RPiRLS: Quantitative Predictions of RNA Interacting with Any Protein of Known Sequence

Abstract
:1. Introduction
2. Results
2.1. Parameter Selection for RLS
2.2. Performance on Predicting RNA-Protein Interactions with Known Structures
2.3. Performance on Predicting ncRNA-Protein Interactions
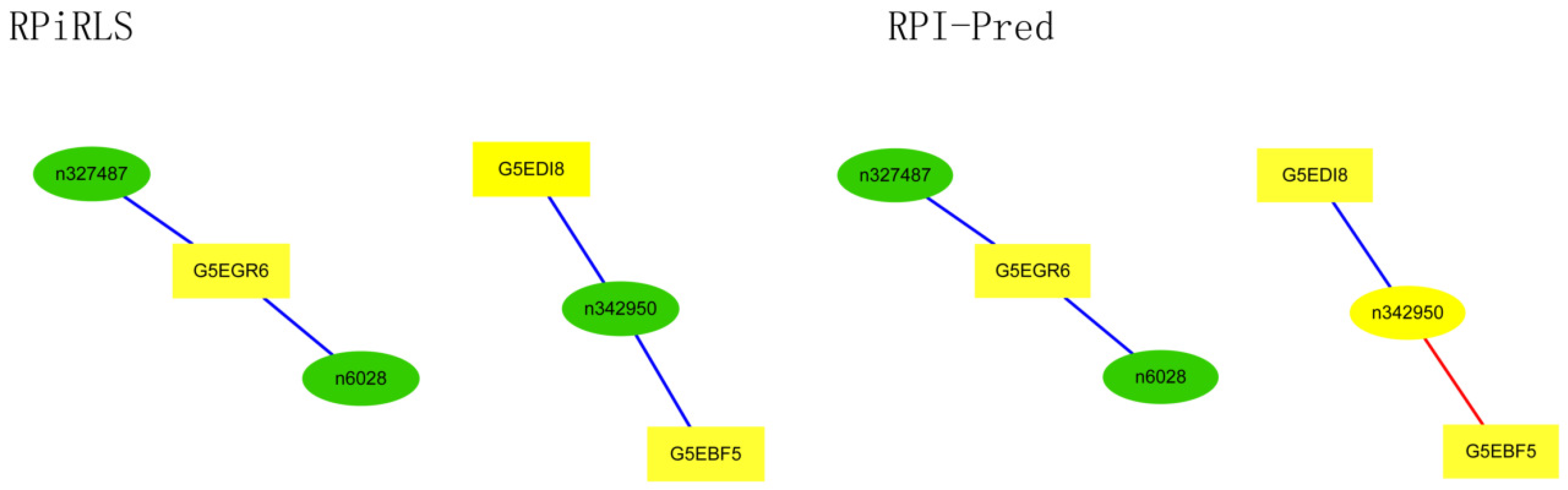
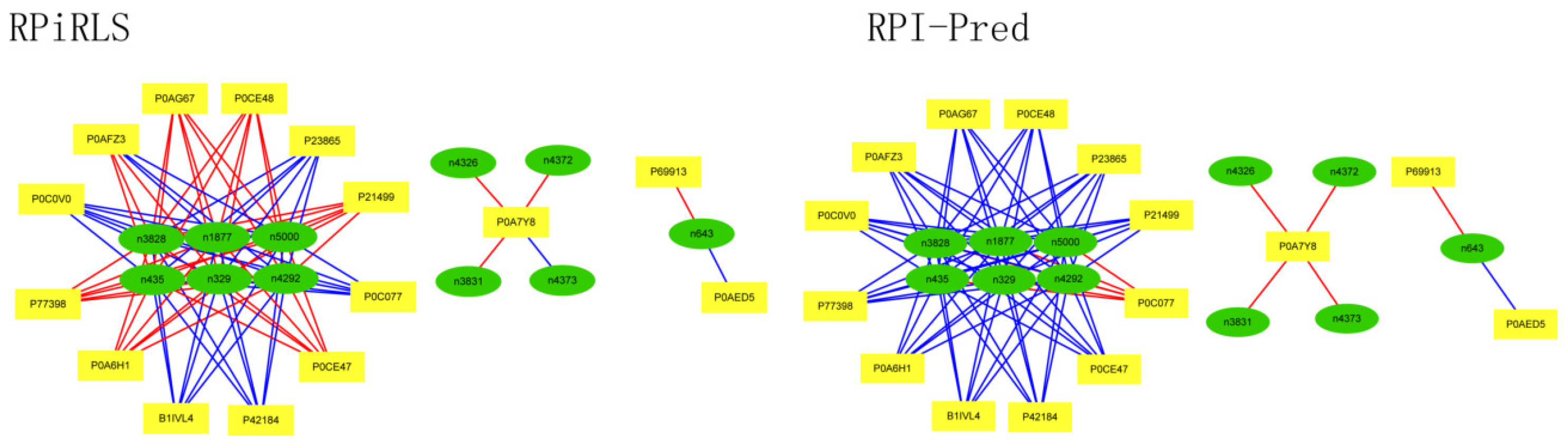
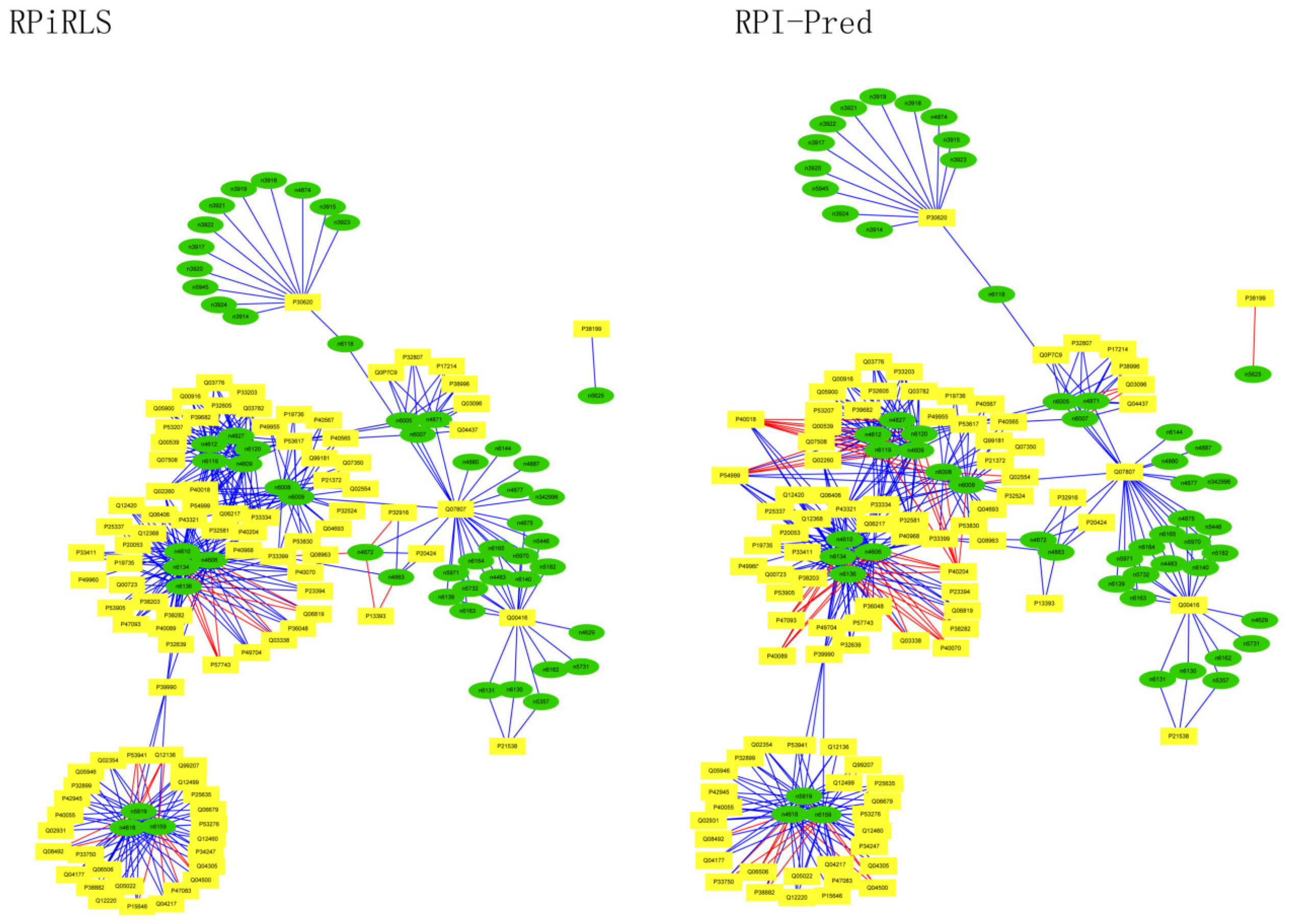
