Inhibitory Effect of Sauchinone on UDP-Glucuronosyltransferase (UGT) 2B7 Activity



Abstract
:1. Introduction
2. Results
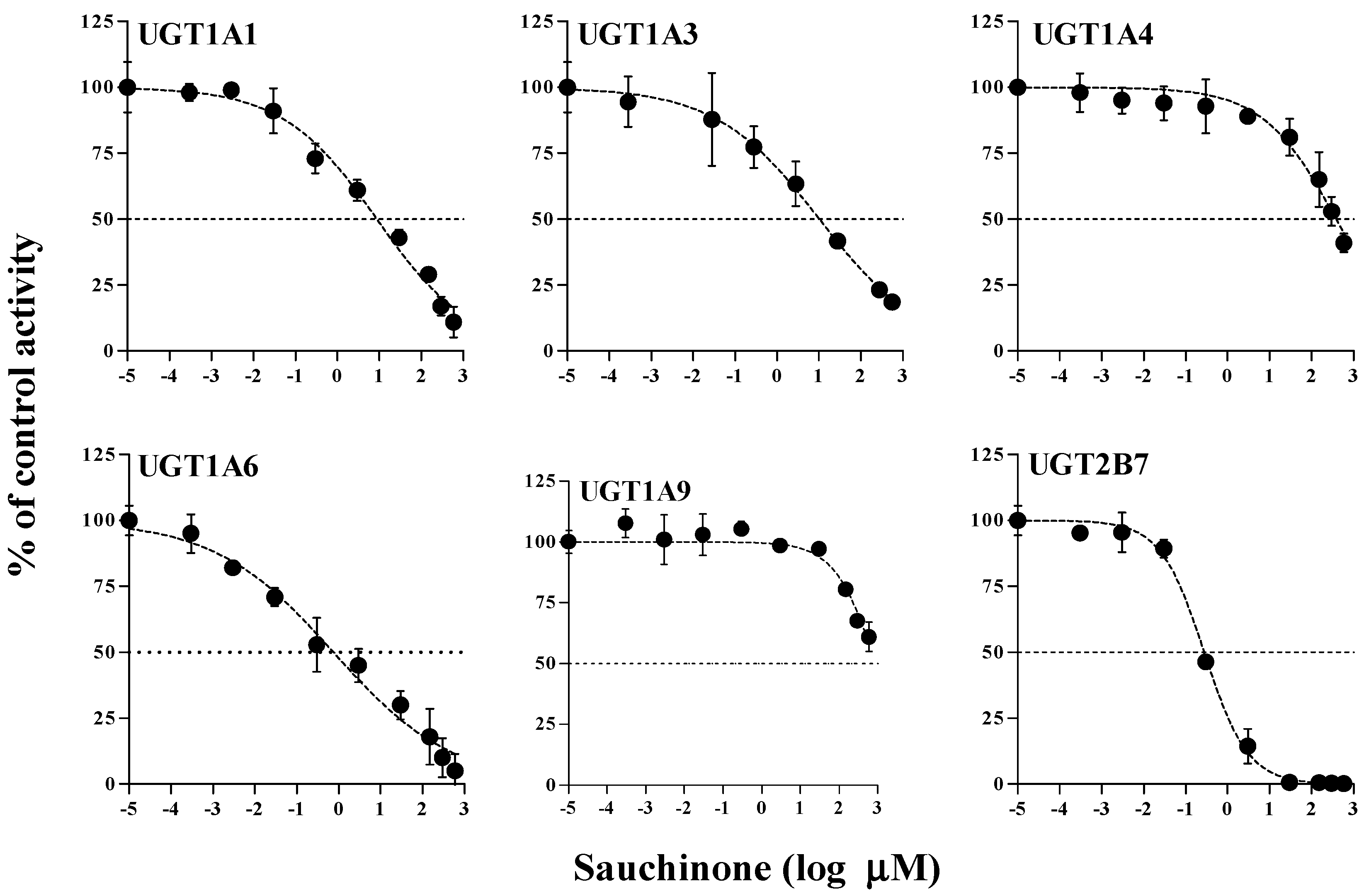
2.1. Inhibitory Effects of Sauchinone on UGT Activities
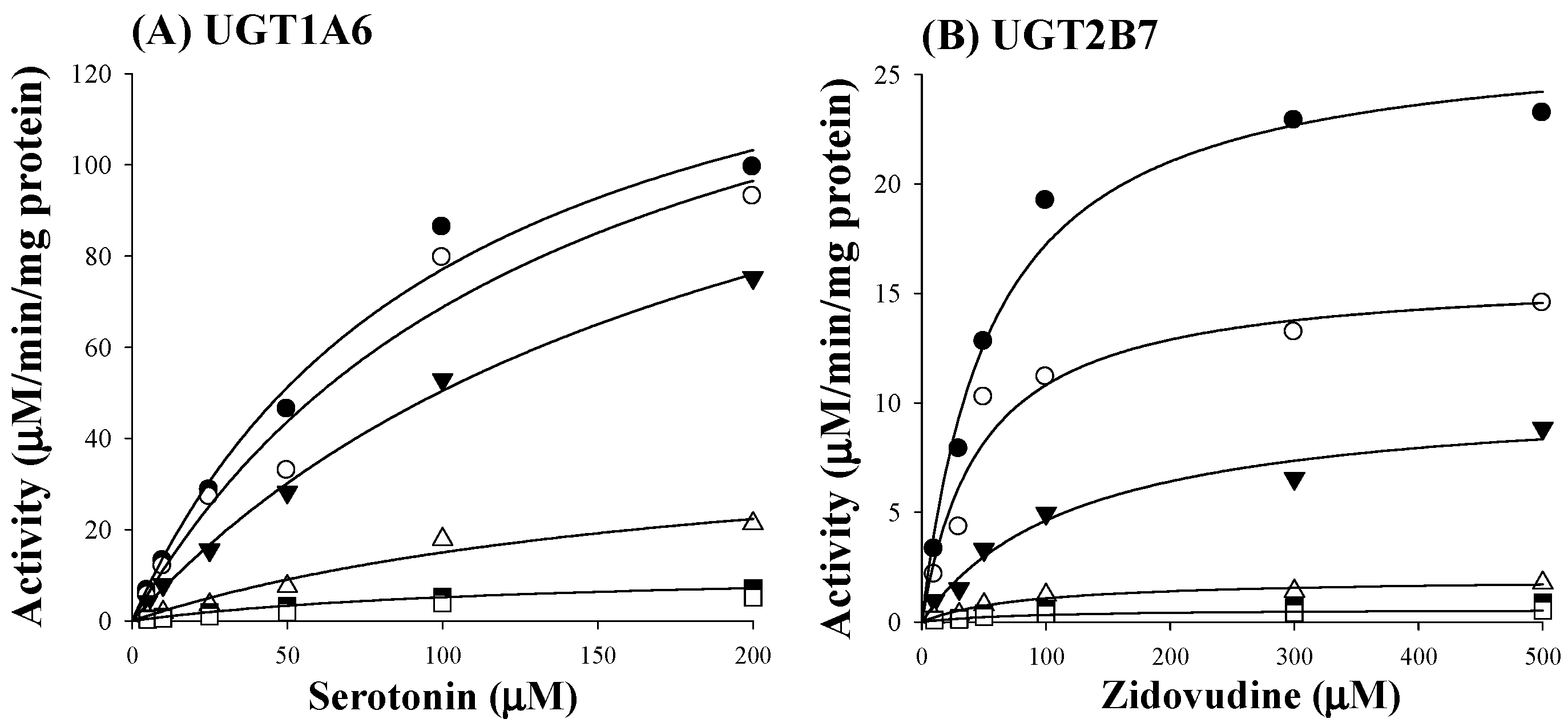
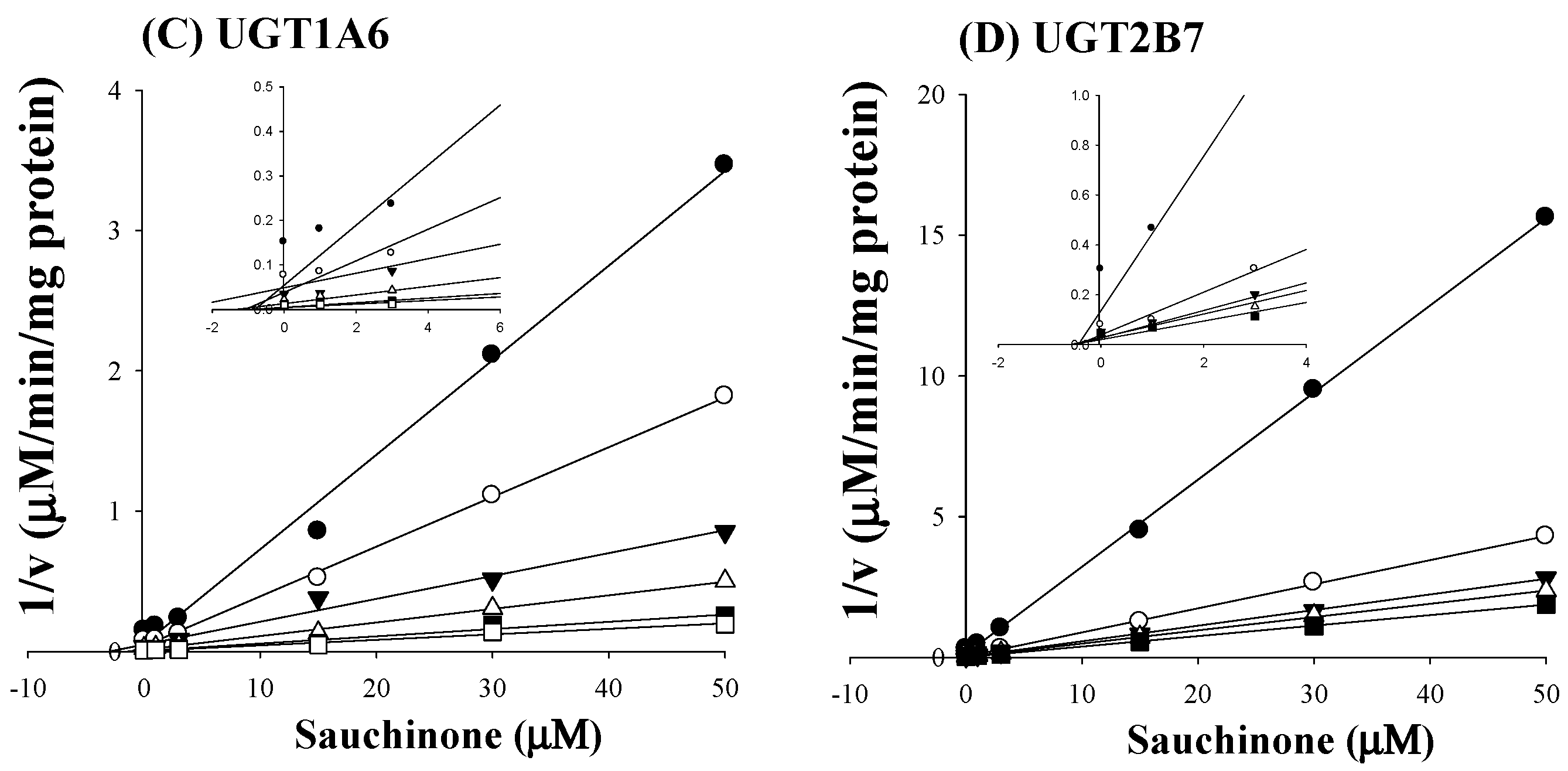
2.2. Ki of Sauchinone on UGT1A6 and 2B7-Mediated Glucuronidation Activities
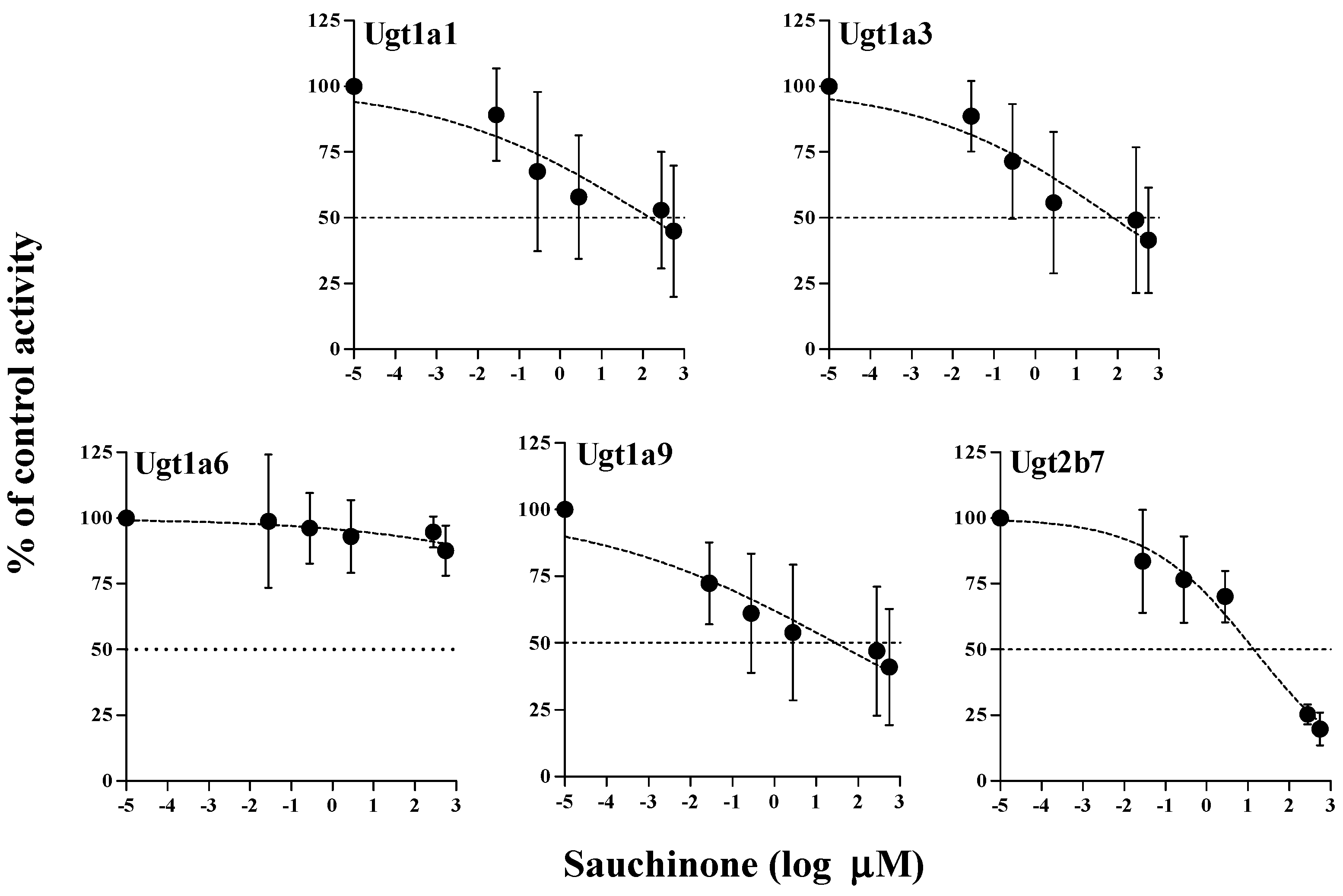
2.3. Inhibitory Effects of Sauchinone on Ugt Activities Using Mice Liver Microsomes (MLMs)
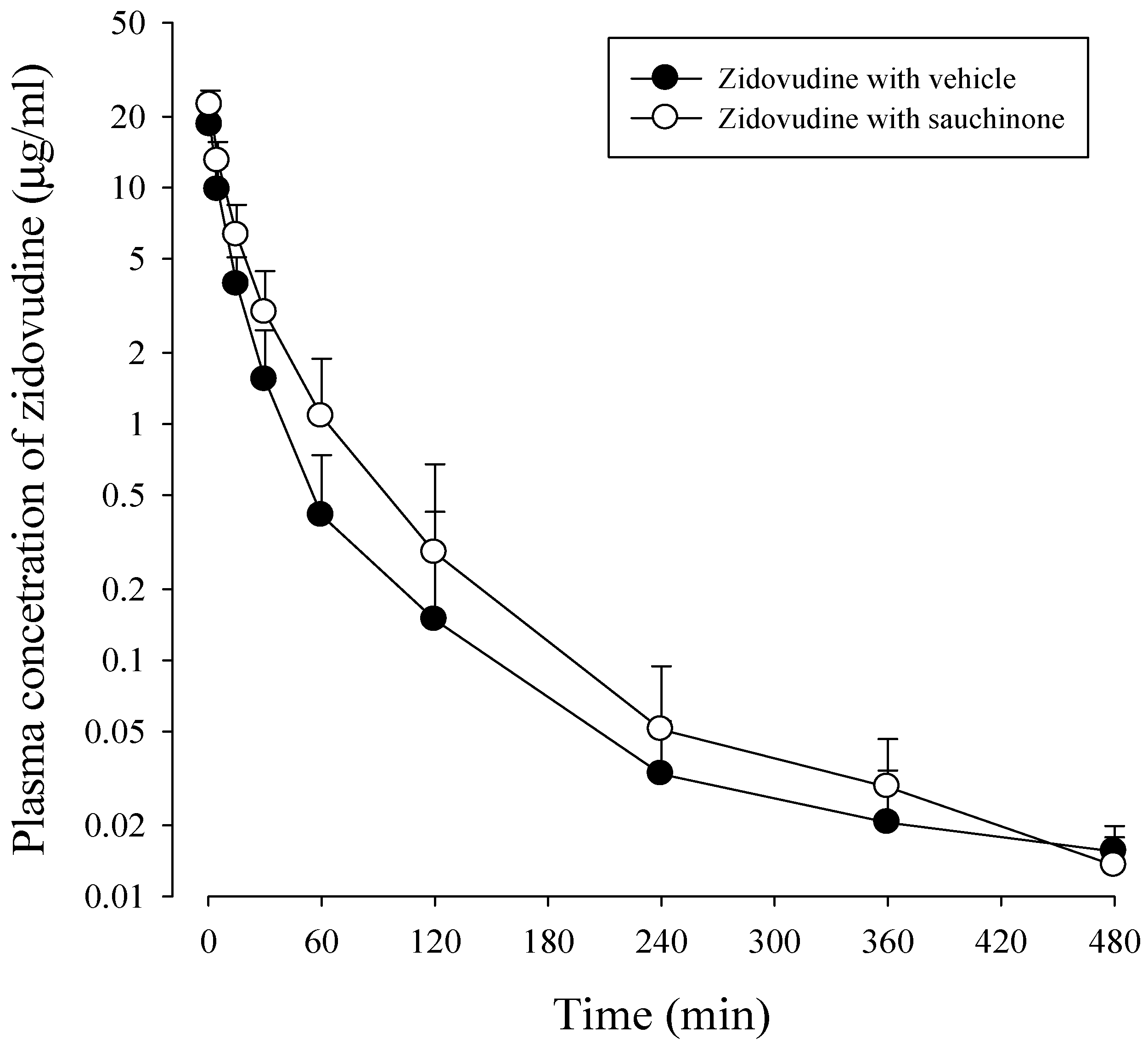
2.4. Pharmacokinetic Study of Zidovudine with or without Sauchinone
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals
4.3. Inhibitory Effects of Sauchinone on UGT Activities
4.4. Inhibition Constant (Ki) of Sauchinone on UGT1A6 and 2B7-Mediated Glucuronidation Activities
4.5. Inhibitory Effects of Sauchinone on UGT Activities Using MLM
4.6. UPLC-MS/MS Analysis for Metabolites of UGTs Substrates
4.7. Data Analysis
4.8. Pharmacokinetic Study of Zidovudine with or without Sauchinone
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qiu, J. China Plans to Modernize Traditional Medicine. Nature 2007, 446, 590–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reynods, K.S.; Zhao, P.; Huang, S.M. Drug Interactions Evaluation: An Integrated Part of Risk Assessment of Therapeutics. Toxicol. Appl. Pharmacol. 2010, 243, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, H.; Xu, S.; Zhou, Q.; Zeng, S. Roles of UDP-Glucuronosyltransferases in Phytochemical Metabolisms of Herbal Medicines and Associated Herb-Drug Interactions. Curr. Drug Metab. 2012, 13, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, H.; Jana, S. Evaluation of Inhibitory Effects of Caffeic Acid and Quercetin on Human Liver Cytochrome p450 Activities. Phytother. Res. 2014, 28, 1873–1878. [Google Scholar] [CrossRef] [PubMed]
