Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents

 , , ,
, , , 
Abstract
:
1. Introduction
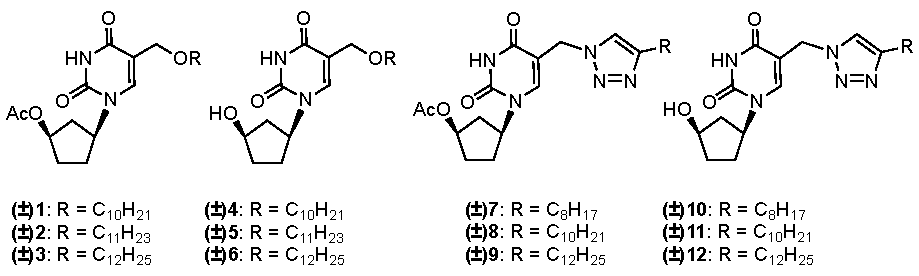
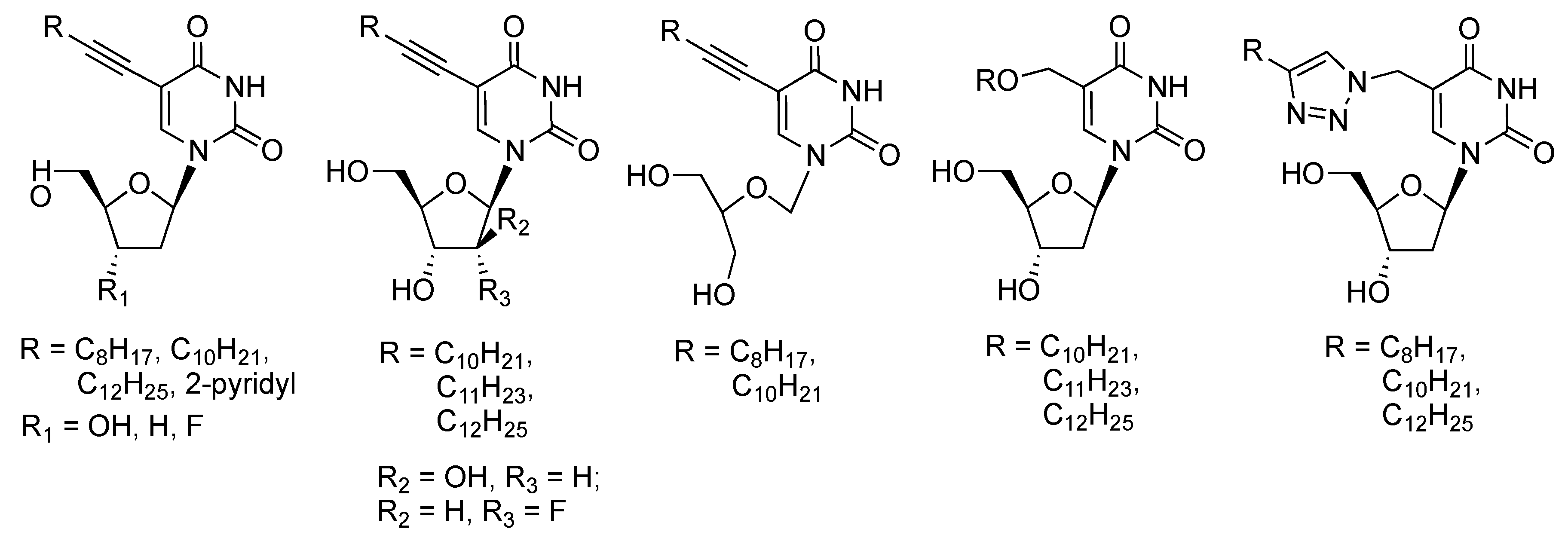
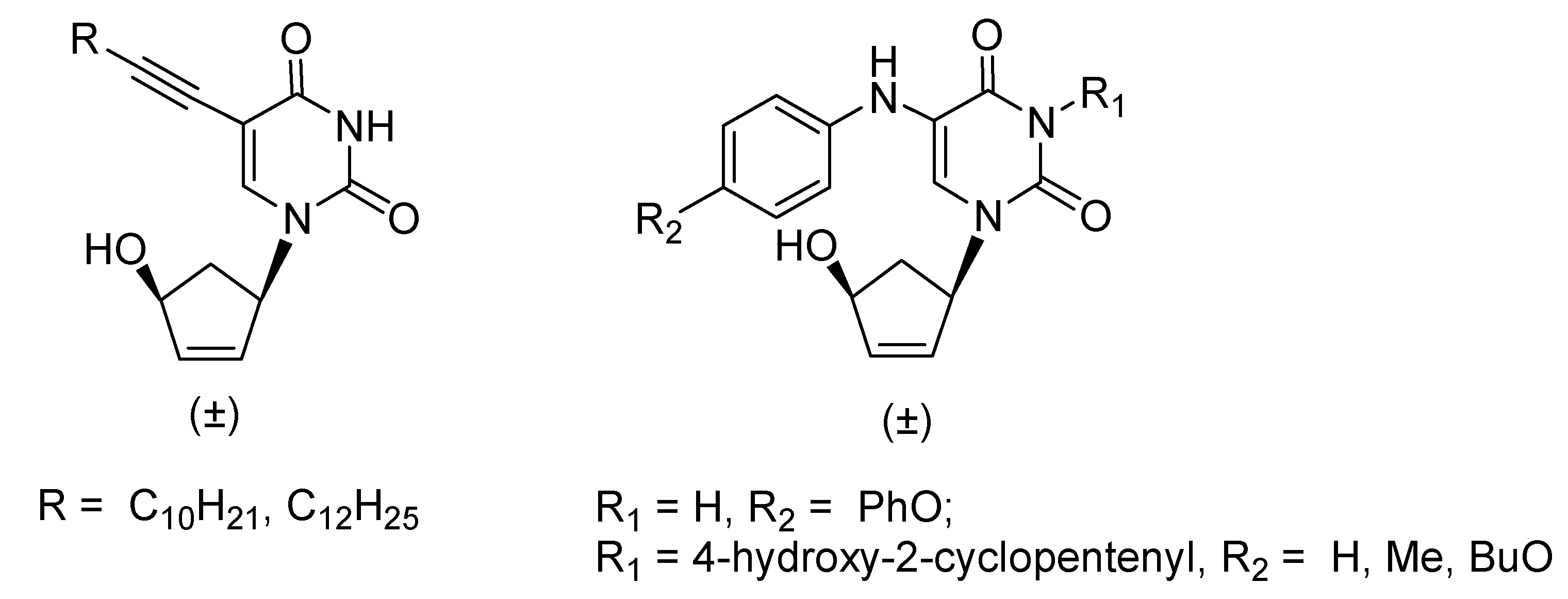
2. Results and Discussion
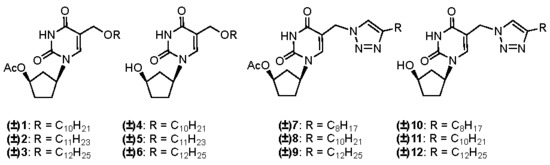
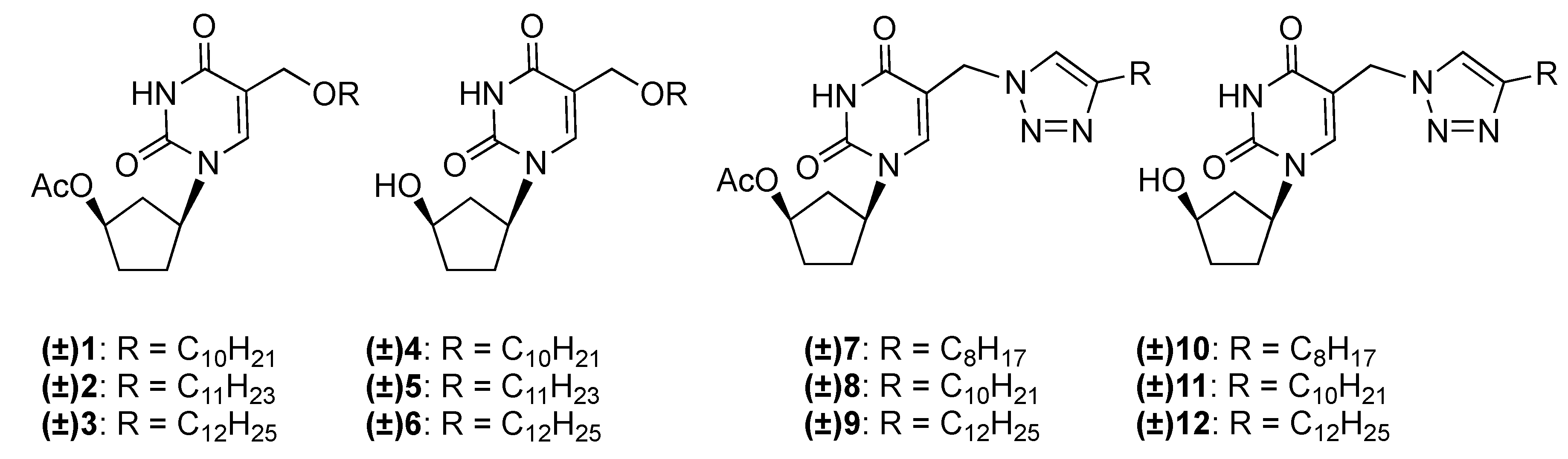
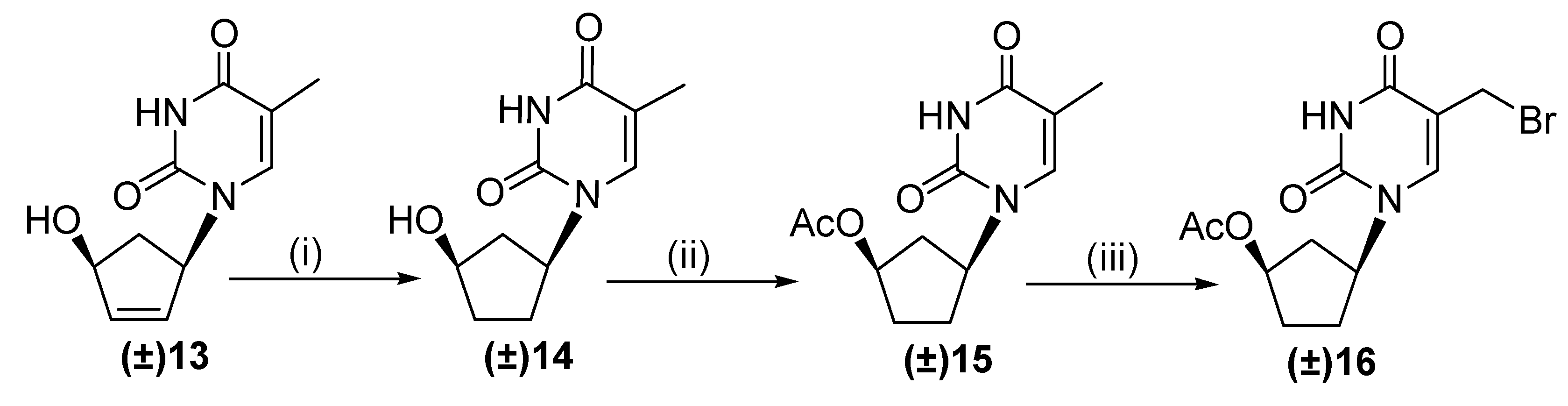
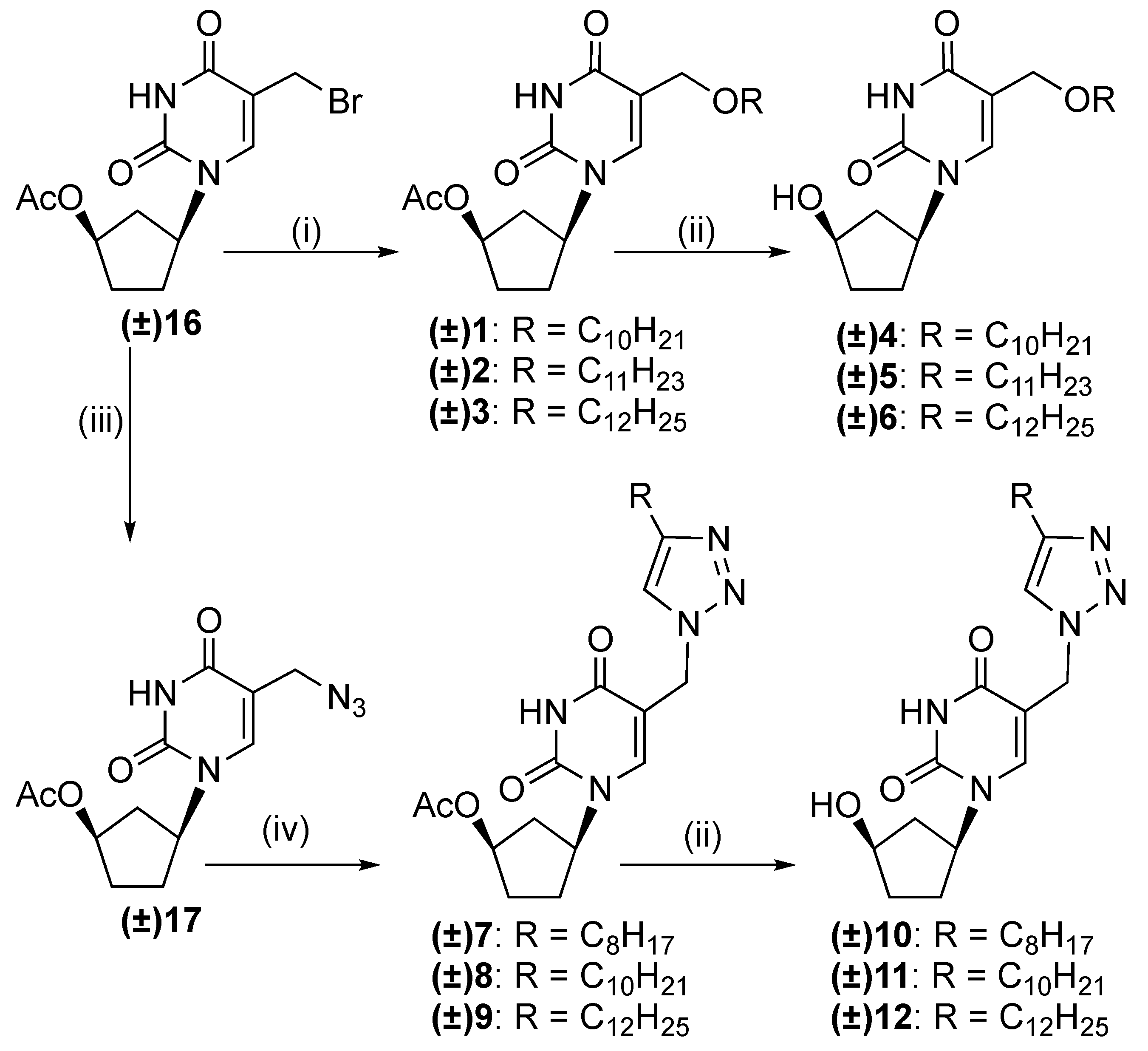
2.1. Chemistry
2.2. Biological Evaluation and Structure-Activity Relationship Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of (±)-1-(4′-Acetoxycyclopent-1′-yl)-5-alkyloxy-methyluracils 1–3
3.1.2. General Procedure for the Synthesis of (±)-1-(4′-Acetoxycyclopent-1′-yl)-5-[(4-alkyl)-1,2,3-triazol-1-yl]methyluracils 7–9
3.1.3. General Procedure for the Synthesis of (±)-1-(4′-Hydroxycyclopent-1′-yl)-5-alkyloxy-methyluracils 4–6 and (±)-1-(4′-hydroxycyclopent-1′-yl)-5-[(4-alkyl)-1,2,3-triazol-1-yl]methyl-uracils 10–12
3.2. Antibacterial Experiments
3.2.1. Bacterial Strains
3.2.2. U-937 Cells
3.2.3. Determination of Minimal Inhibitory Concentration (MIC) against M. tuberculosis ATCC 25177
3.2.4. Determination of MIC against M. bovis ATCC 35737
3.2.5. Antituberculosis Tests with Virulent Strains of M. tuberculosis
3.2.6. Antibacterial Effects
3.2.7. Evaluation of U-937 Cytotoxicity
3.2.8. Electron Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ventola, C.L. The antibiotic resistance crisis: Part 2: Management strategies and new agents. P & T 2015, 40, 344–352. [Google Scholar] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. P & T 2015, 40, 277–283. [Google Scholar] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, D.; Johar, M.; Manning, T.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Design and studies of novel 5-substituted alkynylpyrimidine nucleosides as potent inhibitors of mycobacteria. J. Med. Chem. 2005, 48, 7012–7017. [Google Scholar] [CrossRef] [PubMed]
- Shmalenyuk, E.R.; Chernousova, L.N.; Karpenko, I.L.; Kochetkov, S.N.; Smirnova, T.G.; Andreevskaya, S.N.; Chizhov, A.O.; Efremenkova, O.V.; Alexandrova, L.A. Inhibition of Mycobacterium tuberculosis strains H37Rv and MDR MS-115 by a new set of C5 modified pyrimidine nucleosides. Bioorg. Med. Chem. 2013, 21, 4874–4884. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, B.P.; Wilson, D.J.; Nelson, K.M.; Boshoff, H.I.; Barry, C.E., III; Aldrich, C.C. Development of a selective activity-based probe for adenylating enzymes: Profiling MbtA Involved in siderophore biosynthesis from Mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Van Calenbergh, S.; Pochet, S.; Munier-Lehmann, H. Drug design and identification of potent leads against mycobacterium tuberculosis thymidine monophosphate kinase. Curr. Top Med. Chem. 2012, 12, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, V.; Serpi, M. Nucleoside analogs and tuberculosis: New weapons against an old enemy. Future Med. Chem. 2015, 7, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Dawadi, S.; Boshoff, H.I.M.; Park, S.W.; Schnappinger, D.; Aldrich, C.C. Conformationally Constrained Cinnolinone Nucleoside Analogues as Siderophore Biosynthesis Inhibitors for Tuberculosis. ACS Med. Chem. Lett. 2018, 9, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Bockman, M.R.; Engelhart, C.A.; Dawadi, S.; Larson, P.; Tiwari, D.; Ferguson, D.M.; Schnappinger, D.; Aldrich, C.C. Avoiding Antibiotic Inactivation in Mycobacterium tuberculosis by Rv3406 through Strategic Nucleoside Modification. ACS Infect. Dis. 2018, 4, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Krajczyk, A.; Zeidler, J.; Januszczyk, P.; Dawadi, S.; Boshoff, H.I.; Barry, C.E., III; Ostrowski, T.; Aldrich, C.C. 2-Aryl-8-aza-3-deazaadenosine analogues of 5′-O-[N-(salicyl)sulfamoyl]adenosine: Nucleoside antibiotics that block siderophore biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2016, 24, 3133–3143. [Google Scholar] [CrossRef] [PubMed]
- Dawadi, S.; Kawamura, S.; Rubenstein, A.; Remmel, R.; Aldrich, C.C. Synthesis and pharmacological evaluation of nucleoside prodrugs designed to target siderophore biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2016, 24, 1314–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockman, M.R.; Kalinda, A.S.; Petrelli, R.; De la Mora-Rey, T.; Tiwari, D.; Liu, F.; Dawadi, S.; Nandakumar, M.; Rhee, K.Y.; Schnappinger, D.; et al. Targeting Mycobacterium tuberculosis Biotin Protein Ligase (MtBPL) with Nucleoside-Based Bisubstrate Adenylation Inhibitors. J. Med. Chem. 2015, 58, 7349–7369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somu, R.V.; Boshoff, H.; Qiao, C.; Bennett, E.M.; Barry, C.E., 3rd; Aldrich, C.C. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Shakya, N.; Srivastav, N.C.; Bhavanam, S.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Discovery of novel 5-(ethyl or hydroxymethyl) analogs of 2′-‘up’ fluoro (or hydroxyl) pyrimidine nucleosides as a new class of Mycobacterium tuberculosis, Mycobacterium bovis and Mycobacterium avium inhibitors. Bioorg. Med. Chem. 2012, 20, 4088–4097. [Google Scholar] [CrossRef] [PubMed]
- Shakya, N.; Srivastav, N.C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. 3′-bromo analogues of pyrimidine nucleosides as a new class of potent inhibitors of Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Johar, M.; Manning, T.; Tse, C.; Desroches, N.; Agrawal, B.; Kunimoto, D.Y.; Kumar, R. Growth inhibition of Mycobacterium bovis, Mycobacterium tuberculosis and Mycobacterium avium in vitro: Effect of 1-beta-D-2′-arabinofuranosyl and 1-(2′-deoxy-2′-fluoro-beta-D-2′-ribofuranosyl) pyrimidine nucleoside analogs. J. Med. Chem. 2007, 50, 3696–3705. [Google Scholar] [CrossRef] [PubMed]
- Shmalenyuk, E.R.; Kochetkov, S.N.; Alexandrova, L.A. Novel inhibitors of Mycobacterium tuberculosis growth based on modified pyrimidine nucleosides and their analogues. Russ. Chem. Rev. 2013, 82, 896–915. [Google Scholar] [CrossRef]
- Kusaka, T.; Yamamoto, H.; Shibata, M.; Muroi, M.; Kishi, T. Streptomyces citricolor nov. sp. and a new antibiotic, aristeromycin. J. Antibiot. (Tokyo) 1968, 21, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Yaginuma, S.; Muto, N.; Tsujino, M. Structures of neplanocins, new antitumor antibiotics. Nucleic Acids Symp. Ser. 1980, 8, s65–s67. [Google Scholar]
- Hayashi, M.; Yaginuma, S.; Yoshioka, H.; Nakatsu, K. Studies on neplanocin A, new antitumor antibiotic. II. Structure determination. J. Antibiot. (Tokyo) 1981, 34, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, S.; Muto, N.; Tsujino, M.; Sudate, Y.; Hayashi, M.; Otani, M. Studies on neplanocin A, new antitumor antibiotic. I. Producing organism, isolation and characterization. J. Antibiot. (Tokyo) 1981, 34, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hergueta, A.R.; Fernandez, F.; Lopez, C.; Balzarini, J.; De Clercq, E. Novel carbocyclic nucleosides containing a cyclobutyl ring: Adenosine analogues. Chem. Pharm. Bull. (Tokyo) 2001, 49, 1174–1177. [Google Scholar] [CrossRef] [PubMed]
