4.1. Synthesis of Aminoribosylated Muraymycin Analogues and Reference Compounds
General Methods: All chemicals were purchased from standard suppliers. Reactions involving oxygen and/or moisture sensitive reagents were carried out under an atmosphere of argon using anhydrous solvents. Anhydrous solvents were obtained in the following manner. CH
2Cl
2, MeCN and THF were purchased in HPLC quality, dried with a solvent purification system (MBRAUN MB SPS 800) and stored over activated molecular sieves (3 Å (CH
2Cl
2, MeCN) or 4 Å (THF)). Pyridine was dried over CaH
2, distilled and stored over activated molecular sieves (4 Å). MeOH was degassed, dried and stored over activated molecular sieves (3 Å).
i-PrOH was dried over calcium sulfate hemihydrate and stored over activated molecular sieves (3 Å). All other solvents were of technical quality and distilled prior to use, and deionised water was used throughout. Column chromatography was carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh ASTM, VWR, Darmstadt, Germany) under flash conditions except where indicated. TLC was performed on aluminium plates precoated with silica gel 60 F
254 (VWR, Darmstadt, Germany). Visualisation of the spots was carried out using UV light (254 nm) and/or staining under heating (H
2SO
4 staining solution: 4 g vanillin, 25 mL conc. H
2SO
4, 80 mL AcOH, and 680 mL MeOH; KMnO
4 staining solution: 1 g KMnO
4, 6 g K
2CO
3, and 1.5 mL 1.25 M NaOH solution, all dissolved in 100 mL H
2O; ninhydrin staining solution: 0.3 g ninhydrin, 3 mL AcOH, and 100 mL 1-butanol). Analytical HPLC was performed on a Thermo Scientific system (ThermoFisher Scientific, Dreieich, Germany) equipped with an MWD detector (250.0), an ESI mass spectrometer and an EC 125/3 column (5 × 100 mm) containing reversed phase silica gel NUCLEODUR
TM 100–5 C18ec (5 μm, Macherey-Nagel, Düren, Germany). Method 1: eluent A water (+0.1% TFA), eluent B MeCN (+0.1% TFA); 0–19 min gradient of B (5–100%), 19–22 min 100% B, 22–23 min gradient of B (100–5%), 23–25 min 5% B; flow 0.8 mL/min. Semipreparative HPLC was performed on an Agilent Technologies 1200 Series system (Agilent Technologies, Waldbronn, Germany) equipped with an MWD detector (254.16/280.16) and a LiChroCart
TM column (10 × 250 mm) containing reversed phase silica gel Purospher
TM RP18e (5 μm, VWR). Method 1: eluent A water (+0.1% TFA), eluent B MeCN (+0.1% TFA); 0–36 min gradient of B (5–100%), 36–44 min 100% B, 44–44.1 min gradient of B (100–1%); flow 3 mL/min. Method 2: eluent A water (+0.1% TFA), eluent B MeCN (+0.1% TFA); 0–36 min gradient of B (5–60%), 36–40 min gradient of B (60–100%), 40–44 min 100% B, 44–44.1 min gradient of B (100–1%); flow 3 mL/min. Method 3: eluent A water (+0.1% TFA), eluent B MeCN (+0.1% TFA); 0–36 min gradient of B (5–70%), 36–40 min gradient of B (70–100%), 40–44 min 100% B, 44–44.1 min gradient of B (100–1%); flow 3 mL/min. 500 MHz-
1H, 126 MHz-
13C, as well as 282 MHz-
19F NMR spectra were recorded on Bruker AVANCE-500 and AVANCE-300 spectrometers (Bruker, Bremen, Germany). All
13C and
19F NMR spectra are
1H-decoupled. All spectra were recorded at room temperature except where indicated otherwise and were referenced internally to solvent reference frequencies. Chemical shifts (δ) are quoted in ppm and coupling constants (
J) are reported in Hz. Assignment of signals was carried out using H,H-COSY, HSQC, and HMBC spectra obtained on the spectrometers mentioned above. The numbering of atoms of muraymycin target structures is depicted in the
Supplementary Materials (Figure S1). Mass spectra of small molecules were measured on a Finnigan LCQ ion-trap mass spectrometer or on a Bruker microTOF spectrometer (Bruker, Bremen, Germany). High resolution spectra were measured on a Bruker 7 Tesla Fourier transform ion cyclotron resonance (FTICR) mass spectrometer (Bruker, Bremen, Germany). Infrared spectroscopy (IR) was performed on a Bruker Vertex 70 spectrometer equipped with an integrated ATR unit (PlatinumATR
TM, Bruker). Wavenumbers (ν) are quoted in cm
−1. UV spectroscopy was carried out on an Cary 100 spectrophotometer (Agilent Technologies, Waldbronn, Germany). Wavelengths of maximum absorption (λ
max) are reported in nm. Optical rotations were recorded on a Krüss Optronic polarimeter with a Na source using a 5 cm cell (Krüss Optronic, Hamburg, Germany). Melting points (m.p.) were measured on a Büchi instrument (Büchi, Essen, Germany) and are not corrected.
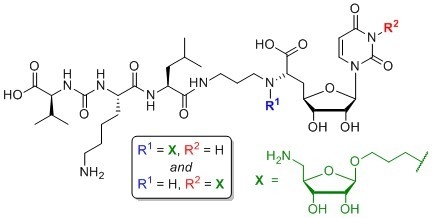
6′-N-Substituted aminoribosylated muraymycin analogue (13): To a solution of protected urea dipeptide 30 (6.1 mg, 14 μmol) in THF (2.5 mL), HOBt (1.9 mg, 14 μmol), PyBOP (7.1 mg, 14 μmol) and DIPEA (4.7 μL, 27 μmol) were added and the mixture was stirred at rt for 30 min. At 0 °C, a solution of protected 6′-N-substituted aminoribosylated truncated muraymycin analogue 29 (15.2 mg, 13.7 μmol) in THF (2 mL) was added dropwise, and the reaction mixture was stirred at 0 °C for 1 h and at rt for 3 h. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (97:3, CH2Cl2-MeOH) to give 31 as a colourless solid (20.4 mg) that was directly used in the subsequent deprotection step. A solution of the protected 6′-N-substituted aminoribosylated muraymycin analogue 31 (16.0 mg) in TFA (80% in CH2Cl2, 5.0 mL) was stirred at rt for 48 h. The mixture was then diluted with CH2Cl2 (20 mL) and the solvent was evaporated under reduced pressure. The resultant residue was dissolved in TFA (10% in water, 5.0 mL), the mixture was stirred at rt for 10 min and then diluted with water (20 mL). The solvent was evaporated under reduced pressure, and the resultant crude product was purified by semipreparative HPLC (method 1) to give 13 (tris-TFA salt) as a colourless solid (5.0 mg, 36% over 2 steps from 29) and 14 (bis-TFA salt) as a colourless solid (0.75 mg, 7% over 2 steps from 29). 13: 1H NMR (500 MHz, D2O): δ [ppm] = 0.90 (d, J = 6.1 Hz, 3 H, 5‴-Ha), 0.95 (d, J = 5.2 Hz, 3H, 5‴-Hb), 0.96 (d, J = 6.2 Hz, 3H, 4v-Ha), 1.00 (d, J = 6.8 Hz, 3H, 4v-Hb), 1.41–1.54 (m, 2H, 4iv-H), 1.56–1.77 (m, 6H, 3‴-H, 4‴-H, 3iv-Ha, 5iv-H), 1.70–1.86 (m, 1H, 3iv-Hb), 1.95–2.06 (m, 3H, 2″-H, 2vi-Ha), 2.06–2.15 (m, 1H, 2vi-Hb), 2.16–2.25 (m, 2H, 5′-Ha, 3v-H), 2.43–2.49 (m, 1H, 5′-Hb), 3.03 (dd, J = 7.6, 7.6 Hz, 2H, 6iv-H), 3.09 (dd, J = 11.5, 11.1 Hz, 1H, 5vii-Ha), 3.16–3.27 (m, 2H, 1″-Ha, 3″-Ha), 3.27–3.34 (m, 2H, 1″-Hb, 1vi-Ha), 3.34–3.40 (m, 2H, 3″-Hb, 1vi-Hb), 3.42 (dd, J = 13.3, 2.4 Hz, 1H, 5vii-Hb), 3.61–3.67 (m, 1H, 3vi-Ha), 3.88 (ddd, J = 6.9, 4.3, 3.9 Hz, 1H, 3vi-Hb), 4.07 (dd, J = 9.6, 3.1 Hz, 1H, 6′-H), 4.10–4.19 (m, 5H, 3′-H, 2iv-H, 2v-H, 2vii-H, 4vii-H), 4.22 (dd, J = 6.3, 4.8 Hz, 1H, 3vii-H), 4.26 (dd, J = 7.7, 6.6 Hz, 1H, 4′-H), 4.30 (dd, J = 10.0, 4.7 Hz, 1H, 2‴-H), 4.55 (dd, J = 4.9, 4.5 Hz, 1H, 2′-H), 5.03 (s, 1H, 1vii-H), 5.76 (d, J = 4.5 Hz, 1H, 1′-H), 5.92 (d, J = 8.1 Hz, 1H, 5-H), 7.69 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O): δ [ppm] = 16.96 (Ca-4v), 18.49 (Cb-4v), 20.58 (Ca-5‴), 21.95 (C-4iv), 22.12 (Cb-5‴), 24.13 (C-2″), 24.19 (C-2vi), 24.40 (C-4‴), 26.25 (C-5iv), 29.46 (C-5′), 29.92 (C-3v), 30.68 (C-3iv), 36.14 (C-3″), 39.21 (C-6iv), 39.52 (C-3‴), 43.11 (C-5vii), 49.65 (C-1″), 50.64 (C-1vi), 52.47 (C-2‴), 54.24 (C-2iv), 58.89 (C-2v), 63.30 (C-6′), 65.62 (C-3vi), 72.36 (C-3vii), 72.42 (C-2′), 73.08 (C-3′), 74.05 (C-2vii), 78.26 (C-4vii), 80.52 (C-4′), 92.19 (C-1′), 102.29 (C-5), 107.52 (C-1vii), 116.33 (q, 1JCF = 291.5 Hz, F3CCOO), 143.23 (C-6), 151.28 (C-2), 159.51 (NC(=O)N), 162.97 (q, 2JCF = 35.5 Hz, F3CCOO), 166.09 (C-4), 171.57 (C-7′), 174.76 (C-1‴), 175.45 (C-1iv), 176.71 (C-1v). 19F NMR (282 MHz, D2O): δ [ppm] = −75.57 (s, CF3). HRMS (ESI+): calcd.: 466.7504 [M+2H]2+, found: 466.7504. IR (ATR): ν = 3066, 2962, 1668, 1556, 1464, 1199, 1131, 1030, 835, 799, 721, 551. UV (MeCN): λmax = 260. optical rotation: [α]D20 = −11.3 (c = 1.3, H2O). m.p. = 145 °C. Analytical HPLC: tR = 6.7 min (method 1). Semipreparative HPLC: tR = 12.3 min (method 1). 14: see below for analytical data.