- Walsky, R.L.; Bauman, J.N.; Bourcier, K.; Giddens, G.; Lapham, K.; Negahban, A.; Ryder, T.F.; Obach, R.S.; Hyland, R.; Goosen, T.C. Optimized Assays for Human UDP-Glucuronosyltransferase (UGT) Activities: Altered Alamethicin Concentration and Utility to Screen for UGT Inhibitors. Drug Metab. Dispos. 2012, 40, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Ensom, M.H.; Chang, T.K. UDP-Glucuronosyltransferases and Clinical Drug-Drug Interactions. Pharmacol. Ther. 2005, 106, 97–132. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Zhang, C.; Li, R.; Chen, B.; Zhang, W.; Zhao, Z.; Fu, Z.; Du, Z.; Du, X.; Yang, X.; et al. Evaluation and Comparison of the Inhibition Effect of Astragaloside IV and Aglycone Cycloastragenol on Various UDP-Glucuronosyltransferase (UGT) Isoforms. Molecules 2016, 21, E1616. [Google Scholar] [CrossRef] [PubMed]
- Nagar, S.; Remmel, R.P. Uridine Diphosphoglucuronosyltransferase Pharmacogenetics and Cancer. Oncogene 2006, 25, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Zhao, Z.Q.; Qin, Z.S.; Wu, K.; Xia, T.F.; Pang, L.Q. Herb-Drug Interaction Between Irinotecan and Psoralidin-Containing Herbs. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Zhao, J.J.; Ma, B.; Roadcap, B.A.; Tang, C.; Qiu, Y.; Liu, L.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Mechanistic Studies on Metabolic Interactions Between Gemfibrozil and Statins. J. Pharmacol. Exp. Ther. 2002, 301, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yang, H.T.; Kong, L.T.; Yu, F.X. In vitro Evidence of Baicalein’s Inhibition of the Metabolism of Zidovudine (AZT). Afr. Health Sci. 2014, 14, 173–177. [Google Scholar] [PubMed]
