, J.T. Heparan sulfate: A heparin in miniature. In Handbook Experimental Pharmacology; Heparin-A century of Progress, Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207, pp. 347–360. [Google Scholar]
- Li, J.P.; Kusche-Gullberg, M. Heparan sulfate: Biosynthesis, structure, and function. Int. Rev. Cell. Mol. Biol. 2016, 325, 215–273. [Google Scholar] [PubMed]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of heparin and related drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Tzanakakis, G.N.; Karamanos, N.K. Proteoglycans in health and disease: Novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 2010, 277, 3904–3923. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knelson, E.H.; Nee, J.C.; Blobe, G.C. Heparan sulfate signaling in cancer. Trends Biochem. Sci. 2014, 39, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parish, C.R. The role of heparan sulphate in inflammation. Nat. Rev. Immunol. 2006, 6, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Chiodelli, P.; Bugatti, A.; Urbinati, C.; Rusnati, M. Heparin/Heparan sulfate proteoglycans glycomic interactome in angiogenesis: Biological implications and therapeutical use. Molecules 2015, 20, 6342–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couchman, J.R.; Multhaupt, H.; Sanderson, R.D. Recent insights into cell surface heparan sulphate proteoglycans and cancer. F1000 Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Heparan sulfate and heparan sulfate proteoglycans in cancer initiation and progression. Front. Endocrinol. 2018, 9, 483. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.; Pejler, G.; Ringvall, M.; Lunderius, C.; Tomasini-Johansson, B.; Kusche-Gullberg, M.; Eriksson, I.; Ledin, J.; Hellman, L.; Kjellén, L. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature 1999, 400, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Humphries, D.E.; Wong, G.W.; Friend, D.S.; Gurish, M.F.; Qiu, W.T.; Huang, C.; Sharpe, A.H.; Stevens, R.L. Heparin is essential for the storage of specific granule proteases in mast cells. Nature 1999, 400, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Lever, R.; Page, C.P. Mast cell glycosaminoglycans. Glycoconj. J. 2017, 34, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Naggi, A.; Torri, G. Re-visiting the structure of heparin. Carbohydr. Res. 2015, 403, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, J.; Kjellén, L. Heparan sulfate biosynthesis: Regulation and variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, S.; Tamba, M.; Ishimine, H.; Araki, K.; Shiomi, K.; Okada, T.; Ohto, T.; Kunita, S.; Takahashi, S.; Wismans, R.G.; et al. Organ-specific sulfation patterns of heparan sulfate generated by extracellular sulfatases Sulf1 and Sulf2 in mice. J. Biol. Chem. 2012, 287, 9579–9590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, E.; Hogwood, J.; Mulloy, B. The anticoagulant and antithrombotic mechanisms of heparin. In Handbook Experimental Pharmacology; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207, pp. 43–61. [Google Scholar]
- Rudd, T.R.; Preston, M.D.; Yates, E.A. The nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis. Mol. Biosyst. 2017, 13, 852–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, M.C.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Powell, A.K.; Yates, E.A.; Lima, M.A. Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 1998, 20, 156–167. [Google Scholar] [CrossRef]
- Kjellén, L.; Lindahl, U. Specificity of glycosaminoglycan-protein interactions. Curr. Opin. Struct. Biol. 2018, 50, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J. Fell-Muir Lecture: Heparan sulphate and the art of cell regulation: A polymer chain conducts the protein orchestra. Int. J. Exp. Pathol. 2015, 96, 203–231. [Google Scholar] [CrossRef] [PubMed]
- Ori, A.; Wilkinson, M.C.; Fernig, D.G. A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J. Biol. Chem. 2011, 286, 19892–19904. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J. Therapeutic use of heparin beyond anticoagulation. Curr. Drug Discov. Technol. 2009, 6, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Naggi, A. Old and new applications of non-anticoagulant heparin. Int. J. Cardiol. 2016, 212, S14–S21. [Google Scholar] [CrossRef] [Green Version]
- Lima, M.; Rudd, T.; Yates, E.A. New applications of heparin and other glycosaminoglycans. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Smorenburg, S.M.; Hettiarachchi, R.J.; Vink, R.; Büller, H.R. The effects of unfractionated heparin on survival in patients with malignancy—A systematic review. Thromb. Haemost. 1999, 82, 1600–1604. [Google Scholar] [PubMed]
- Kakkar, A.K. Thrombosis and cancer. Hematol. J. 2004, 5, S20–S23. [Google Scholar] [CrossRef] [PubMed]
- Kuderer, N.M.; Ortel, T.L.; Francis, C.W. Impact of venous thromboembolism and anticoagulation on cancer and cancer survival. J. Clin. Oncol. 2009, 27, 4902–4911. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Bohlke, K.; Khorana, A.A.; Kuderer, N.M.; Lee, A.Y.; Arcelus, J.I.; Balaban, E.P.; Clarke, J.M.; Flowers, C.R.; Francis, C.W.; et al. American Society of Clinical Oncology. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of clinical oncology clinical practice guideline update 2014. J. Clin. Oncol. 2015, 33, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Tieken, C.; Versteeg, H.H. Anticoagulants versus cancer. Thromb. Res. 2016, 140, S148–S153. [Google Scholar] [CrossRef]
- Läubli, H.; Varki, A.; Borsig, L. Antimetastatic properties of low molecular weight heparin. J. Clin. Oncol. 2016, 34, 2560–2561. [Google Scholar] [CrossRef] [PubMed]
- García-Escobar, I.; Beato-Zambrano, C.; Muñoz Langa, J.; Brozos Vázquez, E.; Obispo Portero, B.; Gutiérrez-Abad, D.; Muñoz Martín, A.J. Cancer and Thrombosis Working Group of the Spanish Society of Medical Oncology (SEOM). Pleiotropic effects of heparins: Does anticoagulant treatment increase survival in cancer patients? Clin. Transl. Oncol. 2018, 20, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Nadir, Y.; Brenner, B. Cancer and thrombosis-new insights. Rambam. Maimonides Med. J. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Bäckström, G.; Höök, M.; Thunberg, L.; Fransson, L.A.; Linker, A. Structure of the antithrombin-binding site in heparin. Proc. Natl. Acad. Sci. USA 1979, 76, 3198–3202. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.D.; Lam, L. Correlation between structure and function of heparin. Proc. Natl. Acad. Sci. USA 1979, 76, 1218–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hejna, M.; Raderer, M.; Zielinski, C.C. Inhibition of metastases by anticoagulants. J. Natl. Cancer Inst. 1999, 91, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Smorenburg, S.M.; Van Noorden, C.J. The complex effects of heparins on cancer progression and metastasis in experimental studies. Pharmacol. Rev. 2001, 53, 93–105. [Google Scholar] [PubMed]
- Stevenson, J.L.; Choi, S.H.; Varki, A. Differential metastasis inhibition by clinically relevant levels of heparins-correlation with selectin inhibition, not antithrombotic activity. Clin. Cancer Res. 2005, 11, 7003–7011. [Google Scholar] [CrossRef] [PubMed]
- Niers, T.M.; Klerk, C.P.; DiNisio, M.; Van Noorden, C.J.; Büller, H.R.; Reitsma, P.H.; Richel, D.J. Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Crit. Rev. Oncol. Hematol. 2007, 61, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L. Antimetastatic activities of heparins and modified heparins. Experimental evidence. Thromb. Res. 2010, 125, S66–S71. [Google Scholar] [CrossRef] [Green Version]