6′-N-Substituted non–aminoribosylated muraymycin analogue (14): Protected 6′-N-substituted aminoribosylated muraymycin analogue 31 was prepared as described above. A solution of 31 (21.2 mg) in TFA (80% in water, 6.0 mL) was stirred at rt for 24 h. The mixture was then diluted with water (20 mL) and the solvent was evaporated under reduced pressure. The resultant crude product was purified by semipreparative HPLC (method 2) to give 14 (tris-TFA salt) as a colourless solid (5.1 mg, 35% over 2 steps from 29). 1H NMR (500 MHz, D2O): δ [ppm] = 0.80 (d, J = 6.1 Hz, 3H, 5‴-Ha), 0.85 (d, J = 4.9 Hz, 3H, 5‴-Hb), 0.86 (d, J = 6.1 Hz, 3H, 4v-Ha), 0.90 (d, J = 6.7 Hz, 3H, 4v-Hb), 1.31–1.43 (m, 2H, 4iv-H), 1.46–1.67 (m, 6H, 3‴-H, 4‴-H, 3iv-Ha, 5iv-H), 1.69–1.76 (m, 1H, 3iv-Hb), 1.84–1.95 (m, 4H, 2″-H, 2vi-H), 2.10 (dqq, J = 6.7, 6.6, 6.1 Hz, 1H, 3v-H), 2.14 (ddd, J = 14.0, 10.7, 3.0 Hz, 1H, 5′-Ha), 2.37–2.42 (m, 1H, 5′-Hb), 3.03 (dd, J = 7.5, 7.5 Hz, 2H, 6iv-H), 3.07–3.19 (m, 2H, 1″-Ha, 3″-Ha), 3.19–3.27 (m, 3H, 1″-Hb, 3″-Hb, 1vi-Ha), 3.29–3.35 (m, 1H, 1vi-Hb), 3.60–3.68 (m, 2H, 3vi-H), 4.00–4.10 (m, 4H, 3′-H, 6′-H, 2iv-H, 2v-H), 4.15–4.22 (m, 1H, 4′-H, 2‴-H), 4.44 (dd, J = 5.1, 4.4 Hz, 1H, 2′-H), 5.67 (d, J = 4.4 Hz, 1H, 1′-H), 5.82 (d, J = 8.1 Hz, 1H, 5-H), 7.59 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O): δ [ppm] = 16.94 (Ca-4v), 18.47 (Cb-4v), 20.61 (Ca-5‴), 22.01 (C-4iv), 22.09 (Cb-5‴), 24.22 (C-2″), 24.38 (C-4‴), 26.21 (C-2vi), 26.24 (C-5iv), 29.34 (C-5′), 29.92 (C-3v), 30.74 (C-3iv), 36.11 (C-3″), 39.21 (C-6iv), 39.54 (C-3‴), 49.87 (C-1″), 50.97 (C-1vi), 52.50 (C-2‴), 54.08 (C-2iv), 58.84 (C-2v), 59.13 (C-3vi), 62.70 (C-6′), 72.47 (C-2′), 73.02 (C-3′), 80.50 (C-4′), 92.01 (C-1′), 102.29 (C-5), 116.33 (q, 1JCF = 291.5 Hz, F3CCOO), 143.11 (C-6), 151.30 (C-2), 159.48 (NC(=O)N), 162.97 (q, 2JCF = 35.4 Hz, F3CCOO), 166.10 (C-4), 171.35 (C-7′), 174.73 (C-1‴), 175.35 (C-1iv), 176.70 (C-1v). 19F NMR (282 MHz, D2O): δ [ppm] = −75.59 (s, CF3). HRMS (ESI+): calcd.: 401.2213 [M+2H]2+, found: 401.2220. IR (ATR): ν = 2962, 1670, 1556, 1464, 1389, 1262, 1199, 1133, 1065, 834, 800, 768. UV (MeCN): λmax = 260. optical rotation: [α]D20 = +6.7 (c = 0.8, H2O). m.p. = 150 °C. Analytical HPLC: tR = 6.9 min (method 1). Semipreparative HPLC: tR = 16.5 min (method 2).