- Sakakibara, Y.; Katoh, M.; Kondo, Y.; Nadai, M. Effects of Phenobarbital on Expression of UDP-Glucuronosyltransferase 1a6 and 1a7 in Rat Brain. Drug Metab. Dispos. 2016, 44, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Mutlib, A.E.; Goosen, T.C.; Bauman, J.N.; Williams, J.A.; Kulkarni, S.; Kostrubsky, S. Kinetics of Acetaminophen Glucuronidation by UDP-Glucuronosyltransferases 1A1, 1A6, 1A9 and 2B15. Potential Implications in Acetaminophen-Induced Hepatotoxicity. Chem. Res. Toxicol. 2006, 19, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Liu, X.; Griffen, S.C.; Lacreta, F.P.; Boulton, D.W. Effects of Rifampin and Mefenamic Acid on the Pharmacokinetics and Pharmacodynamics of Dapagliflozin. Diabetes Obes. Metab. 2013, 15, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Lertora, J.J.; Rege, A.B.; Greenspan, D.L.; Akula, S.; George, W.J.; Hyslop, N.E., Jr.; Agrawal, K.C. Pharmacokinetic Interaction Between Zidovudine and Valproic Acid in Patients Infected with Human Immunodeficiency Virus. Clin. Pharmacol. Ther. 1994, 56, 272–278. [Google Scholar] [CrossRef] [PubMed]
- FDA, Center for Drug Evaluation and Research, Guideline for Industry. Drug Interaction Studies-Study Design, Data Analysis, Implication for Dosing, and Labeling Recommendations; Food and Drug Administration: Rockville, MD, USA, 2012.
- Thelingwani, R.; Masimirembwa, C. Evaluation of Herbal Medicines: Value Addition to Traditional Medicines through Metabolism, Pharmacokinetic and Safety Studies. Curr. Drug Metab. 2014, 15, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.E.; Frye, R.F. Effects of Herbal Supplements on Drug Glucuronidation. Review of clinical, animal, and in vitro studies. Planta Med. 2011, 77, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Kim, I.; Jeong, Y.J.; Cho, Y.M.; Kang, S.C. Anti-Ifflammatory