- Barash, U.; Cohen-Kaplan, V.; Dowek, I.; Sanderson, R.D.; Ilan, N.; Vlodavsky, I. Proteoglycans in health and disease: New concepts for heparanase function in tumor progression and metastasis. FEBS J. 2010, 277, 3890–3903. [Google Scholar] [CrossRef] [PubMed]
- Levy-Adam, F.; Ilan, N.; Vlodavsky, I. Tumorigenic and adhesive properties of heparanase. Semin. Cancer Biol. 2010, 20, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlodavsky, I.; Elkin, M.; Ilan, N. Impact of heparanase and the tumor microenvironment on cancer metastasis and angiogenesis: Basic aspects and clinical applications. Rambam. Maimonides Med. J. 2011, 2, e0019. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Beckhove, P.; Lerner, I.; Pisano, C.; Meirovitz, A.; Ilan, N.; Elkin, M. Significance of heparanase in cancer and inflammation. Cancer Microenviron. 2012, 5, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updat. 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, E.; Khurana, A.; Shridhar, V.; Dredge, K. The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Front. Oncol. 2014, 4, 195. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Vlodavsky, I.; Ilan, N.; Zunino, F. The potential of heparanase as a therapeutic target in cancer. Biochem. Pharmacol. 2014, 89, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassinelli, G.; Zaffaroni, N.; Lanzi, C. The heparanase/heparan sulfate proteoglycan axis: A potential new therapeutic target in sarcomas. Cancer Lett. 2016, 382, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Rivara, S.; Milazzo, F.M.; Giannini, G. Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med. Chem. 2016, 8, 647–680. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, C.; Zaffaroni, N.; Cassinelli, G. Targeting heparan sulfate proteoglycans and their modifying enzymes to enhance anticancer chemotherapy efficacy and overcome drug resistance. Curr. Med. Chem. 2017, 24, 2860–2886. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Ma, S. Recent advances in the discovery of heparanase inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2016, 121, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Kovalszky, I.; Hjerpe, A.; Dobra, K. Nuclear translocation of heparan sulfate proteoglycans and their functional significance. Biochim. Biophys. Acta 2014, 1840, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Vivès, R.R.; Seffouh, A.; Lortat-Jacob, H. Post-Synthetic Regulation of HS Structure: The Yin and Yang of the Sulfs in cancer. Front. Oncol. 2014, 3, 331. [Google Scholar] [CrossRef] [PubMed]
- Fux, L.; Ilan, N.; Sanderson, R.D.; Vlodavsky, I. Heparanase: Busy at the cell surface. Trends Biochem. Sci. 2009, 34, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Mahtouk, K.; Hose, D.; Raynaud, P.; Hundemer, M.; Jourdan, M.; Jourdan, E.; Pantesco, V.; Baudard, M.; De Vos, J.; Larroque, M.; et al. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood 2007, 109, 4914–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, V.C.; Purushothaman, A.; Stewart, M.D.; Thompson, C.A.; Vlodavsky, I.; Au, J.L.; Sanderson, R.D. The heparanase/syndecan-1 axis in cancer: Mechanisms and therapies. FEBS J. 2013, 280, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Ilan, N.; Shteingauz, A.; Vlodavsky, I. Function from within: Autophagy induction by HPSE/heparanase—New possibilities for intervention. Autophagy 2015, 11, 2387–2389. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Hurst, D.R.; Pisano, C.; Mizumoto, S.; Sugahara, K.; Sanderson, R. Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. J. Biol. Chem. 2011, 286, 30377–30383. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Q.; Sutcliffe, E.L.; Bunting, K.L.; Li, J.; Goodall, K.J.; Poon, I.K.A.; Hulett, M.D.; Freeman, C.; Zafar, A.; McInnes, R.L.; et al. The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription 2012, 3, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gorzelanny, C.; Bauer, A.T.; Halter, N.; Komljenovic, D.; Bäuerle, T.; Borsig, L.; Roblek, M.; Schneider, S.W. Nuclear heparanase-1 activity suppresses melanoma progression via its DNA-binding affinity. Oncogene 2015, 34, 5832–5842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, S.D.; Lemjabbar-Alaoui, H. Sulf-2: An extracellular modulator of cell signaling and a cancer target candidate. Expert. Opin. Ther. Targets 2010, 14, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, R.; Meirovitz, A.; Hirshoren, N.; Bulvik, R.; Binder, A.; Rubinstein, A.M.; Elkin, M. Versatile role of heparanase in inflammation. Matrix. Biol. 2013, 32, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Katakam, S.K.; Urbanowitz, A.K.; Gotte, M. Heparan sulphate as a regulator of leukocyte recruitment in inflammation. Curr. Protein Pept. Sci. 2015, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, B.L.; Lord, M.S.; Melrose, J.; Whitelock, J.M. The role of heparan sulfate in inflammation, and the development of biomimetics as anti-inflammatory strategies. J. Histochem. Cytochem. 2018, 66, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fuster, M.; Sriramarao, P.; Esko, J.D. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 2005, 6, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Lortat-Jacob, H.; Grosdidier, A.; Imberty, A. Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. USA. 2002, 99, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Massena, S.; Christoffersson, G.; Hjertström, E.; Zcharia, E.; Vlodavsky, I.; Ausmees, N.; Rolny, C.; Li, J.P.; Phillipson, M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 2010, 116, 1924–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoler-Barak, L.; Barzilai, S.; Zauberman, A.; Alon, R. Transendothelial migration of effector T cells across inflamed endothelial barriers does not require heparan sulfate proteoglycans. Int. Immunol. 2014, 26, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meirovitz, A.; Goldberg, R.; Binder, A.; Rubinstein, A.M.; Hermano, E.; Elkin, M. Heparanase in inflammation and inflammation-associated cancer. FEBS J. 2013, 280, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Elkin, M.; Rapraeger, A.C.; Ilan, N.; Vlodavsky, I. Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. FEBS J. 2017, 284, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Blich, M.; Golan, A.; Arvatz, G.; Sebbag, A.; Shafat, I.; Sabo, E.; Cohen-Kaplan, V.; Petcherski, S.; Avniel-Polak, S.; Eitan, A.; et al. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Goodall, K.J.; Poon, I.K.; Phipps, S.; Hulett, M.D. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS ONE 2014, 9, e109596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunn, G.J.; Bungum, M.K.; Johnson, G.B.; Platt, J.L. Conditional signaling by Toll-like receptor 4. FASEB J. 2005, 19, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Lerner, I.; Hermano, E.; Zcharia, E.; Rodkin, D.; Bulvik, R.; Doviner, V.; Rubinstein, A.M.; Ishai-Michaeli, R.; Atzmon, R.; Sherman, Y.; et al. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J. Clin. Investig. 2011, 121, 1709–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermano, E.; Meirovitz, A.; Meir, K.; Nussbaum, G.; Appelbaum, L.; Peretz, T.; Elkin, M. Macrophage polarization in pancreatic carcinoma: Role of heparanase enzyme. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Rohloff, J.; Zinke, J.; Schoppmeyer, K.; Tannapfel, A.; Witzigmann, H.; Mössner, J.; Wittekind, C.; Caca, K. Heparanase expression is a prognostic indicator for postoperative survival in pancreatic adenocarcinoma. Br. J. Cancer 2002, 86, 1270–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, R.M.; Rao, G.; Plate, J.; Harris, J.E.; Brunn, G.J.; Platt, J.L.; Gattuso, P.; Prinz, R.A.; Xu, X. Elevated serum heparanase-1 levels in patients with pancreatic carcinoma are associated with poor survival. Cancer 2006, 106, 532–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar Galvis, M.L.; Jia, J.; Zhang, X.; Jastrebova, N.; Spillmann, D.; Gottfridsson, E.; van Kuppevelt, T.H.; Zcharia, E.; Vlodavsky, I.; Lindahl, U.; et al. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat. Chem. Biol. 2007, 3, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Naroditsky, I.; Zetser, A.; Ilan, N.; Vlodavsky, I.; Doweck, I. Heparanase induces VEGF C and facilitates tumor lymphangiogenesis. Int. J. Cancer 2008, 123, 2566–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Yang, Y.; Ren, Y.; Nan, L.; Sanderson, R.D. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J. Biol. Chem. 2011, 286, 6490–6499. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Doweck, I.; Naroditsky, I.; Vlodavsky, I.; Ilan, N. Heparanase augments epidermal growth factor receptor phosphorylation: Correlation with head and neck tumor progression. Cancer Res. 2008, 68, 10077–10085. [Google Scholar] [CrossRef] [PubMed]
- Piperigkou, Z.; Mohr, B.; Karamanos, N.; Götte, M. Shed proteoglycans in tumor stroma. Cell. Tissue Res. 2016, 365, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Trapp-Stamborski, V.; Purushothaman, A.; Jin, H.; Wang, H.; Sanderson, R.D.; Rapraeger, A.C. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: Prevention by novel synstatins. Oncogenesis 2016, 5, e202. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Macleod, V.; Miao, H.Q.; Theus, A.; Zhan, F.; Shaughnessy, J.D., Jr.; Sawyer, J.; Li, J.P.; Zcharia, E.; Vlodavsky, I.; et al. Heparanase enhances syndecan-1 shedding: A novel mechanism for stimulation of tumor growth and metastasis. J. Biol. Chem. 2007, 282, 13326–13333. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Chen, L.; Yang, Y.; Sanderson, R.D. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J. Biol. Chem. 2008, 283, 32628–32636. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.A.; Purushothaman, A.; Ramani, V.C.; Vlodavsky, I.; Sanderson, R.D. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J. Biol. Chem. 2013, 288, 10093–10099. [Google Scholar] [CrossRef] [PubMed]
- Roucourt, B.; Meeussen, S.; Bao, J.; Zimmermann, P.; David, G. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell. Res. 2015, 25, 412–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell. Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syn, N.; Wang, L.; Sethi, G.; Theiry, J.P.; Goh, B.C. Exosome-mediated metastasis: From Epithelial-Mesenchymal Transition to escape from immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ramani, V.C.; Sanderson, R.D. Shed syndecan-1 translocates to the nucleus of cells delivering growth factors and inhibiting histone acetylation: A novel mechanism of tumor-host cross-talk. J. Biol. Chem. 2015, 290, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Zong, F.; Fthenou, E.; Wolmer, N.; Hollosi, P.; Kovalszky, I.; Szilak, L.; Mogler, C.; Nilsonne, G.; Tzanakakis, G.; Dobra, K. Syndecan-1 and FGF-2, but not FGF receptor-1, share a common transport route and co-localize with heparanase in the nuclei of mesenchymal tumor cells. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.J.; Martin, G.A.; Barnhart, R.L.; Mano, M.; Cardin, A.D.; Jackson, R.L. Trans-repressor activity of nuclear glycosaminoglycans on Fos and Jun/AP-1 oncoprotein-mediated transcription. J. Cell. Biol. 1992, 116, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalszky, I.; Dudás, J.; Oláh-Nagy, J.; Pogány, G.; Töváry, J.; Timár, J.; Kopper, L.; Jeney, A.; Iozzo, R.V. Inhibition of DNA topoisomerase I activity by heparan sulfate and modulation by basic fibroblast growth factor. Mol. Cell. Biochem. 1998, 183, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Szatmári, T.; Mundt, F.; Kumar-Singh, A.; Möbus, L.; Ötvös, R.; Hjerpe, A.; Dobra, K. Molecular targets and signaling pathways regulated by nuclear translocation of syndecan-1. BMC Cell. Biol. 2017, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sanderson, R.D. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS ONE 2009, 4, e4947. [Google Scholar] [CrossRef] [PubMed]
- Buczek-Thomas, J.A.; Hsia, E.; Rich, C.B.; Foster, J.A.; Nugent, M.A. Inhibition of histone acetyltransferase by glycosaminoglycans. J. Cell. Biochem. 2008, 105, 108–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barash, U.; Zohar, Y.; Wildbaum, G.; Beider, K.; Nagler, A.; Karin, N.; Ilan, N.; Vlodavsky, I. Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia 2014, 28, 2178–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobuhisa, T.; Naomoto, Y.; Okawa, T.; Takaoka, M.; Gunduz, M.; Motoki, T.; Nagatsuka, H.; Tsujigiwa, H.; Shirakawa, Y.; Yamatsuji, T.; et al. Translocation of heparanase into nucleus results in cell differentiation. Cancer Sci. 2007, 98, 535–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doweck, I.; Kaplan-Cohen, V.; Naroditsky, I.; Sabo, E.; Ilan, N.; Vlodavsky, I. Heparanase localization and expression by head and neck cancer: Correlation with tumor progression and patient survival. Neoplasia 2006, 8, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Wichert, A.; Stege, A.; Midorikawa, Y.; Holm, P.S.; Lage, H. Glypican-3 is involved in cellular protection against mitoxantrone in gastric carcinoma cells. Oncogene 2004, 23, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Takahashi, T.; Serada, S.; Fujimoto, M.; Ohkawara, T.; Nakatsuka, R.; Harada, E.; Nishigaki, T.; Takahashi, Y.; Nojima, S.; et al. Overexpression of glypican-1 implicates poor prognosis and their chemoresistance in oesophageal squamous cell carcinoma. Br. J. Cancer 2016, 115, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Barbareschi, M.; Maisonneuve, P.; Aldovini, D.; Cangi, M.G.; Pecciarini, L.; Angelo Mauri, F.; Veronese, S.; Caffo, O.; Lucenti, A.; Palma, P.D.; et al. High syndecan-1 expression in breast carcinoma is related to an aggressive phenotype and to poorer prognosis. Cancer 2003, 98, 474–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.L.; Grizzle, W.E.; Zhang, K.; Hameed, O.; Siegal, G.P.; Wei, S. Syndecan-1 overexpression is associated with nonluminal subtypes and poor prognosis in advanced breast cancer. Am. J. Clin. Pathol. 2013, 140, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Götte, M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 2017, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Zhan, F.; He, J.; Barbieri, P.; Noseda, A.; Tricot, G.; Sanderson, R.D. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget 2016, 7, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Kersting, C.; Ruggiero, M.; Tio, J.; Tulusan, A.H.; Kiesel, L.; Wülfing, P. Predictive value of syndecan-1 expression for the response to neoadjuvant chemotherapy of primary breast cancer. Anticancer Res. 2006, 26, 621–627. [Google Scholar] [PubMed]