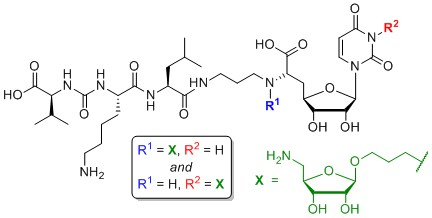
Uracil-N-3-substituted aminoribosylated muraymycin analogue (αβ-15) and uracil-N-3-substituted non-aminoribosylated muraymycin analogue (16): To a solution of the protected uracil-N-3-substituted aminoribosylated truncated N-Cbz-muraymycin analogue 33 (27.4 mg, 22.0 μmol) in i-PrOH (3 mL), 1,4-cyclohexadiene (21.0 μL, 226 μmol) and Pd black (10.0 mg, 94.0 μmol) were added and the mixture was stirred at rt for 1 h. It was then filtered through a syringe filter, and the syringe filter was washed with i-PrOH (4 × 5 mL). The solvent of the combined filtrates was evaporated under reduced pressure to give 34 as a colourless wax (24.0 mg) that was directly used in the subsequent peptide coupling step. To a solution of protected urea dipeptide 30 (6.3 mg, 14 μmol) in THF (2.5 mL), HOBt (2.0 mg, 14 μmol), PyBOP (7.4 mg, 14 μmol) and DIPEA (4.8 μL, 28 μmol) were added and the mixture was stirred at rt for 30 min. At 0 °C, a solution of protected uracil-N-3–substituted aminoribosylated truncated muraymycin analogue 34 (16.0 mg) in THF (2 mL) was added dropwise, and the reaction mixture was stirred at 0 °C for 1 h and at rt for 3 h. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (97:3, CH2Cl2-MeOH) to give 35 as a colourless solid (18.4 mg) that was directly used in the subsequent deprotection step. A solution of the protected uracil-N-3–substituted aminoribosylated muraymycin analogue 35 (20.0 mg) in TFA (80% in CH2Cl2, 6.0 mL) was stirred at rt for 30 h. The mixture was then diluted with CH2Cl2 (20 mL) and the solvent was evaporated under reduced pressure. The resultant residue was dissolved in TFA (10% in water, 5.0 mL), the mixture was stirred at rt for 10 min and then diluted with water (20 mL). The solvent was evaporated under reduced pressure, and the resultant crude product was purified by semipreparative HPLC (method 3) to give αβ-15 (tris-TFA salt) as a colourless solid (5.1 mg, 1:1 mixture of α,β-anomers, 25% over 3 steps from 33) and 16 (tris-TFA salt) as a colourless solid (4.5 mg, 27% over 3 steps from 33). αβ-15: 1H NMR (500 MHz, D2O): δ [ppm] = 0.86 (d, J = 6.0 Hz, 3H, 5‴-Ha), 0.86 (d, J = 6.0 Hz, 3H, 5‴-Ha), 0.92 (d, J = 5.9 Hz, 3H, 5‴-Hb), 0.92 (d, J = 5.9 Hz, 3H, 5‴-Hb), 0.93 (2 d, J = 6.9 Hz, 3H, 4v-Ha), 0.93 (2 d, J = 6.9 Hz, 3H, 4v-Ha), 0.96 (d, J = 6.8 Hz, 3H, 4v-Hb), 0.96 (d, J = 6.8 Hz, 3H, 4v-Hb), 1.35–1.51 (m, 2 × 2 H, 2 × 4iv-H), 1.52–1.74 (m, 2 × 6 H, 2 × 3‴-H, 2 × 4‴-H, 2 × 3iv-Ha, 2 × 5iv-H), 1.74–1.84 (m, 2 × 1 H, 2 × 3iv-Hb), 1.86–1.98 (m, 2 × 4 H, 2 × 2″-H, 2 × 2vi-H), 2.16 (dqq, J = 6.9, 6.8, 5.4 Hz, 1H, 3v-H), 2.16 (dqq, J = 6.9, 6.8, 5.4 Hz, 1H, 3v-H), 2.32 (ddd, J = 15.0, 11.3, 5.7 Hz, 1H, 5′-Ha), 2.32 (ddd, J = 15.0, 11.3, 5.7 Hz, 1H, 5′-Ha), 2.45 (ddd, J = 15.0, 6.4, 2.5 Hz, 1H, 5′-Hb), 2.45 (ddd, J = 15.0, 6.4, 2.5 Hz, 1H, 5′-Hb), 2.99 (dd, J = 7.6, 7.6 Hz, 2H, 6iv-H), 2.99 (dd, J = 7.6, 7.6 Hz, 2H, 6iv-H), 3.04–3.13 (m, 2 × 3H, 2 × 1″-Hb, 2 × 5vii-Ha), 3.21–3.28 (m, 2 × 1H, 2 × 3″-Ha), 3.29–3.42 (m, 2 × 2H, 2 × 3″-Hb, 2 × 5vii-Hb), 3.56 (dt, J = 10.2, 6.0, 1H, β-3vi-Ha), 3.63 (dt, J = 10.4, 6.2 Hz, 1H, α-3vi-Ha), 3.79–3.87 (m, 2 × 1H, 2 × 3vi-Hb), 3.94 (dd, J = 11.3, 6.4 Hz, 1H, 6′-H), 3.94 (dd, J = 11.3, 6.4 Hz, 1H, 6′-H), 3.97–4.04 (m, 2 × 3H, 2 × 1vi-H, α-3vii-H, β-2vii-H), 4.04–4.09 (m, 2 × 2H, 2 × 3′-H, 2 × 2v-H), 4.09–4.20 (m, 1 × 3H, 1 × 4H, 2 × 4′-H, 2 × 2iv-H, α-2vii-H, β-3vii-H, β-4vii-H), 4.20–4.28 (m, 1 × 1H, 1 × 2H, 2 × 2‴-H, α-4vii-H), 4.34–4.39 (m, 2 × 1H, 2 × 2′-H), 4.98 (s, 1H, β-1vii-H), 5.15 (d, J = 4.3 Hz, 1H, α-1vii-H), 5.79 (d, J = 3.5 Hz, 1H, 1′-H), 5.81 (d, J = 3.5 Hz, 1H, 1′-H), 5.94 (d, J = 8.1 Hz, 1H, 5-H), 5.94 (d, J = 8.1 Hz, 1H, 5-H), 7.64 (d, J = 8.1 Hz, 1H, 6-H), 7.64 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O): δ [ppm] = 16.91 (2 × Ca-4v), 18.43 (2 × Cb-4v), 20.58 (2 × Ca-5‴), 21.98 (2 × C-4iv), 22.04 (2 × Cb-5‴), 24.32 (2 × C-4‴), 25.56 (2 × C-2″), 26.20 (2 × C-5iv), 26.65, 26.89 (α-C-2vi, β-C-2vi), 29.87 (2 × C-3v), 30.70 (2 × C-3iv), 32.67 (2 × C-5′), 35.93 (2 × C-3″), 38.96, 39.04 (α-C-1vi, β-C-1vi), 39.16 (2 × C-6iv), 39.42 (2 × C-3‴), 41.37, 43.11 (α-C-5vii, β-C-5vii), 44.24 (2 × C-1″), 52.51 (2 × C-2‴), 54.02 (2 × C-2iv), 58.82 (2 × C-2v), 59.32 (2 × C-6′), 66.53 (β-C-3vi), 66.67 (α-C-3vi), 70.54 (β-C-3vii), 70.69 (α-C-3vii), 72.52 (α-C-2vii) 72.88, 72.92 (2 × C-2′, 2 × C-3′), 74.07 (β-C-2vii), 78.07 (β-C-4vii), 78.79 (α-C-4vii), 79.29, 79,35 (α-C-4′, β-C-4′), 91.79, 92.16 (α-C-1′, β-C-1′), 101.64, 101.68 (α-C-5, β-C-5), 102.31 (α-C-1vii), 107.51 (β-C-1vii), 116.29 (q, 1JCF = 291.5 Hz, F3CCOO), 116.29 (q, 1JCF = 291.5 Hz, F3CCOO), 140.20, 140.31 (α-C-6, β-C-6), 151.52, 151.57 (α-C-2, β-C-2), 159.46 (2 × NC(=O)N), 162.96 (q, 2JCF = 35.5 Hz, F3CCOO), 162.96 (q, 2JCF = 35.5 Hz, F3CCOO), 165.09, 165.15 (α-C-4, β-C-4), 171.80, 171.85 (α-C-7′, β-C-7′), 174.85 (2 × C-1‴), 175.40 (2 × C-1iv), 176.69 (2 × C-1v). 19F NMR (282 MHz, D2O): δ [ppm] = −75.57 (s, CF3), –75.57 (s, CF3). HRMS (ESI+): calcd.: 466.7504 [M+2H]2+, found: 466.7507. IR (ATR): ν = 3293, 3082, 2961, 1649, 1553, 1462, 1182, 1129, 835, 799, 721. UV (MeCN): λmax = 262. optical rotation: [α]D20 = +29.3 (c = 0.7, H2O). m.p. = 156 °C. Analytical HPLC: tR = 6.5 min (method 1). Semipreparative HPLC: tR = 14.4 min (method 3). 16: 1H NMR (500 MHz, D2O): δ [ppm] = 0.90 (d, J = 5.6 Hz, 3H, 5‴-Ha), 0.94–0.97 (m, 6H, 5‴-Hb, 4v-Ha), 0.99 (d, J = 6.5 Hz, 3H, 4v-Hb), 1.41–1.53 (m, 2H, 4iv-H), 1.55–1.76 (m, 6H, 3‴-H, 4‴-H, 3iv-Ha, 5iv-H), 1.78–1.85 (m, 1H, 3iv-Hb), 1.88 (tt, J = 7.1, 6.4 Hz, 2H, 2vi-H), 1.94 (ddt, J = 7.0, 6.9, 6.6 Hz, 2H, 2″-H), 2.19 (dqq, J = 6.7, 6.5, 5.9 Hz, 1H, 3v-H), 2.31 (ddd, J = 15.1, 8.9, 6.3 Hz, 1H, 5′-Ha), 2.47 (ddd, J = 15.1, 5.8, 1.8 Hz, 1H, 5′-Hb), 3.03 (dd, J = 7.5, 7.5 Hz, 2H, 6iv-H), 3.07–3.15 (m, 2H, 1″-H), 3.28 (ddd, J = 13.3, 6.9, 6.4 Hz, 1H, 3″-Ha), 3.34 (ddd, J = 13.3, 7.0, 6.4 Hz, 1H, 3″-Hb), 3.67 (t, J = 6.4 Hz, 2H, 3vi-H), 3.90 (dd, J = 6.3, 5.8 Hz, 1H, 6′-H), 4.01 (t, J = 7.1 Hz, 2H, 1vi-H), 4.08–4.13 (m, 2H, 3′-H, 2v-H), 4.15 (dd, J = 7.6, 6.0 Hz, 1H, 2iv-H), 4.19 (ddd, J = 8.9, 7.2, 1.8 Hz, 1H, 4′-H), 4.28 (dd, J = 9.3, 4.7 Hz, 1H, 2‴-H), 4.43 (dd, J = 3.7, 2.7 Hz, 1H, 2′-H), 5.81 (d, J = 2.7 Hz, 1H, 1′-H), 5.97 (d, J = 8.1 Hz, 1H, 5-H), 7.67 (d, J = 8.1 Hz, 1H, 6-H). 13C NMR (126 MHz, D2O): δ [ppm] = 16.94 (Ca-4v), 18.48 (Cb-4v), 20.64 (Ca-5‴), 22.00 (C-4iv), 22.07 (Cb-5‴), 24.36 (C-4‴), 25.62 (C-2″), 26.23 (C-5iv), 29.34 (C-2vi), 29.94 (C-3v), 30.76 (C-3iv), 32.87 (C-5′), 35.93 (C-3″), 38.67 (C-1vi), 39.20 (C-6iv), 39.50 (C-3‴), 44.26 (C-1″), 52.54 (C-2‴), 54.00 (C-2iv), 58.91 (C-2v), 59.32 (C-3vi), 59.93 (C-6′), 72.81 (C-2′), 72.90 (C-3′), 79.80 (C-4′), 92.49 (C-1′), 101.66 (C-5), 116.33 (q, 1JCF = 291.9 Hz, F3CCOO), 140.55 (C-6), 151.55 (C-2), 159.46 (NC(=O)N), 162.99 (q, 2JCF = 35.5 Hz, F3CCOO), 165.19 (C-4), 172.13 (C-7′), 174.85 (C-1‴), 175.35 (C-1iv), 176.79 (C-1v). 19F NMR (282 MHz, D2O): δ [ppm] = −75.59 (s, CF3). HRMS (ESI+): calcd.: 401.2213 [M+2H]2+, found: 401.2217. IR (ATR): ν = 3306, 2961, 1645, 1554, 1462, 1199, 1181, 1130, 1084, 836, 800, 721. UV (MeCN): λmax = 262. optical rotation: [α]D20 = +27.0 (c = 0.6, H2O). m.p. = 185 °C. Analytical HPLC: tR = 6.8 min (method 1). Semipreparative HPLC: tR = 16.7 min (method 3).