Effects of Saururus chinensis Aerial Parts in Murine Macrophages via Induction of Heme Oxygenase-1. Exp. Biol. Med. (Marywood) 2016, 241, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.H.; Kim, Y.C. Hepatoprotective Diastereomeric Lignans from Saururus chinensis Herbs. J. Nat. Prod. 2000, 63, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.S.; Shin, M.G. Dictionary of Korean Folk Medicine; Young Lim Sa: Seoul, Korea, 1990; pp. 813–814. [Google Scholar]
- Jeong, H.J.; Koo, B.S.; Kang, T.H.; Shin, H.M.; Jung, S.H. Inhibitory Effects of Saururus chinensis and its Components on Stomach Cancer Cells. Phytomedicine 2015, 22, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Won, C.; Yoo, H.; Yi, E.H.; Cho, Y.; Maeng, J.W.; Sung, S.H.; Ye, S.K.; Chung, M.H. Inhibition of Double-Stranded RNA-Induced Inducible Nitric Oxide Synthase Expression by Fraxinellone and Sauchinone in Murine Microglia. Biol. Pharm. Bull. 2009, 32, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Han, S.Y.; Seo, J.S.; Chin, Y.W.; Choi, Y.H. Pharmacokinetics, Tissue Distribution, and Tentative Metabolite Identification of Sauchinone in Mice by Microsampling and HPLC-MS/MS Methods. Biol. Pharm. Bull. 2015, 38, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, P.J. The use of Dixon Plots to Study Enzyme Inhibition. Biochem. Biophys. Acta 1972, 289, 251–253. [Google Scholar] [CrossRef]
- Sun, D.; Zhang, C.Z.; Ran, R.X.; Cao, Y.F.; Du, Z.; Fu, Z.W.; Huang, C.T.; Zhao, Z.Y.; Zhang, W.H.; Fang, Z.Z. In Vitro Comparative Study of the Inhibitory Effects of Mangiferin and Its Aglycone Norathyriol towards UDP-Glucuronosyl Transferase (UGT) Isoforms. Molecules 2017, 22, E1008. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, P.; Phillips, R.; Shaughnessy, A.F. Herbal and Dietary Supplement—Drug Interactions in Patients with Chronic Illnesses. Am. Fam. Physician 2008, 77, 73–78. [Google Scholar] [PubMed]
