Structure–Activity Relationship of Xanthones as Inhibitors of Xanthine Oxidase

Abstract
:1. Introduction
2. Results and Discussion
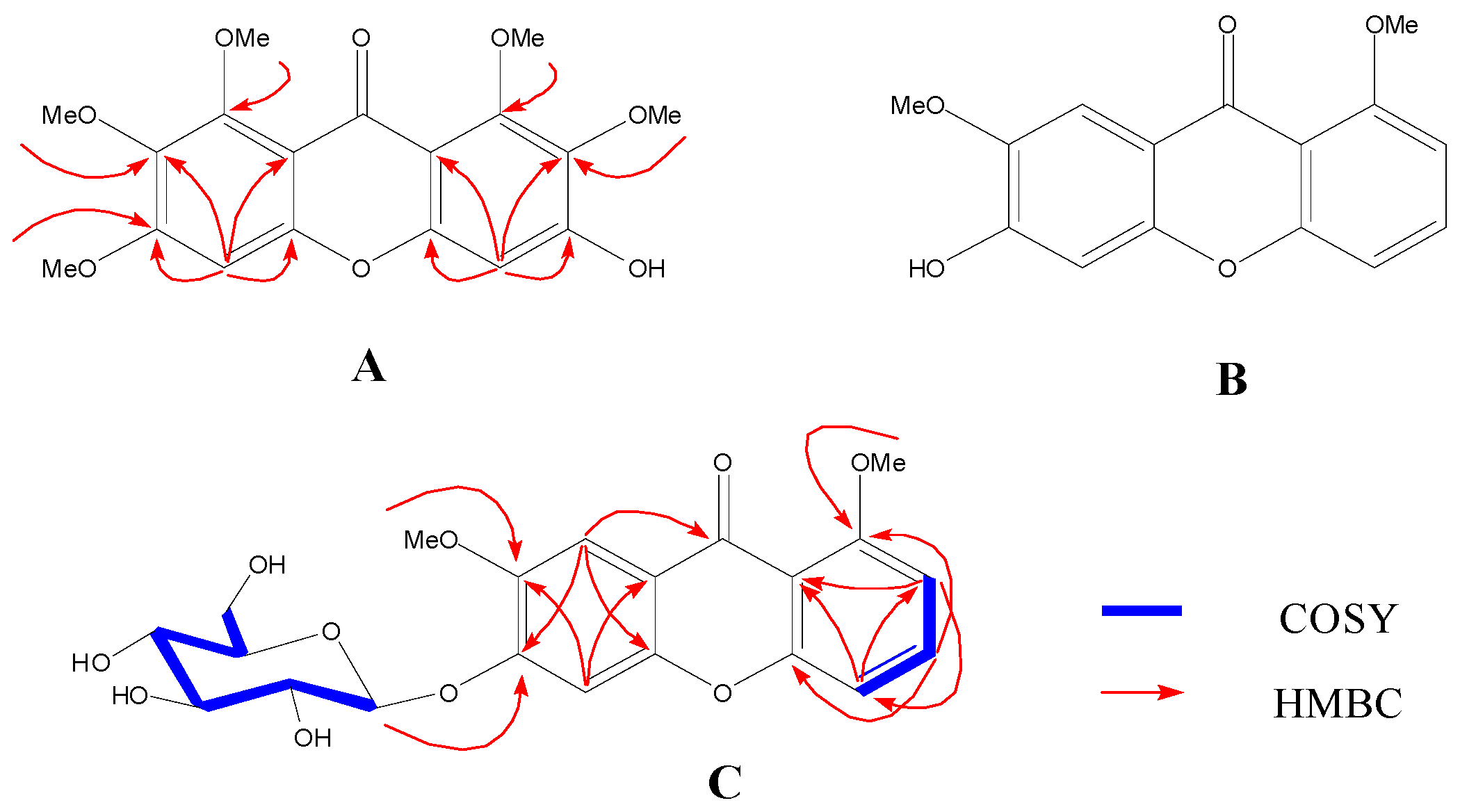
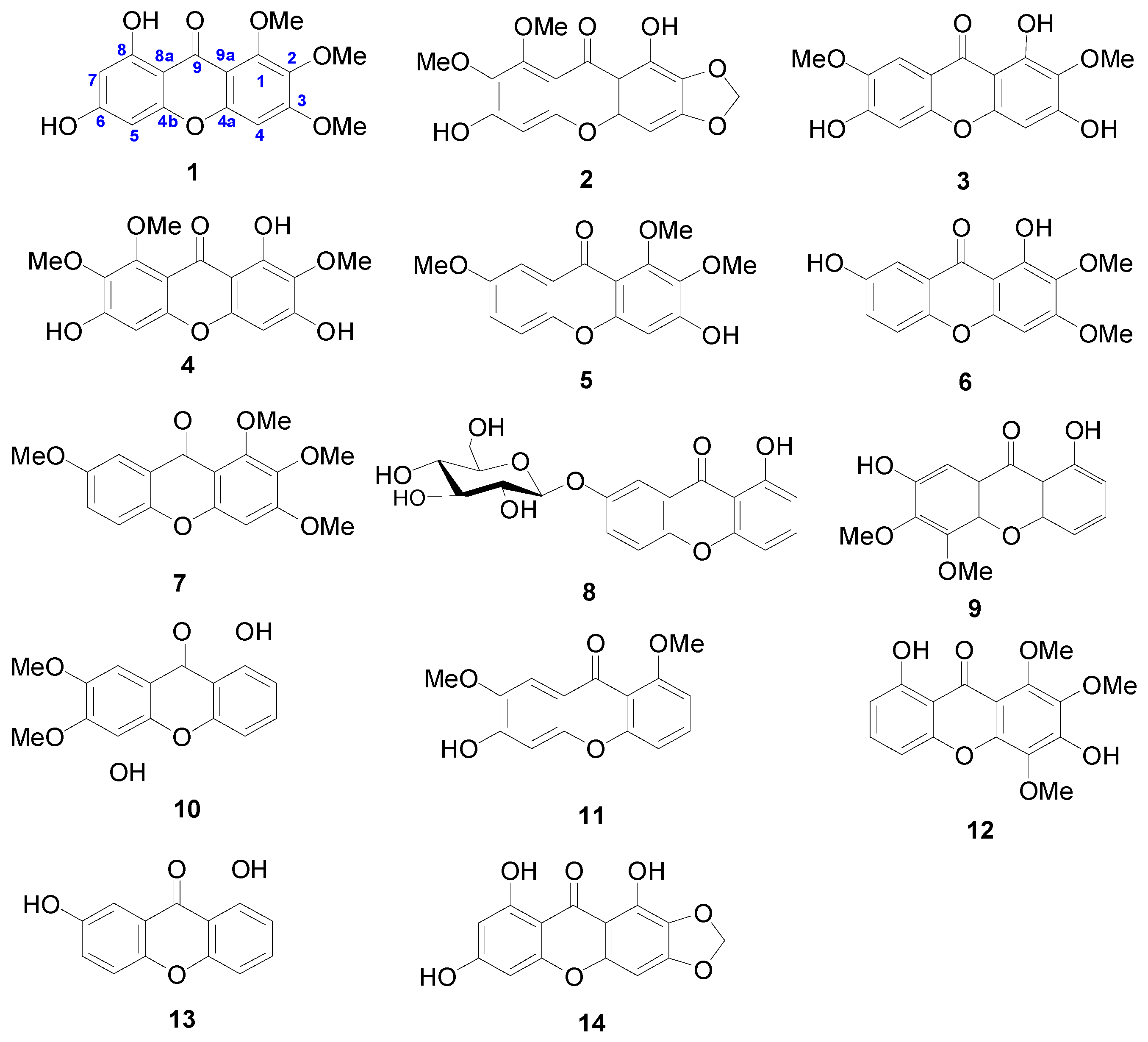
2.1. Identification of New Compounds
2.2. CoMFA and CoMSIA Analysis
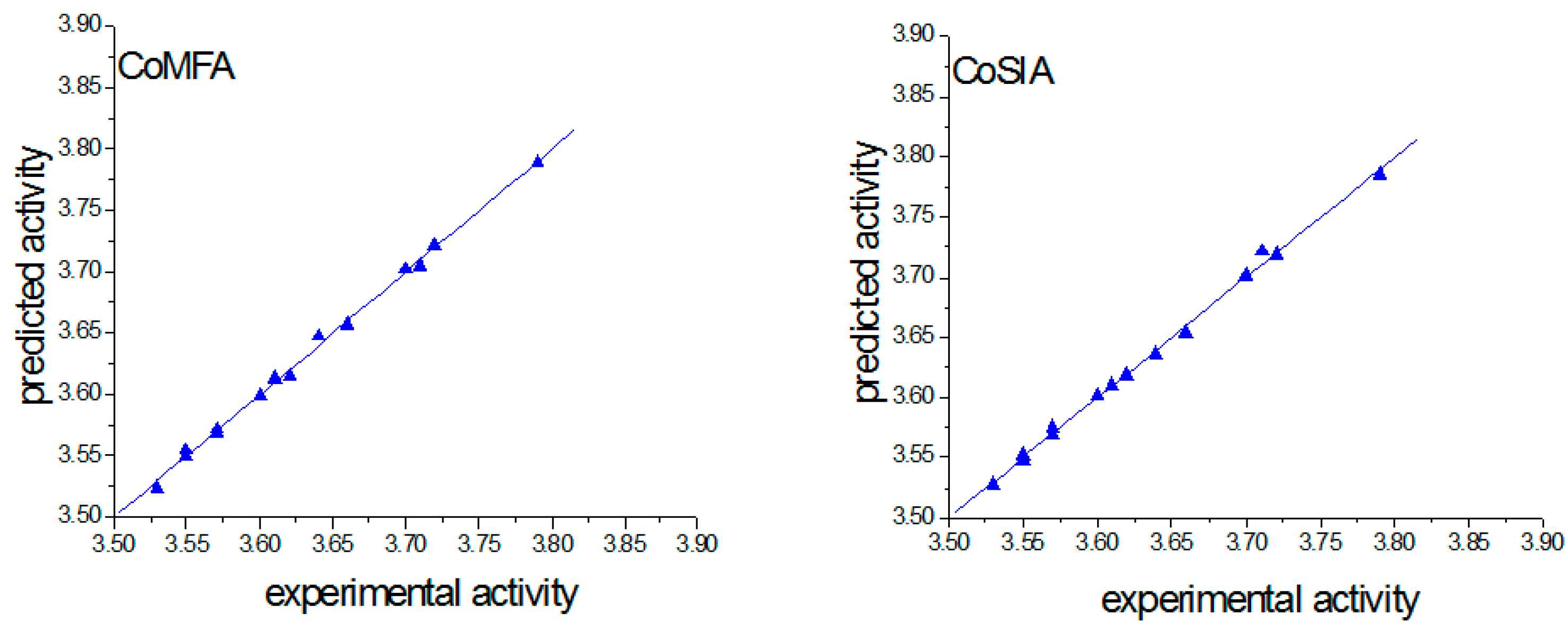
2.3. Verification of Model Reliability
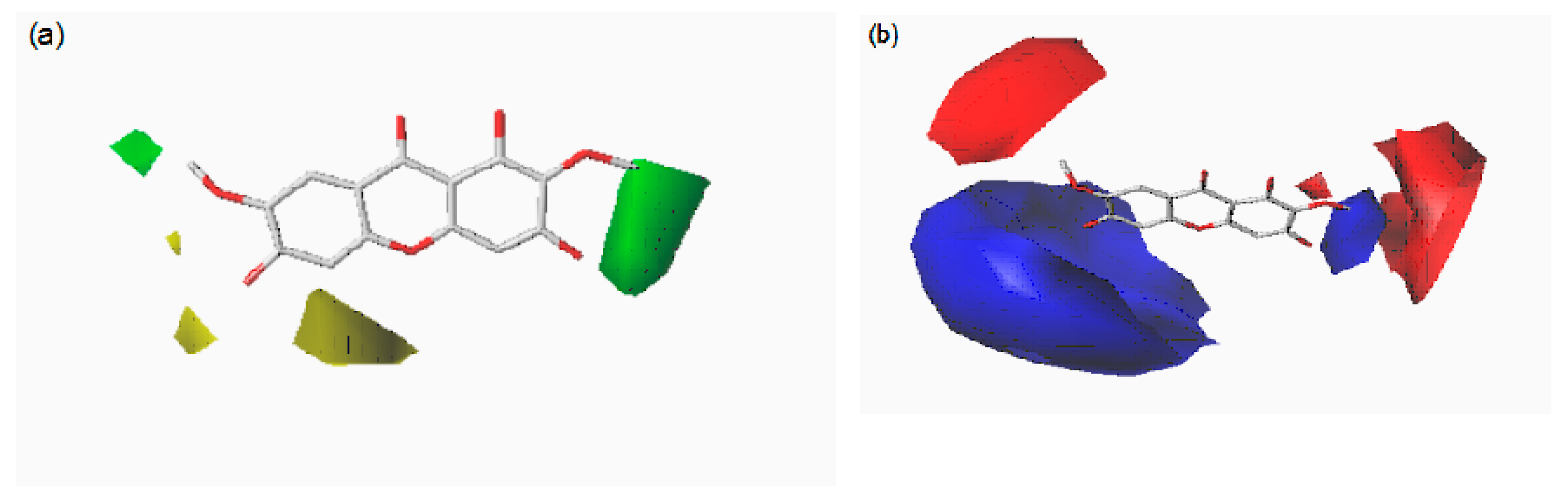
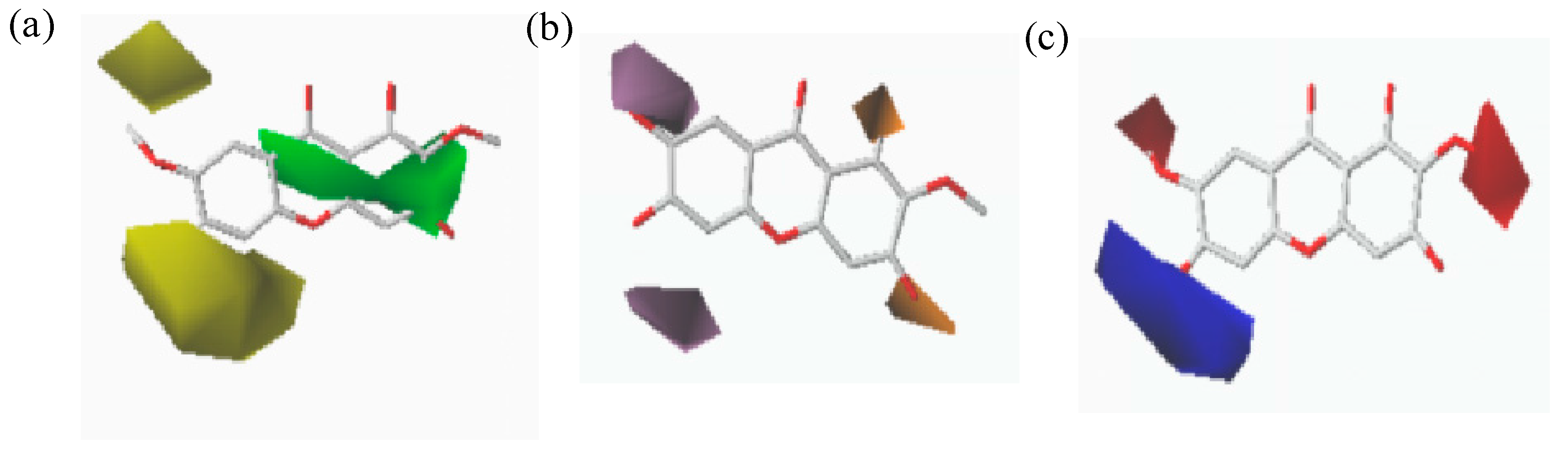
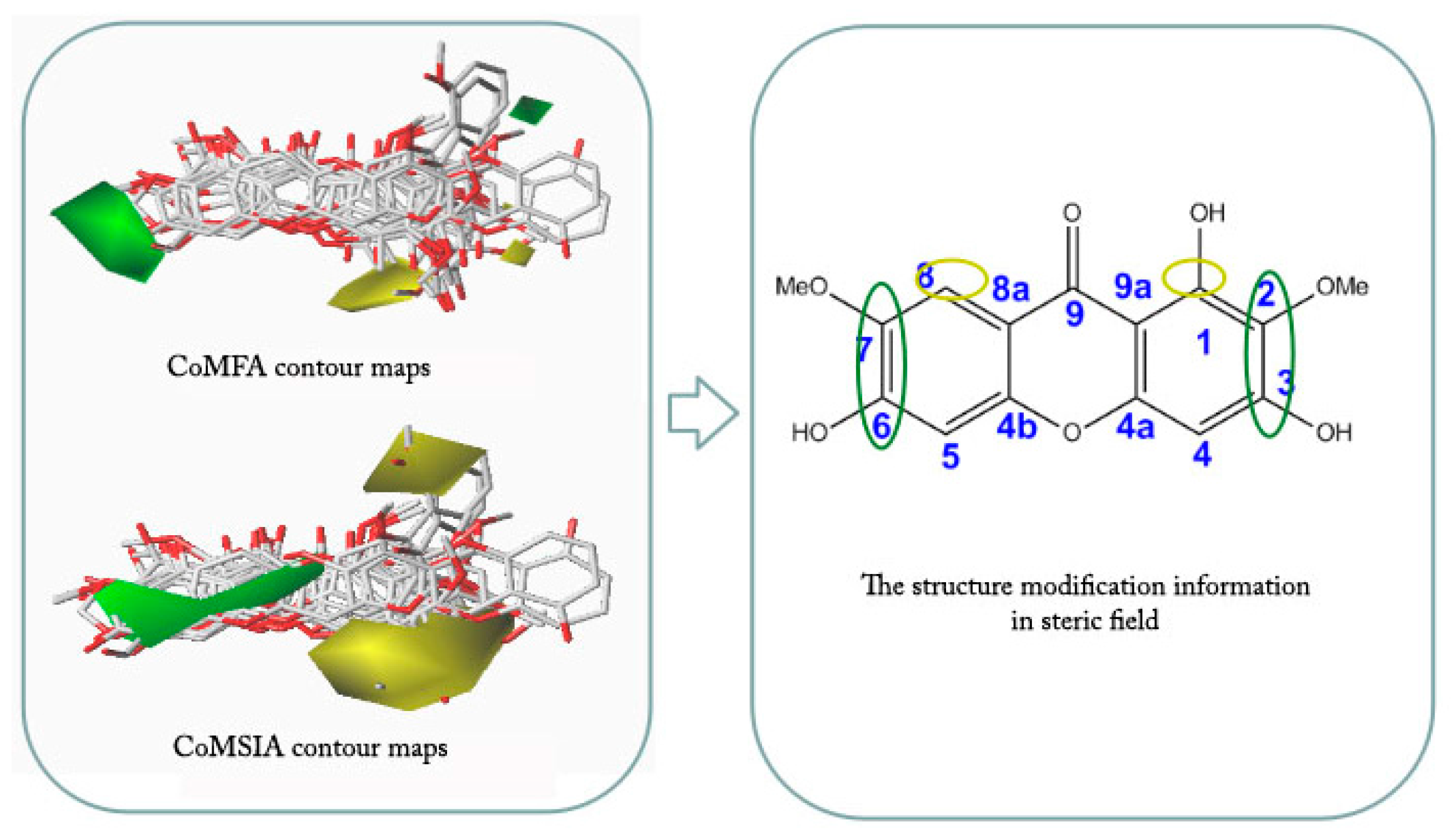
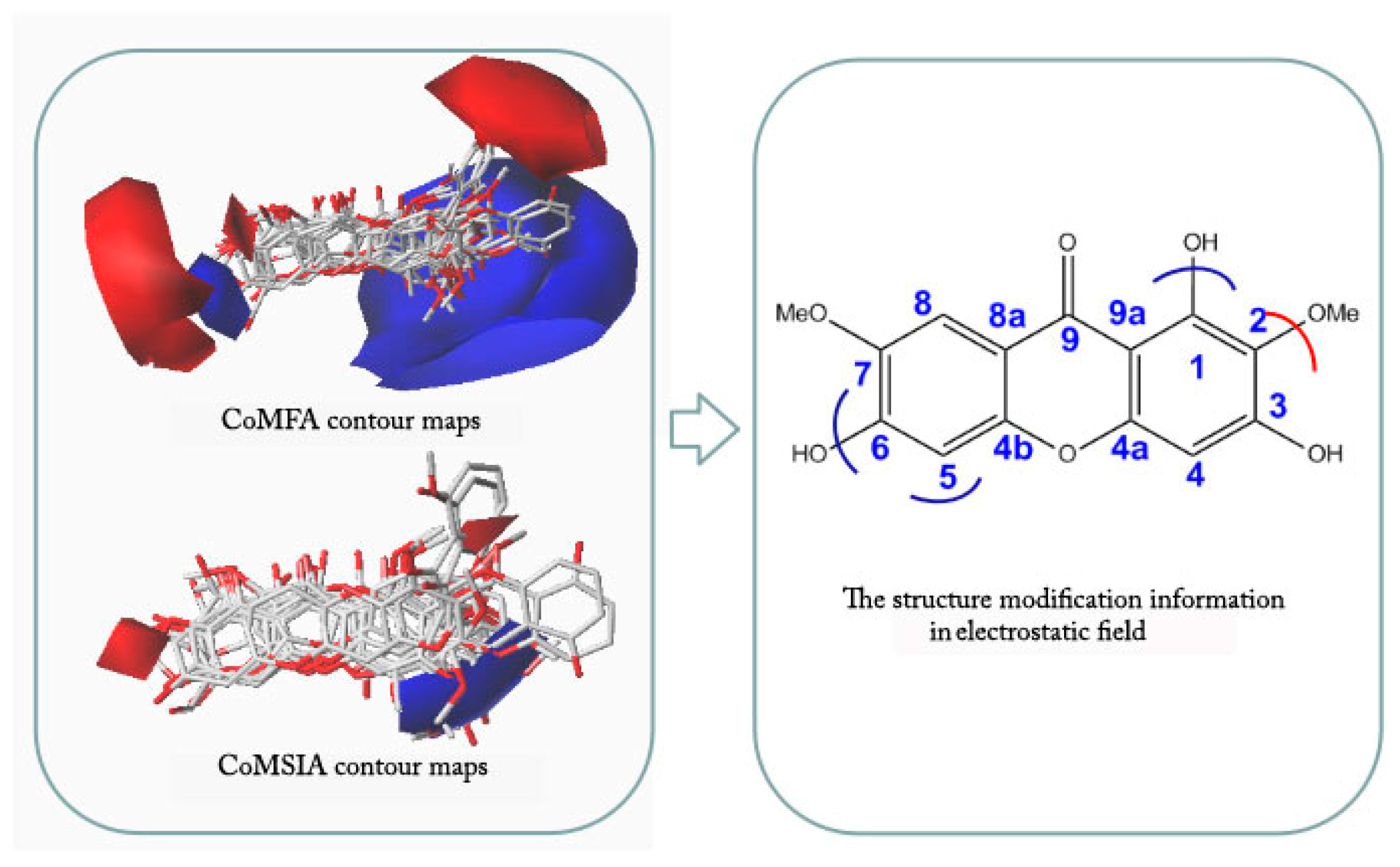
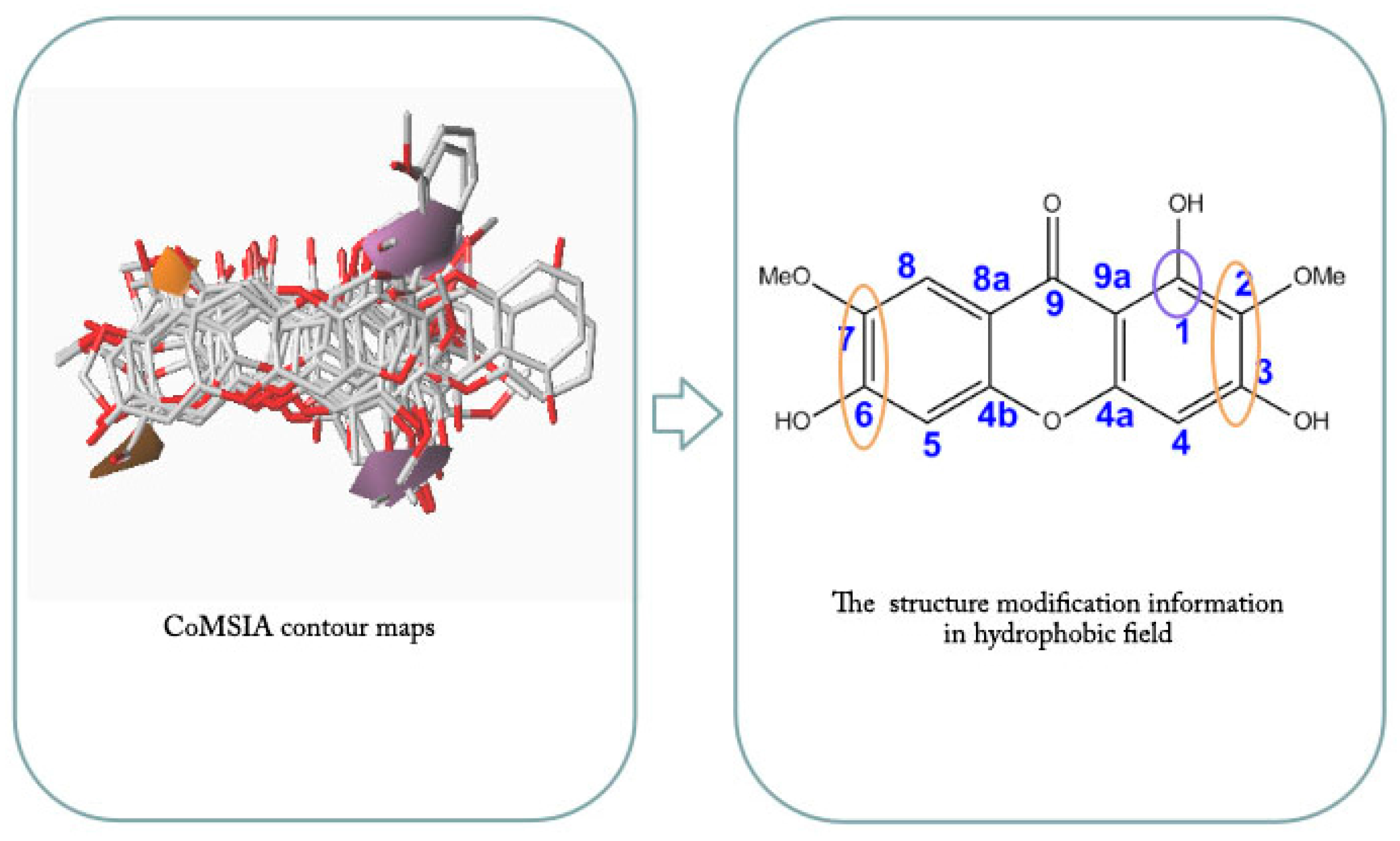
2.4. CoMFA and CoMSIA Contour Maps
3. Experimental Section
3.1. General Information
3.2. Plant Material
3.3. Extraction, Isolation, and Characterization
3.4. Acid Hydrolysis of Compound C
3.5. Bioassay of Xanthine Oxidase Inhibitory Activity
3.6. Dataset Preparation
3.7. CoMFA and CoMSIA Studies
3.8. Validation of 3D-QSAR Models
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cardona, M.L.; Fernándes, I.; Pedro, J.R.; Serrano, A. Xanthones from Hypericum reflexum. Phytochemistry 1990, 29, 3003–3006. [Google Scholar] [CrossRef]