- Isabel Nieto, M.; Manuel Blanco, J.; Caamaño, O.; Fernández, F.; Garcia-Mera, X.; López, C.; Balzarini, J.; De Clercq, E. Synthesis and antiviral activity of carbocyclic nucleosides incorporating a modified cyclopentane ring. Part 3: Adenosine and uridine analogues. Nucleosides Nucleotides 1999, 18, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Nieto, I.; Figueira, M.J.; Blanco, J.M.; Caamano, O.; Fernandez, F.; De Clercq, E.; Balzarini, J. Synthesis of novel carbocyclic nucleosides with a modified cyclopentane ring and evaluation of their antiviral activity. Nucleosides Nucleotides 1999, 18, 641–642. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.I.; Caamano, O.; Fernandez, F.; Gomez, M.; Balzarini, J.; De Clercq, E. Synthesis, antiviral and cytostatic activities, of carbocyclic nucleosides incorporating a modified cyclopentane ring. IV. Adenosine and uridine analogues. Nucleosides Nucleotides Nucleic Acids 2002, 21, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.S.; Khandazhinskaya, A.L.; Kochetkov, S.N. Carbocyclic nucleoside analogues: Classification, target enzymes, mechanisms of action and synthesis. Russ. Chem. Rev. 2012, 81, 729–746. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Andreevskaya, S.N.; Smirnova, T.G.; Khandazhinskaya, A.L. Carbocyclic analogues of inosine-5′-monophosphate: Synthesis and biological activity. Acta Naturae 2012, 4, 73–77. [Google Scholar] [PubMed]
- Alexandrova, L.; Zicari, S.; Matyugina, E.; Khandazhinskaya, A.; Smirnova, T.; Andreevskaya, S.; Chernousova, L.; Vanpouille, C.; Kochetkov, S.; Margolis, L. Dual-targeted anti-TB/anti-HIV heterodimers. Antiviral Res. 2017, 145, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Khandazhinskaya, A.L.; Shirokova, E.A.; Skoblov, Y.S.; Victorova, L.S.; Goryunova, L.Y.; Beabealashvilli, R.S.; Pronyaeva, T.R.; Fedyuk, N.V.; Zolin, V.V.; Pokrovsky, A.G.; et al. Carbocyclic dinucleoside polyphosphonates: Interaction with HIV reverse transcriptase and antiviral activity. J. Med. Chem. 2002, 45, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.S.; Valuev-Elliston, V.T.; Babkov, D.A.; Novikov, M.S.; Ivanov, A.V.; Kochetkov, S.N.; Balzarini, J.; Seley-Radtke, K.L.; Khandazhinskaya, A.L. 5′-Nor carbocyclic nucleosides: Surprising nonnucleoside inhibitors of HIV-1 reverse transcriptase. Med. Chem. Comm. 2013, 4, 741–748. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Valuev-Elliston, V.T.; Geisman, A.N.; Novikov, M.S.; Chizhov, A.O.; Kochetkov, S.N.; Seley-Radtke, K.L.; Khandazhinskaya, A.L. Structure-activity evaluation of new uracil-based non-nucleoside inhibitors of HIV reverse transcriptase. Med. Chem. Comm. 2013, 4, 1443–1451. [Google Scholar] [CrossRef]