- Lee, B.L.; Tauber, M.G.; Sadler, B.; Goldstein, D.; Chambers, H.F. Atovaquone Inhibits the Glucuronidation and Increases the Plasma Concentrations of Zidovudine. Clin. Pharmacol. Ther. 1996, 59, 14–21. [Google Scholar] [CrossRef]
- Sahai, J.; Gallicano, K.; Pakuts, A.; Cameron, D.W. Effect of Fluconazole on Zidovudine Pharmacokinetics in Patients Infected with Human Immunodeficiency Virus. J. Infect. Dis. 1994, 169, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Chen, W.; Shen, H.; Gao, L.; Hong, Y.; Tian, Y.; Li, W.; Zhang, Y.; Tang, Y.; Zhang, H.; et al. Repaglinide-Gemfibrozil Drug Interaction: Inhibition of Repaglinide Glucuronidation as a Potential Additional Contributing Mechanism. Br. J. Clin. Pharmacol. 2010, 70, 870–880. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Fang, Z.Z.; Ge, G.B.; Jiang, P.; Jin, H.Z.; Zhang, W.D.; Yang, L. The Inhibitory Effect of 20(S)-Protopanaxatriol (ppt) Towards UGT1A1 and UGT2B7. Phytother. Res. 2013, 27, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, T.; Wu, Z.; Wu, B. Warfarin is an Effective Modifier of Multiple UDP-Glucuronosyltransferase Enzymes: Evaluation of its Potential to Alter the Pharmacokinetics of Zidovudine. J. Pham. Sci. 2015, 104, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Gwilt, P.R.; Lear, C.L.; Tempero, M.A.; Birt, D.D.; Grandjean, A.C.; Ruddon, R.W.; Nagel, D.L. The Effect of Garlic Extract on Human Metabolism of Acetaminophen. Cancer Epidemiol. Biomark. Prev. 1994, 3, 155–160. [Google Scholar]