Protected but-3-enyl β-
d-5-azido-5-deoxyriboside (
20): To a solution of protected β-
d-ribosyl fluoride
19 (50.0 mg, 20.0 μmol) [
49,
50,
51,
52] in CH
2Cl
2 (2 mL) at 0 °C, molecular sieves (4 Å) and a solution of homoallylic alcohol (18.0 mg, 31.0 μmol) in CH
2Cl
2 (0.5 mL) were added and the mixture was stirred at 0 °C for 1 h. Boron trifluoride etherate (38 μL, 30 mmol, 0.2 M in CH
2Cl
2) was added and the mixture was stirred at 0 °C for further 2 h. It was then quenched with sat. NaHCO
3 solution, warmed to rt, and filtered. The residue was washed with CH
2Cl
2 (3 × 30 mL). The combined organics were washed with brine (100 mL) and dried over Na
2SO
4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (9:1,
iso-hexane–EtOAc) to give
20 as a colourless oil (40.0 mg, 65%).
1H NMR (500 MHz, C
6D
6): δ [ppm] = 0.80 (t,
J = 7.5 Hz, 3H, 1′-H), 0.99 (t,
J = 7.5 Hz, 3H, 5′-H), 1.42 (q,
J = 7.5 Hz, 2H, 2′-H), 1.70 (q,
J = 7.5 Hz, 2H, 4′-H), 2.06–2.17 (m, 2H, 2″-H), 2.70 (dd,
J = 12.5, 6.4 Hz, 1H, 5-H
a), 3.02 (dd,
J = 12.5, 8.1 Hz, 1H, 5-H
b), 3.12 (dt,
J = 9.5, 6.6 Hz, 1H, 1″-H
a), 3.63 (dt,
J = 9.5, 6.7 Hz, 1H, 1″-H
b), 4.17 (dd,
J = 6.0, 0.7 Hz, 1H, 3-H), 4.29 (ddd,
J = 8.1, 6.4, 0.7 Hz, 1H, 4-H), 4.44 (d,
J = 6.0 Hz, 1H, 2-H), 4.98–5.04 (m, 2H, 4″-H), 5.08 (s, 1H, 1-H), 5.68 (ddt,
J = 17.1, 10.3, 6.8 Hz, 1H, 3″-H).
13C NMR (126 MHz, C
6D
6): δ [ppm] = 7.47 (C-5′), 8.29 (C-1′), 29.13 (C-4′), 29.41 (C-2′), 34.03 (C-2″), 53.57 (C-5), 67.03 (C-1″), 82.52 (C-3), 85.62 (C-2), 85.74 (C-4), 109.00 (C-1), 116.43 (C-4″), 116.63 (C-3′), 135.06 (C-3″). HRMS (ESI
+): calcd.: 320.1581 [M+Na]
+, found: 320.1525. IR (ATR):
ν = 2974, 2939, 2882, 2100, 1464, 1360, 1273, 926, 847. optical rotation: [α]
D20 = −47.5 (c = 1.7, CHCl
3). TLC: R
f = 0.49 (9:1,
iso-hexane–Et
2O).
Protected but-3-enyl β-d-5-amino-5-deoxyriboside (21): To a solution of protected but-3-enyl β-d-5-azido-5-deoxyriboside 20 (123 mg, 0.414 mmol) in THF/toluene (1:1, 6 mL), PPh3 (326 mg, 1.24 mmol) and water (373 μL, 20.7 mmol) were added and the mixture was stirred at 50 °C for 13 h. After cooling to rt, di–tert–butyldicarbonate (181 mg, 0.827 mmol) and NaHCO3 (70.0 mg, 0.830 mmol) were added and the mixture was stirred at rt for 1.5 h. It was then diluted with EtOAc (150 mL), washed with water (150 mL) and brine (150 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (9:1, iso-hexane–EtOAc) to give 21 as a colourless oil (150 mg, 98%). 1H NMR (500 MHz, C6D6): δ [ppm] = 0.78 (t, J = 7.5 Hz, 3H, 1′-H), 0.99 (t, J = 7.4 Hz, 3H, 5′-H), 1.41 (q, J = 7.5 Hz, 2H, 2′-H), 1.43 (s, 9H, OC(CH3)3), 1.69 (q, J = 7.4 Hz, 2H, 4′-H), 2.08–2.20 (m, 2H, 2″-H), 3.11 (dt, J = 9.4, 6.5 Hz, 1H, 1″-Ha), 3.20–3.30 (m, 2H, 5-H), 3.53 (dt, J = 9.4, 6.7 Hz, 1H, 1″-Hb), 4.31 (dd, J = 6.0, 6.0 Hz, 1H, 4-H), 4.46 (d, J = 5.9 Hz, 1H, 3-H), 4.53 (d, J = 5.9 Hz, 1H, 2-H), 4.96 (s, 1H, NH), 4.98–5.05 (m, 2H, 4″-H), 5.08 (s, 1H, 1-H), 5.70 (ddt, J = 17.1, 10.3, 6.8 Hz, 1H, 3″-H). 13C NMR (126 MHz, C6D6): δ [ppm] = 7.75 (C-5′), 8.60 (C-1′), 28.49 (OC(CH3)3), 29.38 (C-4′), 29.62 (C-2′), 34.30 (C-2″), 44.18 (C-5), 67.43 (C-1″), 78.88 (OC(CH3)3), 82.79 (C-3), 86.45 (C-2), 86.76 (C-4), 109.32 (C-1), 116.58 (C-3′), 116.79 (C-4″), 135.06 (C-3″), 156.01 (Boc-C=O). HRMS (ESI+): calcd.: 394.2200 [M+Na]+, found: 394.2213. IR (ATR): ν = 2974, 1716, 1699, 1514, 1365, 1248, 1167, 924, 872. optical rotation: [α]D20 = −30.5 (c = 1.9, CHCl3). TLC: Rf = 0.15 (9:1, iso-hexane–Et2O).