- Suarez, E.R.; Paredes-Gamero, E.J.; Del Giglio, A.; Tersariol, I.L.; Nader, H.B.; Pinhal, M.A. Heparan sulfate mediates trastuzumab effect in breast cancer cells. BMC Cancer 2013, 13, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zuo, D.; Chen, Y.; Li, W.; Liu, R.; He, Y.; Ren, L.; Zhou, L.; Deng, T.; Wang, X.; et al. Shed Syndecan-1 is involved in chemotherapy resistance via the EGFR pathway in colorectal cancer. Br. J. Cancer 2014, 11, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ngo, J.A.; Wetzel, M.D.; Marchetti, D. Heparanase mediates a novel mechanism in lapatinib-resistant brain metastatic breast cancer. Neoplasia 2015, 17, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Meirovitz, A.; Hermano, E.; Lerner, I.; Zcharia, E.; Pisano, C.; Peretz, T.; Elkin, M. Role of heparanase in radiation-enhanced invasiveness of pancreatic carcinoma. Cancer Res. 2011, 71, 2772–2780. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meng, X.; Hu, J.; Zhang, Y.; Dang, Y.; Wei, L.; Shi, M. Heparanase promotes radiation resistance of cervical cancer by upregulating hypoxia inducible factor 1. Am. J. Cancer Res. 2017, 7, 234–244. [Google Scholar] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Shteingauz, A.; Boyango, I.; Naroditsky, I.; Hammond, E.; Gruber, M.; Doweck, I.; Ilan, N.; Vlodavsky, I. Heparanase enhances tumor growth and chemoresistance by promoting autophagy. Cancer Res. 2015, 75, 3946–3957. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Sanderson, R.D. Chemotherapy stimulates syndecan-1 shedding: A potentially negative effect of treatment that may promote tumor relapse. Matrix Biol. 2014, 3, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Vlodavsky, I.; Ng, M.; Zhang, Y.; Barbieri, P.; Noseda, A.; Sanderson, R.D. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016, 55, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Alishekevitz, D.; Gingis-Velitski, S.; Kaidar-Person, O.; Gutter-Kapon, L.; Scherer, S.D.; Raviv, Z.; Merquiol, E.; Ben-Nun, Y.; Miller, V.; Rachman-Tzemah, C.; et al. Macrophage-induced lymphangiogenesis and metastasis following Paclitaxel chemotherapy is regulated by VEGFR3. Cell. Rep. 2016, 17, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Guglieri, S.; Casu, B.; Torri, G.; Mourier, P.; Boudier, C.; Viskov, C. Antithrombin-binding octasaccharides and role of extensions of the active pentasaccharide sequence in the specificity and strength of interaction. Evidence for very high affinity induced by an unusual glucuronic acid residue. J. Biol. Chem. 2008, 283, 26662–26675. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Vlodavsky, I.; Sanderson, R.D. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol. Haemost. Thromb. 2007, 36, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. Heparin/heparan sulphate-based drugs. Drug Discov. Today 2010, 15, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Conrad, H.E.; Guo, Y. Structural analysis of periodate-oxidized heparin. Heparin and related polysaccharides. In Advances in Experimental Medicine and Biology; Lane, D.A., Bjork, I., Lindahl, U., Eds.; Springer: Boston, MA, USA, 1991; Volume 313, pp. 31–36. [Google Scholar]
- Bar-Ner, M.; Eldor, A.; Wasserman, L.; Matzner, Y.; Cohen, I.R.; Fuks, Z.; Vlodavsky, I. Inhibition of heparanase-mediated degradation of extracellular matrix heparan sulfate by non-anticoagulant heparin species. Blood 1987, 70, 551–557. [Google Scholar] [PubMed]
- Irimura, T.; Nakajima, M.; Nicolson, G.L. Chemically modified heparins as inhibitors of heparan sulfate specific endo-beta-glucuronidase (heparanase) of metastatic melanoma cells. Biochemistry 1986, 25, 5322–5328. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Mohsen, M.; Lider, O.; Svahn, C.M.; Ekre, H.P.; Vigoda, M.; Ishai-Michaeli, R.; Peretz, T. Inhibition of tumor metastasis by heparanase inhibiting species of heparin. Invasion Metastasis 1994, 14, 290–302. [Google Scholar] [PubMed]
- Bitan, M.; Mohsen, M.; Levi, E.; Wygoda, M.R.; Miao, H.Q.; Lider, O.; Svahn, C.M.; Ekre, H.P.; Ishai-Michaeli, R.; Bar-Shavit, R.; et al. Structural requirements for inhibition of melanoma lung colonization by heparanase inhibiting species of heparin. Isr. J. Med. Sci. 1995, 31, 106–118. [Google Scholar] [PubMed]
- Sciumbata, T.; Caretto, P.; Pirovano, P.; Pozzi, P.; Cremonesi, P.; Galimberti, G.; Leoni, F.; Marcucci, F. Treatment with modified heparins inhibits experimental metastasis formation and leads, in some animals, to long-term survival. Invasion Metastasis 1996, 16, 132–143. [Google Scholar] [PubMed]
- Lapierre, F.; Holme, K.; Lam, L.; Tressler, R.J.; Storm, N.; Wee, J.; Stack, R.J.; Castellot, J.; Tyrrell, D.J. Chemical modifications of heparin that diminish its anticoagulant but preserve its heparanase-inhibitory, angiostatic, anti-tumor and anti-metastatic properties. Glycobiology 1996, 6, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, N.V.; Argyle, B.; Xu, X.; Reynolds, P.R.; Walenga, J.M.; Prechel, M.; Prestwich, G.D.; MacArthur, R.B.; Walters, B.B.; Hoidal, J.R.; et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. Am. J. Physiol. Cell. Physiol. 2010, 299, C97–C110. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Gao, Y.; Tian, M.; Li, N.; Hao, S.; Zeng, X. Selectively desulfated heparin inhibits P-selectin-mediated adhesion of human melanoma cells. Cancer Lett. 2005, 229, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, J.R.; Varki, A.; Esko, J.D. Heparin’s anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J. Clin. Investig. 2002, 110, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Kummarapurugu, A.B.; Afosah, D.K.; Sankaranarayanan, N.V.; Boothello, R.S.; Desai, U.R.; Kennedy, T.; Voynow, J.A. 2-O, 3-O Desulfated heparin blocks high mobility group box 1 release by inhibition of p300 acetyltransferase activity. Am. J. Respir. Cell. Mol. Biol. 2017, 56, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Lai, H.; Zouaoui, R.; Duffner, J.; Zhou, H.P.; Jayaraman, L.; Zhao, G.; Ganguly, T.; Kishimoto, T.K.; Venkataraman, G. Bioactivity screening of partially desulfated low-molecular-weight heparins: A structure/activity relationship study. Glycobiology 2011, 21, 1194–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naggi, A.; Torri, G.; Casu, B.; Pangrazzi, J.; Abbadini, M.; Zametta, M.; Donati, M.B.; Lansen, J.; Maffrand, J.P. “Supersulfated” heparin fragments, a new type of low-molecular weight heparin. Physico-chemical and pharmacological properties. Biochem. Pharmacol. 1987, 36, 1895–1900. [Google Scholar] [CrossRef]