- Lee, L.S.; Wise, S.D.; Chan, C.; Parsons, T.L.; Flexner, C.; Lietman, P.S. Possible Differential Induction of Phase 2 Enzyme and Antioxidant Pathways by American Ginseng, Panax quinquefolius. J. Clin. Pharmacol. 2008, 48, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, N.P.; Baker, S.D.; Zhao, M.; Rudek, M.A.; Guchelaar, H.J.; Nortier, J.W.; Sparreboom, A.; Gelderblom, H. Effect of Milk Thistle (Silybum marianum) on the Pharmacokinetics of Irinotecan. Clin. Cancer Res. 2005, 11, 7800–7806. [Google Scholar] [CrossRef] [PubMed]
- Goey, A.K.; Mooiman, K.D.; Beijnen, J.H.; Schellens, J.H.; Meijerman, I. Relevance of In vitro and Clinical Data for Predicting CYP3A4-Mediated Herb-Drug Interactions in Cancer Patients. Cancer Treat. Rev. 2013, 39, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Filppula, A.M.; Neuvonen, P.J.; Backman, J.T. In vitro Assessment of Time-Dependent Inhibitory Effects on CYP2C8 and CYP3A Activity by Fourteen Protein Kinase Inhibitors. Drug Metab. Dispos. 2014, 42, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Court, M.H.; Krishnaswamy, S.; Hao, Q.; Duan, S.X.; Patten, C.J.; Von Moltke, L.L.; Greenblatt, D.J. Evaluation of 3′-azido-3′-deoxythymidine, Morphine, and Codeine as Probe Substrates for UDP-Glucuronosyltransferase 2B7 (UGT2B7) in Human Liver Microsomes: Specificity and Influence of the UGT2B7*2 Polymorphism. Drug Metab. Dispos. 2003, 31, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, T.; Xie, Y.; Zhao, M.; Che, Y.; Zhang, J.; Liu, H.; Cao, L.; Cheng, X.; Xie, Y.; et al. Mechanism-Based Inhibitory and Peroxisome Proliferator-Activated Receptor α–Dependent Modulating Effects of Silybin on Principal Hepatic Drug-Metabolizing Enzymes. Drug Metab. Dispos. 