- Peres, V.; Nagem, T.J. Trioxygenated naturally occurring xanthones. Phytochemistry 1996, 44, 191–214. [Google Scholar] [CrossRef]
- Peres, V.; Nagem, T.J.; de Oliveira, F.F. Tetraoxygenated naturally occurring xanthones. Phytochemistry 2000, 55, 683–710. [Google Scholar] [CrossRef]
- Rocha, J.L.C.; Pastore, J.F.B.; Brandão, H.N.; Azevedo, A.; David, J.P.; Santos, E.O.; David, J.M. Quantificação de salicilato de metila em quatro gêneros de Polygalaceae, por CLAE-DAD. Quim. Nova 2012, 35, 2263–2266. [Google Scholar] [CrossRef]
- Hua, Y.; Chen, C.X.; Liu, Y.Q.; Chen, S.K.; Zhou, J. Three New Xanthones from Polygala crotalarioides. Chin. Chem. Lett. 2006, 17, 773–775. [Google Scholar]
- Hua, Y.; Chen, C.X.; Liu, Y.Q.; Zhou, J. Two new xanthones from Polygala crotalarioides. J. Asian Nat. Prod. Res. 2007, 9, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Mac Kerell, A.D. Computational ligand-based rational design: Role of conformational sampling and force fields in model development. Med. Chem. Commun. 2011, 2, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Richards, W.G. Computers in drug design. Pure Appl. Chem. 1993, 65, 231–234. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecularfield analysis (CoMFA) 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indexes in a comparative-analysis (CoMSIA) of drug molecules to correlate and predict their biological-activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Comparative molecular similarity indices analysis: CoMSIA. Perspect. Drug Discov. Des. 1998, 12, 87–104. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design: A review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U. Comparative molecular similarity index Analysis (CoMSIA) to study hydrogen-bonding properties and to score combinatorial libraries. J. Comput. Aided Mol. Des. 1999, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.J.; Kellogg, G.E. The effect of physical organic properties on hydrophobic fields. J. Comput. Aided Mol. Des. 1994, 8, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ishikita, H.; Eger, B.T.; Okamoto, K.; Nishino, T.; Pai, E.F. Protein conformational gating of enzymatic activity in xanthine oxidoreductase. J. Am. Chem. Soc. 2012, 134, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Kondo, S.; Pai, E.F.; Nishino, T. An Extremely Potent Inhibitor of Xanthine Oxidoreductase Crystal Structure of the Enzyme-Inhibitor Complex and Mechanism of Inhibition. J. Biol. Chem. 2003, 278, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. USA 2000, 97, 10723–10728. [Google Scholar] [CrossRef] [PubMed]
- Fukunari, A.; Okamoto, K.; Nishino, T.; Eger, B.T.; Pai, E.F.; Kamezawa, M.; Yamada, I.; Kato, N. Y-700 [1-[3-Cyano-4-(2,2-dimethylpropoxy)phenyl]-1H-pyrazole-4-carboxylic acid]: A potent xanthine oxidoreductase inhibitor with hepatic excretion. J. Pharmacol. Exp. Ther. 2004, 311, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, W.G.; Labadie, R.P. Polyoxygenated xanthones of Centaurium littorale. Phytochemistry 1985, 24, 2601–2605. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Rastogi, R.P. 13C-NMR spectroscopy of flavonoids. Heterocycles 1981, 16, 2181–2236. [Google Scholar]
- Hideki, T.; Munekazu, I.; Kohichi, M.; Ito, T.; Tanaka, T.; Chelladurai, V.; Riswan, S. Three xanthones from Poeciloneuron pauciflorum and Mammea acuminata. Phytochemistry 1997, 45, 133–136. [Google Scholar]