- Novikov, M.S.; Valuev-Elliston, V.T.; Babkov, D.A.; Paramonova, M.P.; Ivanov, A.V.; Gavryushov, S.A.; Khandazhinskaya, A.L.; Kochetkov, S.N.; Pannecouque, C.; Andrei, G.; et al. N1,N3-disubstituted uracils as nonnucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. 2013, 21, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T. S-Adenosyl-L-Methionine-Dependent Macromolecule Methyltransferases: Potential Targets for the Design of Chemotherapeutic Agents. J. Med. Chem. 1980, 23, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, R.T.; Creveling, C.R.; Ueland, P.M. Biological Methylation and Drug Design; Humana Press: Clifton, NJ, USA, 1986. [Google Scholar]
- Yuan, C.S.; Liu, S.; Wnuk, S.F.; Robins, M.J.; Borchardt, R.T. Design and synthesis of S-adenosylhomocysteine hydrolase inhibitors as broad-spectrum antiviral agents. In Advances in Antiviral Drug Design; De Clercq, E., Ed.; JAI Press Inc.: Greenwich, CT, USA; London, UK, 1996; Volume 2, pp. 41–88. [Google Scholar]
- Seley, K.L.; Schneller, S.W.; Rattendi, D.; Bacchi, C.J. (+)-7-Deaza-5′-noraristeromycin as an anti-trypanosomal agent. J. Med. Chem. 1997, 40, 622–624. [Google Scholar] [CrossRef] [PubMed]
- Seley, K.L.; Schneller, S.W.; Rattendi, D.; Lane, S.; Bacchi, C.J. Synthesis and antitrypanosomal activities of a series of 7-deaza-5′-noraristeromycin derivatives with variations in the cyclopentyl ring substituents. Antimicrob. Agents Chemother. 1997, 41, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Schneller, S.W.; Seley, K.L.; Hegde, V.R.; Rajappan, V.P. 5′-Norcarbanucleosides in L-Like Configurations. In Recent Advances in Nucleosides: Chemistry and Chemotherapy; Chu, C.K., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2002; pp. 291–297. [Google Scholar]
- Seley, K.L.; Schneller, S.W.; De Clercq, E.; Rattendi, D.; Lane, S.; Bacchi, C.J.; Korba, B. The importance of the 4′-hydroxyl hydrogen for the anti-trypanosomal and antiviral properties of (+)-5′-noraristeromycin and two 7-deaza analogues. Bioorg. Med. Chem. 1998, 6, 797–801. [Google Scholar] [CrossRef]