2015, 43, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-Glucuronosyltransferases: Their Role in Drug Metabolism and Detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Fukami, T.; Yokoi, T.; Nakajima, M. A Comprehensive Review of UDP-Glucuronosyltransferase and Esterases for Drug Development. Drug Metab. Pharmacokinet. 2015, 30, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Lakshmi, Y.S.; C, B.; Golla, K.; Kondapi, A.K. Improved Safety, Bioavailability and Pharmacokinetics of Zidovudine through Lactoferrin Nanoparticles during Oral Administration in Rats. PLoS ONE 2015, 10, e0140399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Squires, K.; Pan-Zhou, X.R.; Bernhard, S.; Agrofoglio, L.; Kirk, M.; Duchin, K.L.; Sommadossi, J.P. Phase I Dose-Escalation Pharmacokinetics of AZT-P-ddI (IVX-E-59) in Patients with Human Immunodeficiency Virus. J. Clin. Pharmacol. 1997, 37, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Uchaipichat, V.; Winner, L.K.; Macjenzie, P.I.; Elliot, D.J.; Williams, J.A.; Miners, J.O. Quantitative Prediction of In vivo Inhibitory Interactions Involving Glucuronidated Drugs from In vitro Data: The Effect of the Fluconazole on Zidovudine Glucuronidation. Br. J. Clin. Pharmacol. 2006, 61, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Systems; Wiley: New York, NY, USA, 1975. [Google Scholar]
- Giri, N.; Shaik, N.; Pan, G.; Terasaki, T.; Mukai, C.; Kitagaki, S.; Miyakoshi, N.; Elmquist, W.F. Investigation of the Role of Breast Cancer Resistance Protin (Bcrp/Abcg2) on Pharmacokinetics and Central Nervous System Penetration of Abacavir and Zidovudine in the Mouse. Drug Metab. Dispos. 2008, 36, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Perrier, D.; Gibaldi, M. General Derivation of the Equation for Time to Reach a Certain Fraction of Steady State. J. Pharm. Sci. 1982, 71, 474–475. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UGTs | Sauchinone | Well-Known Inhibitor | |