- Sissi, C.; Naggi, A.; Torri, G.; Palumbo, M. Effects of sulfation on antithrombin-thrombin/factor Xa interactions in semisynthetic low molecular weight heparins. Semin. Thromb. Hemost. 2001, 27, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Dal Bo, L.; Favini, E.; Cominetti, D.; Pozzi, S.; Tortoreto, M.; De Cesare, M.; Lecis, D.; Scanziani, E.; Minoli, L.; et al. Supersulfated low-molecular weight heparin synergizes with IGF1R/IR inhibitor to suppress synovial sarcoma growth and metastases. Cancer Lett. 2018, 415, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Sissi, C.; Lucatello, L.; Naggi, A.; Torri, G.; Palumbo, M. Interactions of low-molecular-weight semi-synthetic sulfated heparins with human leukocyte elastase and human Cathepsin G. Biochem. Pharmacol. 2006, 71, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.; Asperti, M.; Ruzzenenti, P.; Mandelli, L.; Campostrini, N.; Martini, G.; Di Somma, M.; Maccarinelli, F.; Girelli, D.; Naggi, A.; et al. Oversulfated heparins with low anticoagulant activity are strong and fast inhibitors of hepcidin expression in vitro and in vivo. Biochem. Pharmacol. 2014, 92, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Park, K.; Kim, S.K.; Park, R.W.; Kwon, I.C.; Kim, S.Y.; Byun, Y. Antimetastatic effect of an orally active heparin derivative on experimentally induced metastasis. Clin. Cancer Res. 2008, 14, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, S.W.; Kim, S.K.; Lee, M.; Chang, H.W.; Moon, H.T.; Byun, Y.; Kim, S.Y. Antiangiogenic activity of orally absorbable heparin derivative in different types of cancer cells. Pharm. Res. 2009, 26, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Jeon, O.C.; Kim, S.K.; Al-Hilal, T.A.; Jin, S.J.; Moon, H.T.; Yang, V.C.; Kim, S.Y.; Byun, Y. High antiangiogenic and low anticoagulant efficacy of orally active low molecular weight heparin derivatives. J. Control. Release 2010, 148, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Ishihara, M.; Ishikawa, K.; Ozeki, Y.; Deguchi, H.; Sato, M.; Hashimoto, H.; Saito, Y.; Yura, H.; Kurita, A.; et al. Periodate-treated, non-anticoagulant heparin-carrying polystyrene (NAC-HCPS) affects angiogenesis and inhibits subcutaneous induced tumour growth and metastasis to the lung. Br. J. Cancer 2002, 86, 1803–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshitomi, Y.; Nakanishi, H.; Kusano, Y.; Munesue, S.; Oguri, K.; Tatematsu, M.; Yamashina, I.; Okayama, M. Inhibition of experimental lung metastases of Lewis lung carcinoma cells by chemically modified heparin with reduced anticoagulant activity. Cancer Lett. 2004, 207, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Guerrini, M.; Naggi, A.; Perez, M.; Torri, G.; Ribatti, D.; Carminati, P.; Giannini, G.; Penco, S.; Pisano, C.; et al. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry 2002, 41, 10519–10528. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Guerrini, M.; Guglieri, S.; Naggi, A.; Perez, M.; Torri, G.; Cassinelli, G.; Ribatti, D.; Carminati, P.; Giannini, G.; et al. Undersulfated and glycol-split heparins endowed with antiangiogenic activity. J. Med. Chem. 2004, 47, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Aulicino, C.; Vesci, L.; Casu, B.; Naggi, A.; Torri, G.; Ribatti, D.; Belleri, M.; Rusnati, M.; Presta, M. Undersulfated, low-molecular-weight glycol-split heparin as an antiangiogenic VEGF antagonist. Glycobiology 2005, 15, 1C–6C. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Foderà, R.; Marcellini, M.; Giordano, V.; Cervoni, M.L.; Chiarucci, I.; Penco, S.; Riccioni, T.; Vesci, L.; Carminati, P. Antiangiogenic and antitumoral activity of novel heparin derivatives devoid of anticoagulant effects. In Symposium on Molecular Targets and Cancer Therapeutics, Proceedings of the 14th EORTC NCI-AACR, Frankfurt, Germany, 19–22 November 2002; Abstract A229; European Journal of Cancer: Oxford, UK, 2002. [Google Scholar]
- Zcharia, E.; Zilka, R.; Yaar, A.; Yacoby-Zeevi, O.; Zetser, A.; Metzger, S.; Sarid, R.; Naggi, A.; Casu, B.; Ilan, N.; et al. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. FASEB J. 2005, 19, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Edovitsky, E.; Lerner, I.; Zcharia, E.; Peretz, T.; Vlodavsky, I.; Elkin, M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood 2006, 107, 3609–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [PubMed]
- Naggi, A. Glycol-splitting as a device for modulating inhibition of growth factors and heparanase inhibition by heparin and heparin derivative. In Chemistry and Biology of Heparin and Heparan Sulfate; Garg, H.G., Linhardt, R.J., Hales, C.A., Eds.; Elsevier: Amsterdam, The Netherland, 2005; pp. 461–481. [Google Scholar]
- Alekseeva, A.; Mazzini, G.; Giannini, G.; Naggi, A. Structural features of heparanase-inhibiting non-anticoagulant heparin derivative Roneparstat. Carbohydr. Polym. 2017, 156, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Pala, D.; Rivara, S.; Mor, M.; Milazzo, F.M.; Roscilli, G.; Pavoni, E.; Giannini, G. Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology 2016, 26, 640–654. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, N.; Naggi, A.; Torri, G.; Ishai-Michaeli, R.; Casu, B.; Vlodavsky, I.; Borsig, L. P-selectin- and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. FASEB J. 2007, 21, 3562–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, J.P.; Ramani, V.C.; Ren, Y.; Naggi, A.; Torri, G.; Casu, B.; Penco, S.; Pisano, C.; Carminati, P.; Tortoreto, M.; et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin. Cancer Res. 2011, 17, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Shafat, I.; Ben-Arush, M.W.; Issakov, J.; Meller, I.; Naroditsky, I.; Tortoreto, M.; Cassinelli, G.; Lanzi, C.; Pisano, C.; Ilan, N.; et al. Pre-clinical and clinical significance of heparanase in Ewing’s sarcoma. J. Cell. Mol. Med. 2011, 15, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Lanzi, C.; Tortoreto, M.; Cominetti, D.; Petrangolini, G.; Favini, E.; Zaffaroni, N.; Pisano, C.; Penco, S.; Vlodavsky, I.; et al. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem. Pharmacol. 2013, 85, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Favini, E.; Dal Bo, L.; Tortoreto, M.; De Maglie, M.; Dagrada, G.; Pilotti, S.; Zunino, F.; Zaffaroni, N.; Lanzi, C. Antitumor efficacy of the heparan sulfate mimic roneparstat (SST0001) against sarcoma models involves multi-target inhibition of receptor tyrosine kinases. Oncotarget 2016, 7, 47848–47863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossini, A.; Zunino, F.; Ruggiero, G.; De Cesare, M.; Cominetti, D.; Tortoreto, M.; Lanzi, C.; Cassinelli, G.; Zappasodi, R.; Tripodo, C.; et al. Microenvironment modulation and enhancement of antilymphoma therapy by the heparanase inhibitor roneparstat. Hematol. Oncol. 2018, 36, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, P.; Paoletti, D.; Giannini, G.; Sanderson, R.D.; Noseda, A. Roneparstat and heparanase inhibition: A new toll for cancer treatment. J. Pharmacol. Clin. Toxicol. 2017, 5, 1071. [Google Scholar]
- Zhou, H.; Roy, S.; Cochran, E.; Zouaoui, R.; Chu, C.L.; Duffner, J.; Zhao, G.; Smith, S.; Galcheva-Gargova, Z.; Karlgren, J.; et al. M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS ONE 2011, 6, e21106. [Google Scholar] [CrossRef] [PubMed]
- Kragh, M.; Binderup, L.; Vig Hjarnaa, P.J.; Bramm, E.; Johansen, K.B.; Frimundt Petersen, C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol, Rep. 2005, 14, 99–104. [Google Scholar]
- Mousa, S.A.; Linhardt, R.; Francis, J.L.; Amirkhosravi, A. Anti-metastatic effect of a non-anticoagulant low-molecular-weight heparin versus the standard low-molecular-weight heparin, enoxaparin. Thromb. Haemost. 2006, 96, 816–821. [Google Scholar] [PubMed]
- Sudha, T.; Phillips, P.; Kanaan, C.; Linhardt, R.J.; Borsig, L.; Mousa, S.A. Inhibitory effect of non-anticoagulant heparin (S-NACH) on pancreatic cancer cell adhesion and metastasis in human umbilical cord vessel segment and in mouse model. Clin. Exp. Metastasis 2012, 29, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Sudha, T.; Yalcin, M.; Lin, H.Y.; Elmetwally, A.M.; Nazeer, T.; Arumugam, T.; Phillips, P.; Mousa, S.A. Suppression of pancreatic cancer by sulfated non-anticoagulant low molecular weight heparin. Cancer Lett. 2014, 350, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achour, O.; Poupard, N.; Bridiau, N.; Bordenave Juchereau, S.; Sannier, F.; Piot, J.M.; Fruitier Arnaudin, I.; Maugard, T. Anti-heparanase activity of ultra-low-molecular-weight heparin produced by physicochemical depolymerization. Carbohydr. Polym. 2016, 135, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Parish, C.R.; Freeman, C.; Brown, K.J.; Francis, D.J.; Cowden, W.B. Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res. 1999, 59, 3433–3441. [Google Scholar] [PubMed]