Protected 3-oxopropyl β-d-5-amino-5-deoxyriboside (22): Ozone was bubbled through a solution of protected but-3-enyl β-d-5-amino-5-deoxyriboside 21 (106 mg, 0.269 mmol) in CH2Cl2 (3 mL), MeOH (26 mL) and pyridine (87 μL, 1.1 mmol) at −78 °C for 15 min. Nitrogen was then bubble through the solution at −78 °C for 45 min, and dimethyl sulfide (198 μL, 2.69 mmol) was added. The reaction mixture was stirred overnight and slowly warmed to rt during this period. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (7:3, iso-hexane–EtOAc) to give 22 as a colourless oil (87.0 mg, 86%). 1H NMR (500 MHz, C6D6, 70 °C): δ [ppm] = 0.79 (t, J = 7.4 Hz, 3H, 1′-H), 0.96 (t, J = 7.4 Hz, 3H, 5′-H), 1.40–1.48 (m, 2H, 2′-H), 1.43 (s, 9H, OC(CH3)3), 1.68 (q, J = 7.4 Hz, 2H, 4′-H), 2.01–2.13 (m, 2H, 2″-H), 3.12–3.19 (m, 2H, 5-H), 3.23 (ddd, J = 10.1, 6.6, 5.3 Hz, 1H, 1″-Ha), 3.68 (ddd, J = 10.1, 6.8, 5.4 Hz 1H, 1″-Hb), 4.29 (dd, J = 6.4, 6.3 Hz, 1H, 4-H), 4.42 (d, J = 6.1 Hz, 1H, 3-H), 4.45 (d, J = 6.1 Hz, 1H, 2-H), 4.84 (s, 1H, NH), 4.99 (s, 1H, 1-H), 9.33 (dd, J = 1.6, 1.5 Hz, 1H, 3″-H). 13C NMR (126 MHz, C6D6, 70 °C): δ [ppm] = 7.19 (C-5′), 7.89 (C-1′), 28.06 (OC(CH3)3), 29.21 (C-4′), 29.33 (C-2′), 43.20 (C-2″), 44.04 (C-5), 61.30 (C-1″), 78.66 (OC(CH3)3), 82.47 (C-3), 86.01 (C-2), 86.45 (C-4), 109.15 (C-1), 116.42 (C-3′), 155.61 (Boc-C=O), 198.24 (C-3″). HRMS (ESI+): calcd.: 396.1993 [M+Na]+, found: 396.2005. IR (ATR): ν = 1713, 1695, 1518, 1365, 1257, 1167, 1011, 924, 791. optical rotation: [α]D20 = −17.6 (c = 1.7, CHCl3). TLC: Rf = 0.15 (7:3, iso-hexane–EtOAc).
Protected 3-hydroxypropyl β-d-5-amino-5-deoxyriboside (23): Ozone was bubbled through a solution of protected but-3-enyl β-d-5-amino-5-deoxyriboside 21 (70.0 mg, 19.0 μmol) in CH2Cl2 (3 mL), MeOH (26 mL) and pyridine (61 μL, 0.75 mmol) at −78 °C for 30 min. Dimethyl sulfide (138 μL, 1.88 mmol) was then added. The reaction mixture was stirred overnight and slowly warmed to rt during this period. The solvent was evaporated under reduced pressure, and the resultant crude aldehyde 22 was dissolved in MeOH (10 mL). At 0 °C, sodium borohydride (34.0 mg, 1.90 mmol) was added and the mixture was stirred at rt for 30 min. After addition of sat. NH4Cl solution, the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organics were washed with water (2 × 20 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (65:35, iso-hexane-EtOAc) to give 23 as a colourless oil (66.0 mg, 95%). 1H NMR (500 MHz, C6D6): δ [ppm] = 0.80 (t, J = 7.5 Hz, 3H, 1′-H), 0.99 (t, J = 7.5 Hz, 3H, 5′-H), 1.38–1.47 (m, 2H, 2′-H), 1.43 (s, 9H, OC(CH3)3), 1.51–1.59 (m, 2H, 2″-H), 1.69 (q, J = 7.4 Hz, 2H, 4′-H), 3.14 (ddd, J = 9.6, 5.5, 5.5 Hz, 1H, 1″-Ha), 3.19–3.26 (m, 2H, 5-H), 3.44–3.51 (m, 1H, 3″-Ha), 3.51–3.58 (m, 1H, 3″-Hb), 3.72–3.79 (m, 1H, 1″-Hb), 4.32 (dd, J = 6.6, 5.9 Hz, 1H, 4-H), 4.41 (d, J = 6.0 Hz, 1H, 3-H), 4.50 (d, J = 6.0 Hz, 1H, 2-H), 5.05 (s, 1H, NH), 5.10 (s, 1H, 1-H). 13C NMR (126 MHz, C6D6): δ [ppm] = 7.76 (C-5′), 8.60 (C-1′), 28.48 (OC(CH3)3), 29.39 (C-4′), 29.63 (C-2′), 32.60 (C-2″), 44.42 (C-5), 59.43 (C-3″), 64.87 (C-1″), 79.09 (OC(CH3)3), 82.76 (C-3), 86.42 (C-2), 86.65 (C-4), 109.38 (C-1), 116.64 (C-3′), 156.23 (Boc-C=O). HRMS (ESI+): calcd.: 398.2149 [M+Na]+, found: 398.2130. IR (ATR): ν = 2936, 1689, 1365, 1250, 1167, 1088, 1012, 972, 924. optical rotation: [α]D20 = −30.2 (c = 1.7, CHCl3). TLC: Rf = 0.12 (7:3, iso-hexane–EtOAc).
Protected 3-(tosyloxy)propyl β-d-5-amino-5-deoxyriboside (24): To a solution of protected 3-hydroxypropyl β-d-5-amino-5-deoxyriboside 23 (77.0 mg, 0.205 mmol) in CH2Cl2 (5 mL) at 0 °C, pyridine (38.0 μL, 0.472 mmol) and tosyl chloride (50.8 mg, 0.267 mmol) were added. The mixture was slowly warmed to rt and then stirred at rt for 3 d. Water (20 mL) was added, and the aqueous layer was extracted with CH2Cl2 (2 × 30 mL). The combined organics were washed with brine (30 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (4:1, iso-hexane–EtOAc) to give 24 as a colourless oil (86.0 mg, 79%). 1H NMR (500 MHz, C6D6): δ [ppm] = 0.78 (t, J = 7.5 Hz, 3H, 1′-H), 0.97 (t, J = 7.5 Hz, 3H, 5′-H), 1.41 (q, J = 7.5 Hz, 2H, 2′-H), 1.43 (s, 9H, OC(CH3)3), 1.44–1.52 (m, 2H, 2″-H), 1.67 (q, J = 7.5 Hz, 2H, 4′-H), 1.88 (s, 3H, aryl–CH3), 2.92 (ddd, J = 9.8, 6.3, 6.3 Hz, 1H, 1″-Ha), 3.02–3.10 (m, 1H, 3″-Ha), 3.12–3.20 (m, 1H, 3″-Hb), 3.37 (ddd, J = 9.8, 5.7, 5.7 Hz, 1H, 1″-Hb), 3.90–3.99 (m, 2H, 5-H), 4.22 (dd, J = 6.3, 6.3 Hz, 1H, 4-H), 4.33 (d, J = 6.0 Hz, 1H, 3-H), 4.38 (d, J = 6.0 Hz, 1H, 2-H), 4.67 (dd, J = 5.4, 5.4 Hz, 1H, NH), 4.85 (s, 1H, 1-H), 6.77 (d, J = 8.2 Hz, 2H, 3‴-H, 5‴-H), 7.77 (d, J = 8.2 Hz, 2H, 2‴-H, 6‴-H). 13C NMR (126 MHz, C6D6): δ [ppm] = 7.74 (C-5′), 8.60 (C-1′), 21.18 (aryl–CH3), 28.46 (OC(CH3)3), 29.26 (C-2″), 29.38 (C-4′), 29.65 (C-2′), 44.14 (C-5), 63.25 (C-3″), 67.13 (C-1″), 79.04 (OC(CH3)3), 82.70 (C-3), 86.20 (C-2), 86.75 (C-4), 109.05 (C-1), 116.61 (C-3′), 128.20 (C-2‴, C-6‴), 129.87 (C-3‴, C-5‴), 134.49 (C-4‴), 144.24 (C-1‴), 155.94 (NC(=O)O). HRMS (ESI+): calcd.: 552.2238 [M+Na]+, found: 552.2237. IR (ATR): ν = 1713, 1364, 1174, 1095, 924, 837, 814, 752, 663. UV (MeCN): λmax = 225, 262, 273. optical rotation: [α]D20 = −22.8 (c = 2.0, CHCl3). TLC: Rf = 0.18 (4:1, iso-hexane–EtOAc).