|---|---|---|---|
| UGT1A1 | 8.83 (5.40–14.5) | Chrysin | 28.3 |
| UGT1A3 | 43.9 (18.9–102) | Lithocholic acid | 69.8 |
| UGT1A4 | 391 (253–603) | Hecogenin | 3.99 |
| UGT1A6 | 0.758 (0.383–1.50) | 1-Naphthol | 35.2 |
| UGT1A9 | 919 (517–1632) | Niflumic acid | 0.755 |
| UGT2B7 | 0.279 (0.224–0.347) | Efavirenz | 75.4 |
| Parameters | With Vehicle | With Sauchinone |
|---|---|---|
| Body weight (g) | 30.6 ± 3.74 | 31.7 ± 3.37 |
| AUC480 min (μg min/mL) | 227 ± 61.9 | 344 ± 142 a |
| AUC (μg min/mL) | 228 ± 60.8 | 349 ± 140 a |
| t1/2 (min) | 207 ± 133 | 178 ± 93.1 |
| CL (mL/min/kg) | 68.7 ± 18.7 | 48.6 ± 16.1 a |
| CLR (mL/min/kg) | 34.0 ± 11.2 | 24.0 ± 8.02 a |
| CLNR (mL/min/kg) | 34.8 ± 10.5 | 24.5 ± 9.31 a |
| MRT (min) | 55.8 ± 28.2 | 43.5 ± 16.2 |
| Vss (mL/kg) | 3307 ± 1670 | 2105 ± 1229 |
| Ae0–24 h (% of dose) | 50.3 ± 8.01 | 50.2 ± 8.41 |
| GI24 h (% of dose) | 0.672 ± 0.999 | 0.321 ± 0.646 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, B.H.; Gong, E.C.; Choi, Y.H. Inhibitory Effect of Sauchinone on UDP-Glucuronosyltransferase (UGT) 2B7 Activity. Molecules 2018, 23, 366. https://doi.org/10.3390/molecules23020366
You BH, Gong EC, Choi YH. Inhibitory Effect of Sauchinone on UDP-Glucuronosyltransferase (UGT) 2B7 Activity. Molecules. 2018; 23(2):366. https://doi.org/10.3390/molecules23020366
Chicago/Turabian StyleYou, Byoung Hoon, Eun Chae Gong, and Young Hee Choi. 2018. "Inhibitory Effect of Sauchinone on UDP-Glucuronosyltransferase (UGT) 2B7 Activity" Molecules 23, no. 2: 366. https://doi.org/10.3390/molecules23020366
APA StyleYou, B. H., Gong, E. C., & Choi, Y. H. (2018). Inhibitory Effect of Sauchinone on UDP-Glucuronosyltransferase (UGT) 2B7 Activity. Molecules, 23(2), 366. https://doi.org/10.3390/molecules23020366