- Paul, C.; Li, Y.; Mario, C.; Jia, P.H.; Kanyanga, C.; Bart, V.P.; Luc, P.; Arnold, J.V.; Dirk, V.B. Structure–activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J. Nat. Prod. 1998, 61, 71–76. [Google Scholar]
Sample Availability: Samples of the compounds A and B are not available from the authors because of the limited amount. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH | δC |
|---|---|---|
| 1 | 153.2 s | |
| 2 | 138.6 s | |
| 3 | 156.6 s | |
| 4 | 6.63 (s) | 98.8 d |
| 5 | 6.87 (s) | 96.2 d |
| 6 | 157.7 s | |
| 7 | 138.9 s | |
| 8 | 152.8 s | |
| 9 | 172.8 s | |
| 4a | 152.8 s | |
| 8a | 110.4 s | |
| 9a | 109.6 s | |
| 10a | 152.9 s | |
| OMe-1 | 3.80 (s) | 61.7 q |
| OMe-2 | 3.74 (s) | 61.0 q |
| OMe-6 | 3.89 (s) | 56.5 q |
| OMe-7 | 3.72 (s) | 60.9 q |
| OMe-8 | 3.81 (s) | 61.8 q |
| Position | δH | δC |
|---|---|---|
| 1 | 160.8 s | |
| 2 | 6.97 (d, 8.2) | 110.0 d |
| 3 | 7.51 (t, 8.2) | 134.4 d |
| 4 | 6.76 (d, 8.2) | 105.9 d |
| 5 | 7.59 (s) | 103.8 d |
| 6 | 153.2 s | |
| 7 | 147.3 s | |
| 8 | 7.89 (s) | 106.6 d |
| 9 | 174.7 s | |