- Khachigian, L.M.; Parish, C.R. Phosphomannopentaose sulfate (PI-88): Heparan sulfate mimetic with clinical potential in multiple vascular pathologies. Cardiovasc. Drug Rev. 2004, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ferro, V.; Dredge, K.; Liu, L.; Hammond, E.; Bytheway, I.; Li, C.; Johnstone, K.; Karoli, T.; Davis, K.; Copeman, E.; et al. PI-88 and novel heparan sulfate mimetics inhibit angiogenesis. Semin. Thromb. Hemost. 2007, 33, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Cochran, S.; Li, C.; Fairweather, J.K.; Kett, W.C.; Coombe, D.R.; Ferro, V. Probing the interactions of phosphosulfomannans with angiogenic growth factors by surface plasmon resonance. J. Med. Chem. 2003, 46, 4601–4608. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.; Iqbal, O.; Hoppensteadt, D.A.; Piccolo, P.; Ahmad, S.; Schultz, C.L.; Linhardt, R.J.; Fareed, J. Anticoagulant and antiprotease profiles of a novel natural heparinomimetic mannopentaose phosphate sulfate (PI-88). Clin. Appl. Thromb. Hemost. 2001, 7, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Hosono-Fukao, T.; Tang, R.; Sugaya, N.; van Kuppevelt, T.H.; Jenniskens, G.J.; Kimata, K.; Rosen, S.D.; Uchimura, K. Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology 2010, 20, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Hammond, E.; Davis, K.; Li, C.P.; Liu, L.; Johnstone, K.; Handley, P.; Wimmer, N.; Gonda, T.J.; Gautam, A.; et al. The PG500 series: Novel heparan sulfate mimetics as potent angiogenesis and heparanase inhibitors for cancer therapy. Investig. New Drugs 2010, 28, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dredge, K.; Hammond, E.; Handley, P.; Gonda, T.J.; Smith, M.T.; Vincent, C.; Brandt, R.; Ferro, V.; Bytheway, I. PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. Br. J. Cancer 2011, 104, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmann, M.; Bhattacharya, U.; Feld, S.; Hammond, E.; Ilan, N.; Vlodavsky, I. The heparanase inhibitor PG545 is a potent anti-lymphoma drug: Mode of action. Matrix Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics, T.J.; Mims, A.; Salama, M.E.; Pantin, J.; Rao, N.; Kosak, K.M.; Ahorukomeye, P.; Glenn, M.J.; Deininger, M.W.N.; Boucher, K.M.; et al. Combination of the low anticoagulant heparin CX-01 with chemotherapy for the treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, M.; Chatterjee, M.; Grasso, M.; Specchia, G.; Magen, H.; Einsele, H.; Celeghini, I.; Barbieri, P.; Paoletti, D.; Pace, S.; et al. Phase I study of the heparanase inhibitor Roneparstat: An innovative approach for multiple myeloma therapy. Haematologica 2018. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Roach, J.; Miller, P.; Yu, K.H.; Tjan, C.; Rosano, M.; Krause, S.; Avery, W.; Wolf, J.; Flaherty, K.; et al. Safety, pharmacokinetics, pharmacodynamics, and antitumor activity of necuparanib combined with Nab-Paclitaxel and Gemcitabine in patients with metastatic pancreatic cancer: Phase I results. Oncologist 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Lee, P.H.; Han, K.H.; Fan, J.; Cheung, T.T.; Hu, R.H.; Paik, S.W.; Lee, W.C.; Chau, G.Y.; Jeng, L.B.; et al. A phase III trial of muparfostat (PI-88) as adjuvant therapy in patients with hepatitis virus related hepatocellular carcinoma (HV-HCC) after resection. In Integrating Science into Oncology for a Better Patient Outcome, Proceedings of the 42nd ESMO Congress (ESMO 2017), Madrid, Spain, 8–12 September 2017; Abstract 624PD; Annals of Oncology: Oxford, UK, 2017. [Google Scholar]
- Basche, M.; Gustafson, D.L.; Holden, S.N.; O’Bryant, C.L.; Gore, L.; Witta, S.; Schultz, M.K.; Morrow, M.; Levin, A.; Creese, B.R.; et al. A phase I biological and pharmacologic study of the heparanase inhibitor PI-88 in patients with advanced solid tumors. Clin. Cancer Res. 2006, 12, 5471–5480. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Brennan, T.V.; Hammond, E.; Lickliter, J.D.; Lin, L.; Bampton, D.; Handley, P.; Lankesheer, F.; Morrish, G.; Yang, Y.; et al. A Phase I study of the novel immunomodulatory agent PG545 (pixatimod) in subjects with advanced solid tumours. Br. J. Cancer 2018, 118, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Secchi, M.F.; Gambaro, G.; Onisto, M. Heparanase as a target in cancer therapy. Curr. Cancer Drug Targets 2014, 14, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Heyman, B.; Yang, Y. Mechanisms of heparanase inhibitors in cancer therapy. Exp. Hematol. 2016, 44, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, S.; Coombe, D.R. Heparin mimetics: Their therapeutic potential. Pharmaceuticals 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Tkaczynski, E.; Arulselvan, A.; Tkaczynski, J.; Avery, S.; Xiao, L.; Torok-Storb, B.; Abrams, K.; Rao, N.V.; Johnson, G.; Kennedy, T.P.; et al. 2-O, 3-O desulfated heparin mitigates murine chemotherapy- and radiation-induced thrombocytopenia. Blood Adv. 2018, 2, 754–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, M.; Franqui-Machin, R.; Xu, H.; Shaughnessy, J., Jr.; Barlogie, B.; Roodman, D.; Quelle, D.E.; Janz, S.; Tomasson, M.H.; Sanderson, R.D.; et al. NEK2 induces osteoclast differentiation and bone destruction via heparanase in multiple myeloma. Leukemia 2017, 31, 1648–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultes, B.C.; Lolkema, M.P.J.K.; Chu, C.L.; Zhou, H.; Long, A.; Lockley, M.; Avery, W.; Kurtagic, E.; Galcheva-Gargova, Z.; Miller, P.; et al. M402, a heparan sulfate mimetic and novel candidate for the treatment of pancreatic cancer. In Proceedings of the 2012 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 1–5 June 2012; Abstract 4056. Journal of Clinical Oncology: Alexandria, VA, USA, 2012. [Google Scholar]
- Galcheva-Gargova, Z.; Chu, C.L.; Long, A.; Duffner, J.; Holte, K.; Schultes, B.C. Role of M402, a novel heparan sulfate mimetic, in pancreatic cancer cell invasion and metastasis: Inhibition of the Sonic Hedgehog pathway and heparanase activity. In Proceedings of the 2012 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 1–5 June 2012; Abstract 25. Journal of Clinical Oncology: Alexandria, VA, USA, 2012. [Google Scholar]
- Chu, C.L.; Long, A.; Honan, C.M.; Duffner, J.; Avery, W.; D’Alessandro, J.; Kishimoto, T.K.; Schultes, B.C. M402, a novel heparan sulfate mimetic, inhibits pancreatic tumor growth and desmoplasia potentially via sonic hedgehog signaling in an orthotopic mouse model. In Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research, Chicago, IL, USA, 31 March–4 April 2012; Abstract 1524. Cancer Research: Philadelphia, PA, USA, 2012. [Google Scholar]
- Liu, C.J.; Chang, J.; Lee, P.H.; Lin, D.Y.; Wu, C.C.; Jeng, L.B.; Lin, Y.J.; Mok, K.T.; Lee, W.C.; Yeh, H.Z.; et al. Adjuvant heparanase inhibitor PI-88 therapy for hepatocellular carcinoma recurrence. World J. Gastroenterol. 2014, 20, 11384–11393. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.Y.; Wang, Z.; Hu, J.; Liu, W.F.; Shen, Z.Z.; Zhang, X.; Yu, L.; Fan, J.; Zhou, J. PI-88 inhibits postoperative recurrence of hepatocellular carcinoma via disrupting the surge of heparanase after liver resection. Tumor Biol. 2016, 37, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.; Brandt, R.; Dredge, K. PG545, a heparan sulfate mimetic, reduces heparanase expression in vivo, blocks spontaneous metastases and enhances overall survival in the 4T1 breast carcinoma model. PLoS ONE 2012, 7, e52175. [Google Scholar] [CrossRef] [PubMed]