Protected 6′
-N-substituted aminoribosylated (6′
S)-nucleosyl amino acid (
26): To a solution of the protected (6′
S)-nucleosyl amino acid
25 (83.0 mg, 0.142 mmol) [
35,
36] in THF (6 mL) over molecular sieves (4 Å), aldehyde
22 (63.0 mg, 0.169 mmol) was added and the mixture was stirred at rt for 24 h. Amberlyst™ 15 (6.6 mg, 31 μmol) and sodium triacetoxyborohydride (60.0 mg, 0.284 mmol) were added, and the mixture was stirred at rt for 22 h. It was then filtered, the filtrate was diluted with EtOAc (100 mL) and washed with sat. Na
2CO
3 solution (100 mL). The aqueous layer was extracted with EtOAc (3 × 50 mL), and the combined organics were dried over Na
2SO
4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (97.5:2.5, CH
2Cl
2-MeOH) to give
26 as a colourless solid (101 mg, 76%).
1H NMR (500 MHz, CD
3OD): δ [ppm] = 0.09 (s, 3H, SiCH
3), 0.10 (s, 3H, SiCH
3), 0.12 (s, 3H, SiCH
3), 0.13 (s, 3H, SiCH
3), 0.87 (t,
J = 7.5 Hz, 3H, 1
iv-H), 0.89 (t,
J = 7.5 Hz, 3H, 5
iv-H), 0.91 (s, 9H, SiC(CH
3)
3), 0.93 (s, 9H, SiC(CH
3)
3), 1.44 (s, 9H, NC(=O)OC(CH
3)
3), 1.49 (s, 9H, C(=O)OC(CH
3)
3), 1.58 (q,
J = 7.5 Hz, 2H, 2
iv-H), 1.67 (q,
J = 7.5 Hz, 2H, 4
iv-H), 1.70–1.82 (m, 2H, 2″-H), 1.86–1.94 (m, 1H, 5′-H
a), 2.00–2.08 (m, 1H, 5′-H
b), 2.58–2.65 (m, 1H, 1″-H
a), 2.65–2.72 (m, 1H, 1″-H
b), 3.17 (d,
J = 7.2 Hz, 2H, 5‴-H), 3.34 (dd,
J = 9.2, 4.7 Hz, 1H, 6′-H), 3.41–3.47 (m, 1H, 3″-H
a), 3.74–3.80 (m, 1H, 3″-H
b), 3.91 (dd,
J = 4.6, 4.4 Hz, 1H, 3′-H), 4.04–4.10 (m, 1H, 4′-H), 4.16 (dd,
J = 7.2, 7.2 Hz, 1H, 4‴-H), 4.33 (dd,
J = 4.4, 4.1 Hz, 1H, 2′-H), 4.60 (d,
J = 6.0 Hz, 1H, 3‴-H), 4.67 (d,
J = 6.0 Hz, 1H, 2‴-H), 5.01 (s, 1H, 1‴-H), 5.75 (d,
J = 8.1 Hz, 1H, 5-H), 5.78 (d,
J = 4.1 Hz, 1H, 1′-H), 7.65 (d,
J = 8.1 Hz, 1H, 6-H).
13C NMR (126 MHz, CD
3OD): δ [ppm] = −4.40 (SiCH
3), –4.39 (SiCH
3), –4.39 (SiCH
3), −3.98 (SiCH
3), 7.75 (C-5
iv), 8.72 (C-1
iv), 18.88 (Si
C(CH
3)
3), 18.94 (Si
C(CH
3)
3, 26.40 (SiC(
CH
3)
3), 26.47 (SiC(
CH
3)
3), 28.47 (NC(=O)OC(
CH
3)
3), 28.79 (C(=O)OC(
CH
3)
3), 29.92 (C-4
iv), 30.31 (C-2
iv), 30.54 (C-2″), 38.00 (C-5′), 44.63 (C-5‴), 46.10 (C-1″), 60.70 (C-6′), 67.13 (C-3″), 75.92 (C-2′), 76.60 (C-3′), 80.28 (NC(=O)O
C(CH
3)
3), 82.57 (C(=O)O
C(CH
3)
3), 82.92 (C-4′), 83.83 (C-3‴), 87.09 (C-2‴), 87.20 (C-4‴), 91.87 (C-1′), 103.04 (C-5), 110.00 (C-1‴), 117.68 (C-3
iv), 142.68 (C-6), 152.15 (C-2), 158.31 (Boc-C=O), 166.01 (C-4), 174.84 (C-7′). HRMS (ESI
+): calcd.: 943.5490 [M+H]
+, found: 943.5486. IR (ATR):
ν = 2931, 2858, 1692, 1561, 1366, 1251, 1157, 1090, 836, 776. UV (MeCN): λ
max = 261. optical rotation: [α]
D20 = −15.0 (c = 1.0, CHCl
3). m.p. = 62 °C. TLC: R
f = 0.30 (94:6, CH
2Cl
2-MeOH).
Protected 6′
-N-substituted aminoribosylated truncated
N-Cbz-muraymycin analogue (
28): To a solution of the protected 6′
-N-substituted aminoribosylated (6′
S)–nucleosyl amino acid
26 (88.0 mg, 93.3 μmol) in THF (6 mL) over molecular sieves (4 Å), aldehyde
27 (33.0 mg, 103 μmol) [
20] was added and the mixture was stirred at rt for 39 h. Amberlyst™ 15 (4.3 mg, 21 μmol) and sodium triacetoxyborohydride (39.3 mg, 187 μmol) were added, and the mixture was stirred at rt for 15 h. More aldehyde
27 (33.0 mg, 103 μmol) and, after 4 h, more sodium triacetoxyborohydride (39.3 mg, 187 μmol) was added. The mixture was stirred at rt for 18 h, then more sodium triacetoxyborohydride (39.3 mg, 187 μmol) was added and stirring at rt was continued for 2 h. The mixture was then filtered, the filtrate was diluted with EtOAc (100 mL) and washed with sat. Na
2CO
3 solution (70 mL). The aqueous layer was extracted with EtOAc (3 × 50 mL), and the combined organics were dried over Na
2SO
4. The solvent was evaporated under reduced pressure, and the resultant crude product was purified by column chromatography (1. 98.2:1.8→98:2, CH
2Cl
2-MeOH, 2. 98:2, CH
2Cl
2-MeOH) to give
28 as a colourless solid (81.6 mg, 70%).
1H NMR (500 MHz, CD
3OD): δ [ppm] = 0.06 (s, 3H, SiCH
3), 0.10 (s, 3H, SiCH
3), 0.13 (s, 3H, SiCH
3), 0.15 (s, 3H, SiCH
3), 0.86 (t,
J = 7.5 Hz, 3H, 1
vii-H), 0.88–0.93 (m, 3H, 5
vii-H), 0.90 (s, 9H, SiC(CH
3)
3), 0.94 (s, 9H, SiC(CH
3)
3), 0.92–0.97 (m, 6H, 5‴-H), 1.44 (s, 9H, NC(=O)OC(CH
3)
3), 1.48 (s, 9H, C(=O)OC(CH
3)
3), 1.51–1.57 (m, 2H, 3‴-H), 1.57 (q,
J = 7.5 Hz, 2H, 2
vii-H), 1.67 (q,
J = 7.5 Hz, 2H, 4
vii-H), 1.62–1.75 (m, 5H, 2″-H, 4‴-H, 2
v-H), 1.77–1.85 (m, 1H, 5′-H
a), 2.17–2.27 (m, 1H, 5′-H
b), 2.52–2.62 (m, 2H, 1″-H
a, 1
v-H
a), 2.64–2.67 (m, 2H, 1″-H
b, 1
v-H
b), 3.13–3.19 (m, 2H, 5
vi-H), 3.22 (dd,
J = 6.7, 6.7 Hz, 2H, 3″-H), 3.40–3.47 (m, 1H, 3
v-H
a), 3.50 (dd,
J = 10.6, 3.5 Hz, 1H, 6′-H), 3.66–3.73 (m, 1H, 3
v-H
b), 3.94 (dd,
J = 4.5, 4.1 Hz, 1H, 3′-H), 3.96–4.02 (m, 1H, 4′-H), 4.12 (dd,
J = 8.9, 7.7 Hz, 1H, 2‴-H), 4.15 (dd,
J = 7.3, 6.8 Hz, 1H, 4
vi-H), 4.40 (dd,
J = 4.8, 4.5 Hz, 1H, 2′-H), 4.59 (d,
J = 6.0 Hz, 1H, 3
vi-H), 4.67 (d,
J = 6.0 Hz, 1H, 2
vi-H), 5.01 (s, 1H, 1
vi-H), 5.07 (d,
J = 12.5 Hz, 1H, 1
iv-H
a), 5.11 (d,
J = 12.5 Hz, 1H, 1
iv-H
b), 5.76 (d,
J = 8.1 Hz, 1H, 5-H), 5.78 (d,
J = 4.8 Hz, 1H, 1′-H), 7.24–7.38 (m, 5H, 3
iv-H, 4
iv-H, 5
iv-H, 6
iv-H, 7
iv-H,), 7.61 (d,
J = 8.1 Hz, 1H, 6-H).