| 4a | 158.3 s | |
| 8a | 117.2 s | |
| 9a | 112.5 s | |
| 10a | 150.8 s | |
| OMe-1 | 3.83 (s) | 56.1 q |
| OMe-7 | 3.67 (s) | 55.7 q |
| Glc-1 | 5.88 (d, 7.5) | 101.6 d |
| 2 | 4.42 (m) | 74.5 d |
| 3 | 4.44 (m) | 78.3 d |
| 4 | 4.40 (m) | 70.9 d |
| 5 | 4.35 (m) | 79.2 d |
| 6 | 4.57 (d, 11.7) 4.40 (m) | 62.2 t |
| Number | pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|
| pre.pIC50 | Residue | pre.pIC50 | Residue | ||
| 1 | 3.66 | 3.657 | −0.003 | 3.654 | −0.006 |
| 2 | 3.64 | 3.648 | 0.008 | 3.636 | −0.004 |
| 3 | 3.79 | 3.789 | −0.001 | 3.785 | −0.005 |
| 4 | 3.71 | 3.705 | −0.005 | 3.721 | 0.011 |
| 5 | 3.55 | 3.551 | 0.001 | 3.547 | −0.003 |
| 6 | 3.60 | 3.599 | −0.001 | 3.601 | 0.001 |
| 7 | 3.62 | 3.615 | −0.005 | 3.619 | −0.001 |
| 8 | 3.72 | 3.722 | 0.002 | 3.719 | −0.001 |
| 9 | 3.57 | 3.569 | −0.001 | 3.57 | 0 |
| 10 | 3.57 | 3.572 | 0.002 | 3.575 | 0.005 |
| 11 | 3.55 | 3.555 | 0.005 | 3.553 | 0.003 |
| 12 | 3.61 | 3.613 | 0.003 | 3.61 | 0 |
| 13 | 3.53 | 3.524 | −0.006 | 3.528 | −0.002 |
| 14 | 3.70 | 3.702 | 0.002 | 3.701 | 0.001 |
| Method | CoMFA | CoMSIA |
|---|---|---|