- Ferro, V.; Liu, L.; Johnstone, K.D.; Wimmer, N.; Karoli, T.; Handley, P.; Rowley, J.; Dredge, K.; Li, C.P.; Hammond, E.; et al. Discovery of PG545: A highly potent and simultaneous inhibitor of angiogenesis, tumor growth, and metastasis. J. Med. Chem. 2012, 55, 3804–3813. [Google Scholar] [CrossRef] [PubMed]
- Barash, U.; Lapidot, M.; Zohar, Y.; Loomis, C.; Moreira, A.; Feld, S.; Goparaju, C.; Yang, H.; Hammond, E.; Zhang, G.; et al. Involvement of heparanase in the pathogenesis of mesothelioma: Basic aspects and clinical applications. J. Natl. Cancer Inst. 2018, 110. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, A.; Kundu, S.; Haseeb, L.; Yu, D.; Olofsson, T.; Dredge, K.; Hammond, E.; Barash, U.; Vlodavsky, I.; Forsberg-Nilsson, K. Inhibition of heparanase in pediatric brain tumor cells attenuates their proliferation, invasive capacity, and in vivo tumor growth. Mol. Cancer Ther. 2017, 16, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Winterhoff, B.; Freyer, L.; Hammond, E.; Giri, S.; Mondal, S.; Roy, D.; Teoman, A.; Mullany, S.A.; Hoffmann, R.; von Bismarck, A.; et al. PG545 enhances anti-cancer activity of chemotherapy in ovarian models and increases surrogate biomarkers such as VEGF in preclinical and clinical plasma samples. Eur. J. Cancer 2015, 51, 879–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.B.; Yun, M.; Kim, E.O.; Kim, J.; Kim, B.; Jung, J.H.; Wang, E.; Mukhopadhyay, D.; Hammond, E.; Dredge, K.; et al. The heparan sulfate mimetic PG545 interferes with Wnt/β-catenin signaling and significantly suppresses pancreatic tumorigenesis alone and in combination with gemcitabine. Oncotarget 2015, 6, 4992–5004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, T.V.; Lin, L.; Brandstadter, J.D.; Rendell, V.R.; Dredge, K.; Huang, X.; Yang, Y. Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J. Clin. Investig. 2016, 126, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Ostapoff, K.T.; Awasthi, N.; Cenik, B.K.; Hinz, S.; Dredge, K.; Schwarz, R.E.; Brekken, R.A. PG545, an angiogenesis and heparanase inhibitor, reduces primary tumor growth and metastasis in experimental pancreatic cancer. Mol. Cancer Ther. 2013, 12, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.; Haynes, N.M.; Cullinane, C.; Brennan, T.V.; Bampton, D.; Handley, P.; Karoli, T.; Lanksheer, F.; Lin, L.; Yang, Y.; et al. Immunomodulatory activities of pixatimod: Emerging nonclinical and clinical data, and its potential utility in combination with PD-1 inhibitors. J. Immunother. Cancer 2018, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.; Barash, U.; Boyango, I.; Feld, S.; Zohar, Y.; Hammond, E.; Ilan, N.; Kremer, R.; Vlodavsky, I. Patient derived xenografts (PDX) predict an effective heparanase-based therapy for lung cancer. Oncotarget 2018, 9, 19294–19306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meer, J.Y.; Kellenbach, E.; van den Bos, L.J. From farm to pharma: An overview of industrial heparin manufacturing methods. Molecules 2017, 22, 1025. [Google Scholar] [CrossRef] [PubMed]
- Pavão, M.S. Glycosaminoglycans analogs from marine invertebrates: Structure, biological effects, and potential as new therapeutics. Front. Cell. Infect. Microbiol. 2014, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Oreste, P.; Zoppetti, G. Semi-synthetic heparinoids. In Handbook of Experimental Pharmacology; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207, pp. 403–422. [Google Scholar]
- Colliec-Jouault, S.; Bavington, C.; Delbarre-Ladrat, C. Heparin-like entities from marine organisms. In Handbook of Experimental Pharmacology; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207, pp. 423–449. [Google Scholar]
- Gomes, A.M.; Kozlowski, E.O.; Pomin, V.H.; de Barros, C.M.; Zaganeli, J.L.; Pavão, M.S. Unique, extracellular matrix heparan sulfate from the bivalve Nodipecten nodosus (Linnaeus, 1758) safely inhibits arterial thrombosis after photochemically induced endothelial lesion. J. Biol. Chem. 2010, 285, 7312–7323. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.M.; Kozlowski, E.O.; Borsig, L.; Teixeira, F.C.; Vlodavsky, I.; Pavão, M.S. Antitumor properties of a new non-anticoagulant heparin analog from the mollusk Nodipecten nodosus: Effect on P-selectin, heparanase, metastasis and cellular recruitment. Glycobiology 2015, 25, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Yang, H.O.; Shin, K.H.; Choi, H.S.; Jung, S.H.; Kim, Y.M.; Oh, D.K.; Linhardt, R.J.; Kim, Y.S. Suppression of tumor growth by a new glycosaminoglycan isolated from the African giant snail Achatina fulica. Eur. J. Pharmacol. 2003, 465, 191–198. [Google Scholar] [CrossRef]
- Joo, E.J.; Yang, H.; Park, Y.; Park, N.Y.; Toida, T.; Linhardt, R.J.; Kim, Y.S. Induction of nucleolin translocation by acharan sulfate in A549 human lung adenocarcinoma. J. Cell. Biochem. 2010, 110, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Oreste, P.; Zoppetti, G.; Presta, M. Biotechnological engineering of heparin/heparan sulphate: A novel area of multi-target drug discovery. Curr. Pharm. Des. 2005, 11, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Sheng, J.; Liu, Y.; Li, J.; Liu, J.; Wang, F. Heparosan-derived heparan sulfate/heparin-like compounds: One kind of potential therapeutic agents. Med. Res. Rev. 2013, 33, 665–692. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Grazioli, G.; Razi, N.; Guerrini, M.; Naggi, A.; Torri, G.; Oreste, P.; Tursi, F.; Zoppetti, G.; Lindahl, U. Heparin-like compounds prepared by chemical modification of capsular polysaccharide from E. coli K5. Carbohydr. Res. 1994, 263, 271–284. [Google Scholar] [CrossRef]
- Fu, L.; Suflita, M.; Linhardt, R.J. Bioengineered heparins and heparan sulfates. Adv. Drug Deliv. Rev. 2016, 97, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poggi, A.; Rossi, C.; Casella, N.; Bruno, C.; Sturiale, L.; Dossi, C.; Naggi, A. Inhibition of B16-BL6 melanoma lung colonies by semisynthetic sulfaminoheparosan sulfates from E. coli K5 polysaccharide. Semin. Thromb. Hemost. 2002, 28, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Leali, D.; Belleri, M.; Urbinati, C.; Coltrini, D.; Oreste, P.; Zoppetti, G.; Ribatti, D.; Rusnati, M.; Presta, M. Fibroblast growth factor-2 antagonist activity and angiostatic capacity of sulfated Escherichia coli K5 polysaccharide derivatives. J. Biol. Chem. 2001, 276, 37900–37908. [Google Scholar] [PubMed]