- Seley, K.L.; Schneller, S.W.; Korba, B. Does the anti-hepatitis B virus activity of (+)-5′-noraristeromycin exist in its 4′-epimer and 4′-deoxygenated derivatives? J. Med. Chem. 1998, 41, 2168–2170. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.D.; Koga, M.; Schneller, S.W.; Snoeck, R.; De Clercq, E. (+-)-carbocyclic 5′-nor-2′-deoxyguanosine and related purine derivatives: Synthesis and antiviral properties. J. Med. Chem. 1992, 35, 2191–2195. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.D.; Schneller, S.W.; Hosoya, M.; Snoeck, R.; Andrei, G.; Balzarini, J.; De Clercq, E. Synthesis and antiviral properties of (+/-)-5′-noraristeromycin and related purine carbocyclic nucleosides. A new lead for anti-human cytomegalovirus agent design. J. Med. Chem. 1992, 35, 3372–3377. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.R.; Seley, K.L.; Schneller, S.W. Carbocyclic 5′-noruridine. Nucleosides Nucleotides Nucleic Acids 2000, 19, 269–273. [Google Scholar]
- Hegde, V.R.; Seley, K.L.; Schneller, S.W.; Elder, T.J. 5′-Amino-5′-deoxy-5′-noraristeromycin. J. Org. Chem. 1998, 63, 7092–7094. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.M.; Chen, X.; Schneller, S.W.; Ikeda, S.; Snoeck, R.; Andrei, G.; Balzarini, J.; De Clercq, E. Antiviral enantiomeric preference for 5′-noraristeromycin. J. Med. Chem. 1994, 37, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.; Khandazhinskaya, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Chizhov, A.; Karpenko, I.; Kochetkov, S.; Alexandrova, L. The synthesis and antituberculosis activity of 5′-nor carbocyclic uracil derivatives. Bioorg. Med. Chem. 2012, 20, 6680–6686. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.; Novikov, M.; Babkov, D.; Ozerov, A.; Chernousova, L.; Andreevskaya, S.; Smirnova, T.; Karpenko, I.; Chizhov, A.; Murthu, P.; et al. 5-Arylaminouracil Derivatives: New Inhibitors of Mycobacterium tuberculosis. Chem. Biol. Drug Des. 2015, 86, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Kuo, G.-H.; Benneche, T. A Transition-metal Controlled Synthesis of (+/-)-Aristeromycin and (+/-)-2′,3′-diepe-Aristeromycin. An Unusual Directive Effect in Hydroxylations. J. Am. Chem. Soc. 1988, 110, 621–622. [Google Scholar] [CrossRef]