13C NMR (126 MHz, CD
3OD): δ [ppm] = −4.43 (SiCH
3), –4.31 (SiCH
3), –4.23 (SiCH
3), −3.98 (SiCH
3), 7.80 (C-5
vii), 8.74 (C-1
vii), 18.86 (Si
C(CH
3)
3), 19.00 (Si
C(CH
3)
3), 22.06 (C
a-5‴), 23.48 (C
b-5‴), 25.95 (C-4‴), 26.39 (SiC(
CH
3)
3), 26.49 (SiC(
CH
3)
3), 28.68 (C(=O)OC(
CH
3)
3), 28.81 (NC(=O)OC(
CH
3)
3), 29.26 (C-2″), 29.93 (C-4
vii), 30.13 (C-2
v), 30.33 (C-2
vii), 35.38 (C-5′), 38.59 (C-3″), 42.36 (C-3‴), 44.67 (C-5
vi), 49.28 (C-1
v), 49.97 (C-1″), 55.12 (C-2‴), 61.91 (C-6′), 66.82 (C-3
v), 67.73 (C-1
iv), 75.63 (C-2′), 76.96 (C-3′), 80.27 (NC(=O)O
C(CH
3)
3), 82.60 (C(=O)O
C(CH
3)
3), 83.35 (C-4′), 83.88 (C-2
vi), 87.03 (C-3
vi), 87.10 (C-4
vi), 91.76 (C-1′), 103.23 (C-5), 110.00 (C-1
vi), 117.67 (C-3
vii), 128.88, 129.04, 129.49 (C-3
iv, C-4
iv, C-5
iv, C-6
iv C-7
iv), 138.18 (C-2
iv), 142.84 (C-6), 152.15 (C-2), 158.28, 158.34 (Cbz–C=O, Boc-C=O), 165.94 (C-4), 172.89, 175.19 (C-7′, C-1‴). HRMS (ESI
+): calcd.: 1247.7277 [M+H]
+, found: 1247.7252. IR (ATR):
ν = 2929, 2857, 1692, 1366, 1258, 1153, 1087, 1043, 836, 777. UV (MeCN): λ
max = 260. optical rotation: [α]
D20 = −5.0 (c = 1.0, CHCl
3). m.p. = 75 °C. TLC: R
f = 0.26 (96:4, CH
2Cl
2-MeOH).
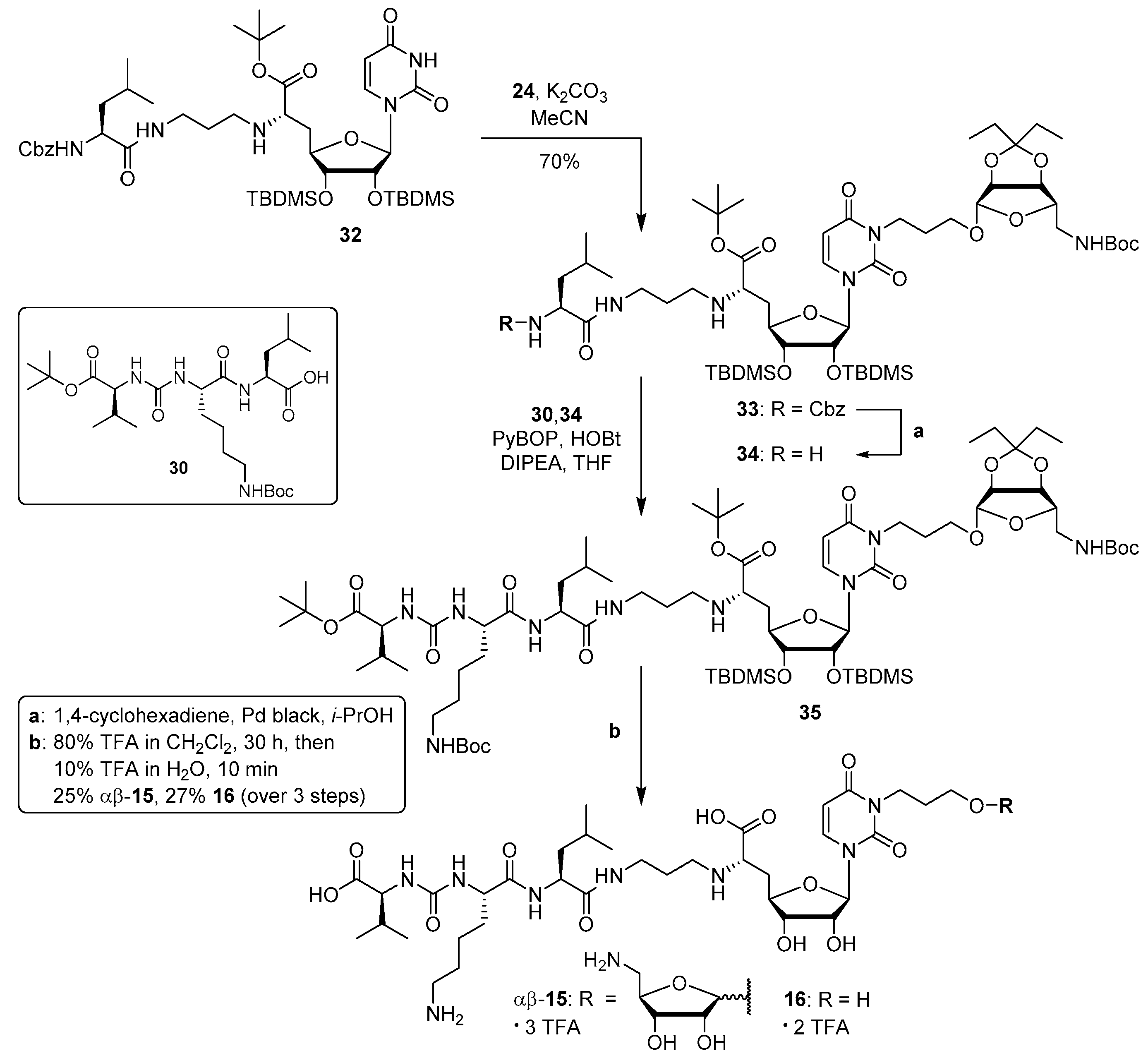
Protected 6′-N-substituted aminoribosylated truncated muraymycin analogue (29): To a solution of the protected 6′-N-substituted aminoribosylated truncated N-Cbz–muraymycin analogue 28 (28.2 mg, 22.6 μmol) in i-PrOH (3 mL), 1,4–cyclohexadiene (21.0 μL, 226 μmol) and Pd black (10.0 mg, 94.0 μmol) were added and the mixture was stirred at rt for 1.5 h. It was then filtered through a syringe filter, and the syringe filter was washed with i-PrOH (4 × 5 mL). The solvent of the combined filtrates was evaporated under reduced pressure to give 29 as a colourless wax (25.2 mg, quant.). 1H NMR (500 MHz, CD3OD): δ [ppm] = 0.07 (s, 3 H, SiCH3), 0.10 (s, 3H, SiCH3), 0.13 (s, 3H, SiCH3), 0.15 (s, 3H, SiCH3), 0.87 (t, J = 7.5 Hz, 3H, 1vi-H), 0.89 (t, J = 7.5 Hz, 3H, 5vi-H), 0.90 (s, 9H, SiC(CH3)3), 0.95 (s, 9H, SiC(CH3)3), 0.95–1.02 (m, 6H, 5‴-H), 1.45 (s, 9H, NC(=O)OC(CH3)3), 1.49 (s, 9H, C(=O)OC(CH3)3), 1.58 (q, J = 7.5 Hz, 2H, 2vi-H), 1.67 (q, J = 7.5 Hz, 2H, 4vi-H), 1.63–1.77 (m, 7H, 2″-H, 3‴-H, 4‴-H, 2iv-H), 1.77–1.85 (m, 1H, 5′-Ha), 2.17–2.26 (m, 1H, 5′-Hb), 2.56–2.65 (m, 2H, 1″-Ha, 1iv-Ha), 2.66–2.78 (m, 2H, 1″-Hb, 1iv-Hb), 3.13–3.26 (m, 3H, 3″-Ha, 5v-H), 3.33–3.41 (m, 1H, 3″-Hb), 3.42–3.49 (m, 1H, 3iv-Ha), 3.51 (dd, J = 10.5, 3.5 Hz, 1H, 6′-H), 3.68–3.75 (m, 1H, 3iv-Hb), 3.76–3.81 (m, 1H, 2‴-H), 3.95 (dd, J = 4.4, 4.1 Hz, 1H, 3′-H), 3.96–4.01 (m, 1H, 4′-H), 4.13–4.19 (m, 1H, 4v-H), 4.43 (dd, J = 4.8, 4.4 Hz, 1H, 2′-H), 4.58–4.62 (m, 1H, 3v-H), 4.68 (d, J = 6.0 Hz, 1H, 2v-H), 4.99–5.02 (m, 1H, 1v-H), 5.76 (d, J = 4.8 Hz, 1H, 1′-H), 5.77 (d, J = 8.0 Hz, 1H, 5-H), 7.60 (d, J = 8.0 Hz, 1H, 6-H). 13C NMR (126 MHz, CD3OD): δ [ppm] = −4.43 (SiCH3), –4.34 (SiCH3), –4.27 (SiCH3), –3.98 (SiCH3), 7.79 (C-5vi), 8.73 (C-1vi), 18.86 (SiC(CH3)3), 19.00 (SiC(CH3)3), 22.29 (Ca-5‴), 23.08 (Cb-5‴), 25.56 (C-4‴), 26.38 (SiC(CH3)3), 26.48 (SiC(CH3)3), 28.67 (C(=O)OC(CH3)3), 28.81 (NC(=O)OC(CH3)3), 29.42 (C-2″), 29.93 (C-4vi), 30.06 (C-2iv), 30.32 (C-2vi), 35.10 (C-5′), 39.07 (C-3″), 41.99 (C-3‴), 44.63 (C-5v), 50.05 (C-1iv), 50.27 (C-1″), 53.25 (C-2‴), 62.04 (C-6′), 66.83 (C-3iv), 75.61 (C-2′), 76.99 (C-3′), 80.30 (NC(=O)OC(CH3)3), 82.69 (C(=O)OC(CH3)3), 83.29 (C-4′), 83.89 (C-2v), 87.04 (C-3v), 87.12 (C-4v), 92.13 (C-1′), 103.15 (C-5), 109.91 (C-1v), 117.71 (C-3vi), 142.95 (C-6), 152.09 (C-2), 158.32 (Boc-C=O), 166.04 (C-4), 170.68, 172.93 (C-7′, C-1‴). HRMS (ESI+): calcd.: 1113.6897 [M+H]+, found: 1113.6909. IR (ATR): ν = 2961, 2929, 1695, 1366, 1258, 1149, 1085, 1014, 867, 797. UV (MeCN): λmax = 260. optical rotation: [α]D20 = −53.8 (c = 1.3, CHCl3). TLC: Rf = 0.10 (95:5, CH2Cl2-MeOH).