- Borgenström, M.; Jalkanen, M.; Salmivirta, M. Sulfated derivatives of Escherichia coli K5 polysaccharides as modulators of fibroblast growth factor signaling. J. Biol. Chem. 2003, 278, 49882–49889. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Paradigms in the structural biology of the mitogenic ternary complex FGF:FGFR:heparin. Biochimie 2016, 127, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Borgenström, M.; Wärri, A.; Hiilesvuo, K.; Käkönen, R.; Käkönen, S.; Nissinen, L.; Pihlavisto, M.; Marjamäki, A.; Vlodavsky, I.; Naggi, A.; et al. O-sulfated bacterial polysaccharides with low anticoagulant activity inhibit metastasis. Semin. Thromb. Hemost. 2007, 33, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Pollari, S.; Käkönen, R.S.; Mohammad, K.S.; Rissanen, J.P.; Halleen, J.M.; Wärri, A.; Nissinen, L.; Pihlavisto, M.; Marjamäki, A.; Perälä, M.; et al. Heparin-like polysaccharides reduce osteolytic bone destruction and tumor growth in a mouse model of breast cancer bone metastasis. Mol. Cancer Res. 2012, 10, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Pikas, D.S.; Li, J.P.; Vlodavsky, I.; Lindahl, U. Substrate specificity of heparanases from human hepatoma and platelets. J. Biol. Chem. 1998, 273, 18770–18777. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.B.; Liu, J. Unraveling the specificity of heparanase utilizing synthetic substrates. J. Biol. Chem. 2010, 285, 14504–14513. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.B.; Liu, J. Multi-faceted substrate specificity of heparanase. Matrix Biol. 2013, 32, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jones, C.L.; Liu, J. Using an enzymatic combinatorial approach to identify anticoagulant heparan sulfate structures. Chem. Biol. 2007, 14, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Fu, H.; Deng, C.; Chen, J. Modulating the SDF-1/CXCL12-induced cancer cell growth and adhesion by sulfated K5 polysaccharides in vitro. Biomed. Pharmacother. 2015, 73, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Sadir, R.; Imberty, A.; Baleux, F.; Lortat-Jacob, H. Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 2004, 279, 43854–43860. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Fu, H.; Wang, M.; Deng, C.; Song, Z.; Chen, J. Immunomodulatory activity of heparan sulfate mimetics from Escherichia coli K5 capsular polysaccharide in vitro. Carbohydr. Polym. 2015, 115, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Mencio, C.; Desai, U.R.; Kuberan, B. Sulfation patterns determine cellular internalization of heparin-like polysaccharides. Mol. Pharm. 2013, 10, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.L.; Hansen, S.U.; Baráth, M.; Rushton, G.; Gardiner, J.M.; Avizienyte, E.; Jayson, G.C. Synthetic heparan sulfate oligosaccharides inhibit endothelial cell functions essential for angiogenesis. PLoS ONE 2010, 5, e11644. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Hansen, S.U.; Miller, G.J.; Cole, C.L.; Rushton, G.; Avizienyte, E.; Gardiner, J.M. Synthetic heparan sulfate dodecasaccharides reveal single sulfation site interconverts CXCL8 and CXCL12 chemokine biology. Chem. Commun. 2015, 51, 13846–13849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avizienyte, E.; Cole, C.L.; Rushton, G.; Miller, G.J.; Bugatti, A.; Presta, M.; Gardiner, J.M.; Jayson, G.C. Synthetic site-selectively mono-6-O-sulfated heparan sulfate dodecasaccharide shows anti-angiogenic properties in vitro and sensitizes tumors to cisplatin in vivo. PLoS ONE 2016, 11, e0159739. [Google Scholar] [CrossRef] [PubMed]
- Kuhnast, B.; El Hadri, A.; Boisgard, R.; Hinnen, F.; Richard, S.; Caravano, A.; Nancy-Portebois, V.; Petitou, M.; Tavitian, B.; Dollé, F. Synthesis, radiolabeling with fluorine-18 and preliminary in vivo evaluation of a heparan sulphate mimetic as potent angiogenesis and heparanase inhibitor for cancer applications. Org. Biomol. Chem. 2016, 14, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; El Hadri, A.; Richard, S.; Denis, F.; Holte, K.; Duffner, J.; Yu, F.; Galcheva-Gargova, Z.; Capila, I.; Schultes, B.; et al. Synthesis and biological evaluation of a unique heparin mimetic hexasaccharide for structure-activity relationship studies. J. Med. Chem. 2014, 57, 4511–4520. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Purunomo, E.; Takeda, N.; Tamura, J.; Kitagawa, H. Heparan sulfate containing unsubstituted glucosamine residues: Biosynthesis and heparanase-inhibitory activity. J. Biol. Chem. 2014, 289, 15231–15243. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.; Friand, V.; Papy-Garcia, D.; Dagouassat, M.; Martin, L.; Vassy, R.; Haddad, O.; Sainte-Catherine, O.; Kraemer, M.; Saffar, L.; et al. Glycosaminoglycans and their synthetic mimetics inhibit RANTES-induced migration and invasion of human hepatoma cells. Mol. Cancer Ther. 2007, 6, 2948–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, G.J.; Oh, Y.I.; Chang, S.K.; Hsieh-Wilson, L.C. Tunable heparan sulfate mimetics for modulating chemokine activity. J. Am. Chem. Soc. 2013, 135, 10898–10901. [Google Scholar] [CrossRef] [PubMed]
- Raiber, E.A.; Wilkinson, J.A.; Manetti, F.; Botta, M.; Deakin, J.; Gallagher, J.; Lyon, M.; Ducki, S.W. Novel heparin/heparan sulfate mimics as inhibitors of HGF/SF-induced MET activation. Bioorg. Med. Chem. Lett. 2007, 17, 6321–6325. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.A.; Blaum, B.S.; Gallagher, J.T.; Uhrín, D.; Lyon, M. The binding properties of minimal oligosaccharides reveal a common heparan sulfate/dermatan sulfate-binding site in hepatocyte growth factor/scatter factor that can accommodate a wide variety of sulfation patterns. J. Biol. Chem. 2009, 284, 6311–6321. [Google Scholar] [CrossRef] [PubMed]
- Serin, G.; Mirjolet, J.F.; Nancy-Portebois, V.; Cabannes, E.; Petitou, M. Antitumor activity of EP80061, a small-glycodrug in preclinical studies. In Proceedings of the 101st Annual Meeting of the American Association for Cancer Research (AACR-2010), Washington, DC, USA, 17–21 April 2010; Abstract 5459. Cancer Research: Philadelphia, PA, USA, 2010. [Google Scholar]
- Vlodavsky, I.; Gross-Cohen, M.; Weissmann, M.; Ilan, N.; Sanderson, R.D. Opposing functions of heparanase-1 and heparanase-2 in cancer progression. Trends Biochem. Sci. 2018, 43, 18–31. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
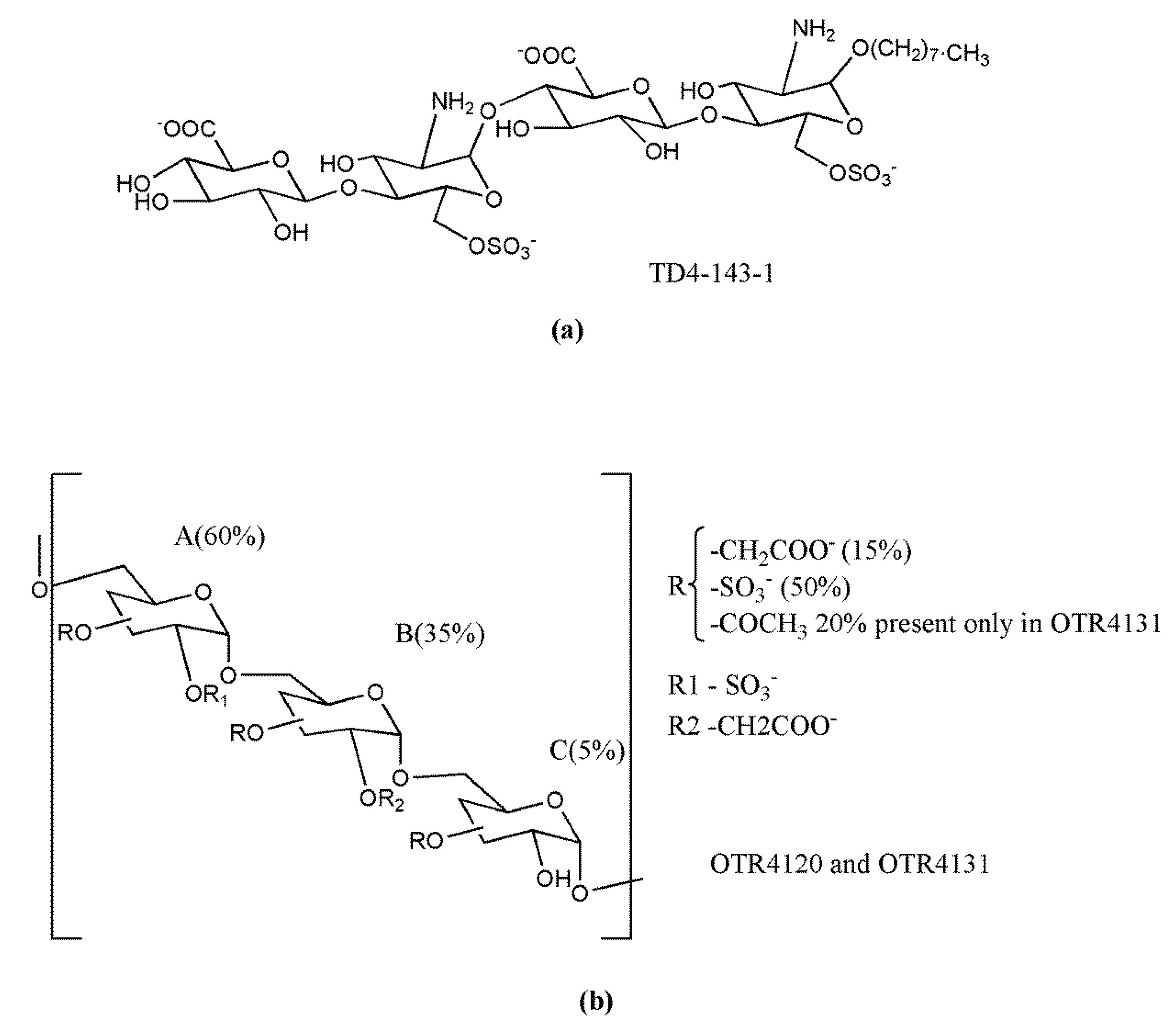{kind=link}
| Drug | Company | Phase | Tumors | Half-Life | Administration Route and Dose | Ref. |
|---|---|---|---|---|---|---|
| CX-01 (ODSH) | Cantex Pharmaceuticals | II a | Acute myeloid leukemia | ~3 h | i.v.4 mg/kg | [139,186] |
| Roneparstat (SST0001) | Leadiant Biosciences | I | Multiple myeloma | 14–20 h | s.c.25–400 mg/day | [187] |
| Necuparanib (M402) | Momenta Pharmaceuticals | I/II b | Pancreatic cancer | N/A c | s.c.0.5–5 mg/kg/day | [188] |
| Muparfostat (PI-88) | Progen Pharmaceuticals Medigen Biotechnology | III | Hepatocellular carcinoma | ~8 h | s.c.160 mg/day | [189,190] |
| Pixatimod (PG545) | Progen Pharmaceuticals | I | Solid tumors | 141 h | i.v.25–150 mgonce weekly | [191] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanzi, C.; Cassinelli, G. Heparan Sulfate Mimetics in Cancer Therapy: The Challenge to Define Structural Determinants and the Relevance of Targets for Optimal Activity. Molecules 2018, 23, 2915. https://doi.org/10.3390/molecules23112915
Lanzi C, Cassinelli G. Heparan Sulfate Mimetics in Cancer Therapy: The Challenge to Define Structural Determinants and the Relevance of Targets for Optimal Activity. Molecules. 2018; 23(11):2915. https://doi.org/10.3390/molecules23112915
Chicago/Turabian StyleLanzi, Cinzia, and Giuliana Cassinelli. 2018. "Heparan Sulfate Mimetics in Cancer Therapy: The Challenge to Define Structural Determinants and the Relevance of Targets for Optimal Activity" Molecules 23, no. 11: 2915. https://doi.org/10.3390/molecules23112915
APA StyleLanzi, C., & Cassinelli, G. (2018). Heparan Sulfate Mimetics in Cancer Therapy: The Challenge to Define Structural Determinants and the Relevance of Targets for Optimal Activity. Molecules, 23(11), 2915. https://doi.org/10.3390/molecules23112915