- Barwolff, D.; Langer, P. “2′-deoxy-5(hydroxymethy1)uridine and 2′-deoxy-5-formyluridine selective bromination of the 5-methyl group of 5-methylpyrimidine nucleosides”. In Nucleic Acid Chemistry; Townsend, L.B., Tipson, R.S., Eds.; Wiley: New York, NY, USA, 1978; Volume 1, pp. 359–366. [Google Scholar]
- Levina, A.S.; Tabatadse, D.R.; Khalimskaya, L.M.; Prichodko, T.A.; Shishkin, G.V.; Alexandrova, L.A.; Zarytova, V.P. Oligonucleotide derivatives bearing reactive and stabilizing groups attached to C5 of deoxyuridine. Bioconjug. Chem. 1993, 4, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Park, S.R.; Jeon, H.B.; Kim, R.S. A new solvent system for efficient synthesis of 1,2,3-triazoles. Tetrahedron Lett. 2006, 47, 5105–5109. [Google Scholar] [CrossRef]
- Shakya, N.; Garg, G.; Agrawal, B.; Kumar, R. Chemotherapeutic interventions against tuberculosis. Pharmaceuticals (Basel) 2012, 5, 690–718. [Google Scholar] [CrossRef] [PubMed]
- Sadders, B.; Stoker, N.C. (Eds.) Mycobacteria: Biology; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Cook, G.M.; Berney, M.; Gebhard, S.; Heinemann, M.; Cox, R.A.; Danilchanka, O.; Niederweis, M. Physiology of mycobacteria. Adv. Microb. Physiol. 2009, 55, 81–182. [Google Scholar] [PubMed]
- Zhang, Y.; Yew, W.W. Mechanisms of drug resistance in Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2009, 13, 1320–1330. [Google Scholar] [PubMed]
- Taneja, N.K.; Tyagi, J.S. Resazurin reduction assays for screening of anti-tubercular compounds against dormant and actively growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2007, 60, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomino, J.C.; Martin, A.; Camacho, M.; Guerra, H.; Swings, J.; Portaels, F. Resazurin microtiter assay plate: Simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Palomino, J.C. Drug susceptibility testing for Mycobacterium tuberculosis. In Procedure Manual; Available online: http://citeseerx.ist.psu.edu/viewdoc/download;jsessionid=69E596855D1D287F85065BF8EA086DBA?doi=10.1.1.663.5920&rep=rep1&type=pdf (accessed on 30 September 2018).
- Systems, T.D. Sensititre. Broth Microdilution (MIC) Method for Rapidly Growing Mycobacteria (RGM), Slowly Growing Nontuberculosis Mycobacteria, Nocardia and other Aerobic Acitomycetes. Available online: http://www.trekds.com/techDocs/techInsert/010-NONTBMYCO_US_GB_V2.8_-_CID8643.pdf (accessed on 30 September 2018).