Protected uracil
-N-3-substituted aminoribosylated truncated
N-Cbz-muraymycin analogue (
33): To a solution of protected truncated muraymycin analogue
32 (81.0 mg, 91.0 μmol) [
36] in MeCN (10 mL), tosylate
24 (48.2 mg, 91.0 μmol) and K
2CO
3 (13.8 mg, 100 μmol) were added. Over a period of 8 h, the mixture was stirred and slowly heated to reflux, and then it was stirred under reflux for 16 h. After cooling to rt, it was diluted with EtOAc (150 mL) and washed with water (100 mL). The organic layer was dried over Na
2SO
4, and the solvent was evaporated under reduced pressure. The resultant crude product was purified by column chromatography (98:2, CH
2Cl
2-MeOH) to give
33 as a colourless solid (79.1 mg, 70%).
1H NMR (500 MHz, CD
3OD): δ [ppm] = 0.11 (s, 3H, SiCH
3), 0.11 (s, 3H, SiCH
3), 0.11 (s, 3H, SiCH
3), 0.13 (s, 3H, SiCH
3), 0.87–0.92 (m, 3H, 5
vii-H), 0.88 (t,
J = 7.5 Hz, 3H, 1
vii-H), 0.89–0.96 (m, 6H, 5‴-H), 0.91 (s, 9H, SiC(CH
3)
3), 0.93 (s, 9H, SiC(CH
3)
3), 1.44 (s, 9H, NC(=O)OC(CH
3)
3), 1.49 (s, 9H, C(=O)OC(CH
3)
3), 1.51–1.56 (m, 2H, 3‴-H), 1.58 (q,
J = 7.5 Hz, 2H, 2
vii-H), 1.64–1.72 (m, 3H, 2″-H, 4‴-H), 1.67 (q,
J = 7.5 Hz, 2H, 4
vii-H), 1.83–1.90 (m, 2H, 2
v-H), 1.88–1.96 (m, 1H, 5′-H
a), 2.02–2.10 (m, 1H, 5′-H
b), 2.52–2.59 (m, 1H, 1″-H
a), 2.60–2.68 (m, 1H, 1″-H
b), 3.13–3.18 (m, 2H, 5
vi-H), 3.21–3.29 (m, 2H, 3″-H), 3.25 (dd,
J = 9.7, 4.3 Hz, 1H, 6′-H), 3.39–3.46 (m, 1H, 3
v-H
a), 3.72–3.78 (m, 1H, 3
v-H
b), 3.85 (dd,
J = 5.3, 4.3 Hz, 1H, 3′-H), 4.00 (dd,
J = 6.9, 6.9 Hz, 2H, 1
v-H) 4.07–4.13 (m, 2H, 4′-H, 2‴-H), 4.16 (dd,
J = 7.1, 7.1 Hz, 1H, 4
vi-H), 4.31 (dd,
J = 4.3, 3.6 Hz, 1H, 2′-H), 4.55 (d,
J = 6.0 Hz, 1H, 3
vi-H), 4.66 (d,
J = 6.0 Hz, 1H, 2
vi-H), 5.00 (s, 1H, 1
vi-H), 5.05–5.12 (m, 2H, 1
iv-H), 5.80 (d,
J = 3.6 Hz, 1H, 1′-H), 5.84 (d,
J = 8.0 Hz, 1H, 5-H), 7.25–7.38 (m, 5H, C-3
iv, C-4
iv, C-5
iv, C-6
iv, C-7
iv), 7.64 (d,
J = 8.1 Hz, 1H, 6-H).
13C NMR (126 MHz, CD
3OD): δ [ppm] = −4.47 (SiCH
3), –4.41 (SiCH
3), –4.17 (SiCH
3), –3.93 (SiCH
3), 7.77 (C-5
vii), 8.81 (C-1
vii), 18.91 (Si
C(CH
3)
3), 18.93 (Si
C(CH
3)
3), 22.01 (C
a-5‴), 23.44 (C
b-5‴), 25.94 (C-4‴), 26.47 (SiC(
CH
3)
3), 26.48 (SiC(
CH
3)
3), 28.47 (C(=O)OC(
CH
3)
3), 28.67 (C-2
v), 28.78 (NC(=O)OC(
CH
3)
3), 29.91 (C-4
vii), 30.35 (C-2
vii), 30.73 (C-2″), 37.95 (C-5′), 38.20 (C-3″), 40.04 (C-1
v), 42.28 (C-3‴), 44.62 (C-5
vi), 46.09 (C-1″), 55.23 (C-2‴), 60.75 (C-6′), 67.11 (C-3
v), 67.72 (C-1
iv), 75.21 (C-2′), 76.52 (C-3′), 80.27 (NC(=O)O
C(CH
3)
3), 82.24 (C-4′), 83.03 (C(=O)O
C(CH
3)
3), 83.84 (C-2
vi), 87.05 (C-3
vi), 87.15 (C-4
vi), 92.75 (C-1′), 102.41 (C-5), 110.14 (C-1
vi), 117.64 (C-3
vii), 128.88, 129.04, 129.48 (C-3
iv, C-4
iv, C-5
iv, C-6
iv, C-7
iv), 138.18 (C-2
iv), 140.52 (C-6), 152.33 (C-2), 158.32, 158.35 (Boc-C=O, Cbz–C=O), 164.83 (C-4), 174.56, 175.49 (C-7′, C-1‴). HRMS (ESI
+): calcd.: 1247.7277 [M+H]
+, found: 1247.7292. IR (ATR):
ν = 2959, 2930, 2857, 1708, 1663, 1258, 1156, 1091, 802, 778. UV (MeCN): λ
max = 262. optical rotation: [α]
D20 = −4.5 (c = 1.1, CHCl
3). m.p. = 66 °C. TLC: R
f = 0.13 (96:4, CH
2Cl
2-MeOH).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