Performance on Predicting LncRNA-Protein Interaction Networks
3. Discussion
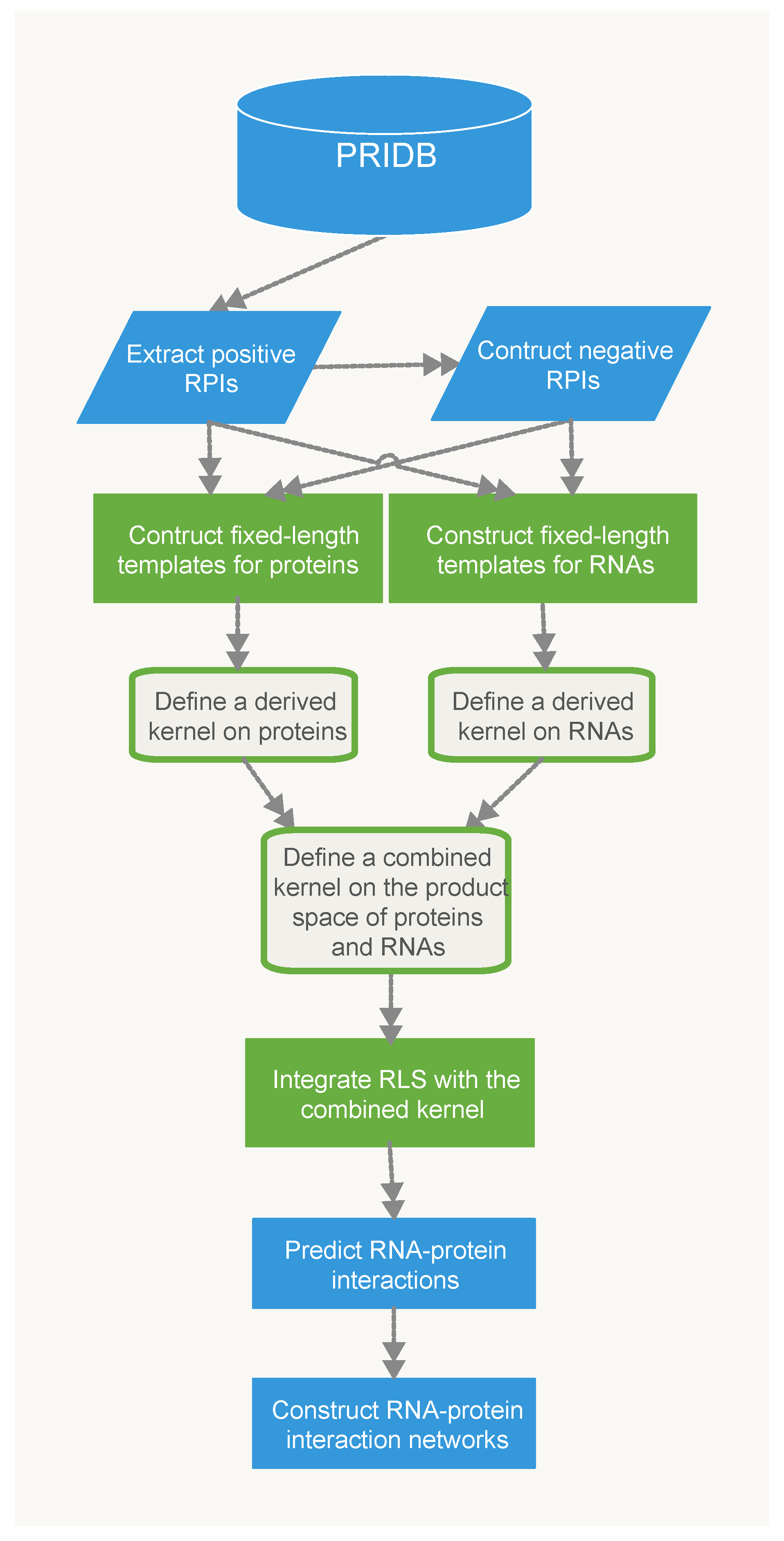
4. Materials and Methods
4.1. Training Data Set
4.2. Test Data Sets
4.3. Methods
4.3.1. Derived Kernel
- Step 1.