- Andreevskaya, S.N.; Chernousova, L.N.; Smirnova, T.G.; Larinova, E.E.; Kuzmin, A.V. Examining the ex vivo growth in the macrophages of Mycobacterium Tuberculosis of various genotypic clusters. Probl. Tuberk. Bolezn. Legk. 2006, 12, 43–48. [Google Scholar]
- Siddiqui, S.H.; Rusch-Gerdes, S. MGIT Procedure Manual; Foundation for Innovative New Diagnostics: Geneva, Switzerland, 2006. [Google Scholar]
- Engelkirk, P.; Duben-Engelkirk, J. Laboratory Diagnosis of Infectious Diseases; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008; pp. 168–169. [Google Scholar]
- Popenko, V.I.; Potekhin, A.A.; Karajan, B.P.; Skarlato, S.O.; Leonova, O.G. The size of DNA molecules and chromatin organization in the macronucleus of the ciliate Didinium nasutum (Ciliophora). J. Eukaryot Microbiol. 2015, 62, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Miller, O.L., Jr.; Beatty, B.R. Visualization of nucleolar genes. Science 1969, 164, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.J. Negative Staining of Thinly Spread Biological Particulates. In Electron Microscopy Methods and Protocols. Methods in Molecular Biology; Kuo, J., Ed.; Springer: Berlin, Germany, 1999. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | M. tuberculosis ATCC 25177 MIC99 a (μg/mL) | M. bovis ATCC 35737 MIC99 a (μg/mL) | M. smegmatis mc2155 MIC99 a (μg/mL) | M. smegmatis VKPM Ac 1339 MIC99 a (μg/mL) |
|---|---|---|---|---|
| (±) 1 | 409 | 409 | >100 | >100 |
| (±) 2 | 423 | 423 | >100 | >100 |
| (±) 3 | 54 | 54 | 67 | 67 |
| (±) 4 | 183 | 366 | >100 | >100 |
| (±) 5 | 191 | 381 | >100 | >100 |
| (±) 6 | 198 | >395 | 67 | 67 |
| (±) 7 | 54 | 54 | >100 | >100 |
| (±) 8 | 29 | 57 | 67 | 6.7 |
| (±) 9 | 61 | 488 | >100 | >100 |
| (±) 10 | 98 | 195 | >100 | >100 |
| (±) 11 | 53 | 105 | 67 | 6.7 |
| (±) 12 | 56 | 446 | >100 | >100 |
| Rifampicin | <0.2 | <0.2 | >100 | 10 |
| Streptomycin | 0.4 | 0.4 | - | - |
| Kanamycin | 1.6 | 1.6 | - | - |
| Ciprofloxacin | - | - | 1 | 2 |
| Amikacin | - | - | <3.8 | <3.8 |
| Isoniazid | - | - | 64 | 1 |
| Compound | M. tuberculosis H37Rv MIC99 (μg/mL) | M. tuberculosis MS-115 MIC99 (μg/mL) | U-937 TC50 (μg/mL) |
|---|---|---|---|
| (±) 3 | 20 | 20 | 20.6 |
| (±) 7 | 50 | 20 | 23.7 |
| (±) 8 | 50 | 50 | 15.1 |
| Rifampicin | 1 | >50 | >500 |
| Isoniazid | 0.1 | >100 | - |
| Levofloxacin | 1.5 | 1.5 | - |
| Nonoxynol-9 | - | - | 5.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khandazhinskaya, A.L.; Alexandrova, L.A.; Matyugina, E.S.; Solyev, P.N.; Efremenkova, O.V.; Buckheit, K.W.; Wilkinson, M.; Buckheit, R.W., Jr.; Chernousova, L.N.; Smirnova, T.G.; et al. Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules 2018, 23, 3069. https://doi.org/10.3390/molecules23123069
Khandazhinskaya AL, Alexandrova LA, Matyugina ES, Solyev PN, Efremenkova OV, Buckheit KW, Wilkinson M, Buckheit RW Jr., Chernousova LN, Smirnova TG, et al. Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules. 2018; 23(12):3069. https://doi.org/10.3390/molecules23123069
Chicago/Turabian StyleKhandazhinskaya, Anastasia L., Liudmila A. Alexandrova, Elena S. Matyugina, Pavel N. Solyev, Olga V. Efremenkova, Karen W. Buckheit, Maggie Wilkinson, Robert W. Buckheit, Jr., Larisa N. Chernousova, Tatiana G. Smirnova, and et al. 2018. "Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents" Molecules 23, no. 12: 3069. https://doi.org/10.3390/molecules23123069
APA StyleKhandazhinskaya, A. L., Alexandrova, L. A., Matyugina, E. S., Solyev, P. N., Efremenkova, O. V., Buckheit, K. W., Wilkinson, M., Buckheit, R. W., Jr., Chernousova, L. N., Smirnova, T. G., Andreevskaya, S. N., Leonova, O. G., Popenko, V. I., Kochetkov, S. N., & Seley-Radtke, K. L. (2018). Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules, 23(12), 3069. https://doi.org/10.3390/molecules23123069