| PLS statistics | ||
| q2 | 0.613 | 0.608 |
| SEP | 0.065 | 0.066 |
| Optimum components | 6 | 6 |
| r2 | 0.997 | 0.997 |
| SEE | 0.005 | 0.006 |
| F-ratio | 426 | 0.997 |
| Field distribution | ||
| Steric | 0.632 | 0.152 |
| Electrostatic | 0.368 | 0.316 |
| Hydrophobic | 0.532 |
| Number | pIC50 | coMFA-pre. pIC50 | coMSIA-pre. pIC50 | Original Activity Order | CoMFA pre. Activity Order | CoMSIA pre. Activity Order | Docking Score | Docking Score Order |
|---|---|---|---|---|---|---|---|---|
| 1 | 3.66 | 3.657 | 3.654 | 3 | 8 | 4 | 72.23 | 2 |
| 2 | 3.64 | 3.648 | 3.636 | 8 | 4 | 8 | 71.41 | 8 |
| 3 | 3.79 | 3.789 | 3.785 | 4 | 14 | 14 | 60.48 | 9 |
| 4 | 3.71 | 3.705 | 3.721 | 14 | 2 | 2 | 60.59 | 14 |
| 5 | 3.55 | 3.551 | 3.547 | 1 | 3 | 3 | 53.7 | 4 |
| 6 | 3.6 | 3.599 | 3.601 | 2 | 7 | 7 | 54.48 | 3 |
| 7 | 3.62 | 3.615 | 3.619 | 7 | 12 | 12 | 45.98 | 6 |
| 8 | 3.72 | 3.722 | 3.719 | 12 | 6 | 6 | 64.35 | 10 |
| 9 | 3.57 | 3.569 | 3.570 | 6 | 10 | 1 | 62.93 | 5 |
| 10 | 3.57 | 3.572 | 3.575 | 9 | 9 | 10 | 54.32 | 1 |
| 11 | 3.55 | 3.555 | 3.553 | 10 | 11 | 9 | 50.12 | 11 |
| 12 | 3.61 | 3.613 | 3.610 | 5 | 5 | 11 | 49.86 | 12 |
| 13 | 3.53 | 3.524 | 3.528 | 11 | 1 | 5 | 38.95 | 7 |
| 14 | 3.7 | 3.702 | 3.701 | 13 | 13 | 13 | 61.05 | 13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.-Y.; Peng, J.-L.; Wang, J.-M.; Geng, Y.-Y.; Zuo, Z.-L.; Hua, Y. Structure–Activity Relationship of Xanthones as Inhibitors of Xanthine Oxidase. Molecules 2018, 23, 365. https://doi.org/10.3390/molecules23020365
Zhou L-Y, Peng J-L, Wang J-M, Geng Y-Y, Zuo Z-L, Hua Y. Structure–Activity Relationship of Xanthones as Inhibitors of Xanthine Oxidase. Molecules. 2018; 23(2):365. https://doi.org/10.3390/molecules23020365
Chicago/Turabian StyleZhou, Ling-Yun, Jia-Le Peng, Jun-Ming Wang, Yuan-Yuan Geng, Zhi-Li Zuo, and Yan Hua. 2018. "Structure–Activity Relationship of Xanthones as Inhibitors of Xanthine Oxidase" Molecules 23, no. 2: 365. https://doi.org/10.3390/molecules23020365
APA StyleZhou, L. -Y., Peng, J. -L., Wang, J. -M., Geng, Y. -Y., Zuo, Z. -L., & Hua, Y. (2018). Structure–Activity Relationship of Xanthones as Inhibitors of Xanthine Oxidase. Molecules, 23(2), 365. https://doi.org/10.3390/molecules23020365