- Set an initial kernel at the first layer. Here the initial kernel is defined as:where . and are substrings of the same length k. if and only if for .
- Step 2.
- Let , Denote by the length of f, so here . Then define the second layer neural response of f at t :where is the template set at the first layer, here we consider all possible substrings of length k making up the template set , so here . is the transformation set at the first layer. , , for . The second layer neural response of f, denoted as , defines a map as .
- Step 3.
- Compute the second layer derived kernel by normalizing the inner product of two neural responses:where denotes the inner product with respect to the uniform measure , where is the cardinality of the template set and is the Dirac measure; .With correlation normalization:
4.3.2. Regularized Least Squares
4.3.3. Integrate RLS with the Combined Kernel
4.4. Prediction Measures
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Akbaripour-Elahabad, M.; Zahiri, J.; Rafeh, R.; Eslami, M.; Azari, M. rpiCOOL: A tool for In Silico RNA–protein interaction detection using random forest. J. Theor. Biol. 2016, 402, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.; Cameron, V.; de Haseth, P.L. Uhlenbeck, O.C.Sequence-specific interaction of R17 coat protein with its ribonucleic acid binding site. Biochemistry 1983, 22, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, S. RNA-RNA and RNA-rotein interactions in coronavirus replication and transcription. Rna Biol. 2011, 8, 237–248. [Google Scholar]
- Li, Z.; Nagy, P.D. Diverse roles of host RNA binding proteins in RNA virus replication. Rna Biol. 2011, 8, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Rinn, J.L. RNA–protein interactions in human health and disease. Semin. Cell Dev. Biol. 2011, 22, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.B. The three-dimensional Structure of the ribosome and its components. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 35–58. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V.; White, S.W. Ribosomal protein structures: Insights into the architecture, machinery and evolution of the ribosome. Trends Biochem. Sci. 1998, 23, 208–212. [Google Scholar] [CrossRef]
- Kim, M.Y.; Hur, J.; Jeong, S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep. 2009, 42, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Darnell, R.B. RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet. 2010, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Mohamadkhani, A. Long Noncoding RNAs in Interaction With RNA Binding Proteins in Hepatocellular Carcinoma. Hepat. Mon. 2014, 14, e18794. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human, HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; De, S.; Srikantan, S.; Abdelmohsen, K.; Grammatikakis, I.; Kim, J.; Kim, K.M.; Noh, J.H.; White, E.J.; Martindale, J.L.; et al. PAR-CLIP analysis uncovers, AUF1 impact on target RNA fate and genome integrity. Nat. Commun. 2013, 5, 5248. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, S.T.; Boeva, V.; Escamilla-Del-Arenal, M.; Ancelin, K.; Granier, C.; Matias, N.R.; Sanulli, S.; Chow, J.; Schulz, E.; Picard, C.; et al. Jarid2 Is Implicated in the Initial Xist-Induced Targeting of, PRC2 to the Inactive X Chromosome. Mol. Cell 2014, 53, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Chen, X.; Ping, Z.; Qi, Z. Long non-coding RNA GAS5 inhibited hepatitis C virus replication by binding viral NS3 protein. Virology 2016, 492, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. Electron. Ed. 2006, 1, 302. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Anna-Carina Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.J.; Mark, B.; Susan, J. Protein-RNA interactions: Structural analysis and functional classes. Proteins Struct. Funct. Bioinform. 2007, 66, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Baroni, T.E.; Chittur, S.V.; George, A.D.; Tenenbaum, S.A. Advances in RIP-Chip Analysis: RNA-Binding Protein Immunoprecipitation-Microarray Profiling. Methods Mol. Biol. 2008, 419, 93–108. [Google Scholar] [PubMed]
- Buenrostro, J.D.; Araya, C.L.; Chircus, L.M.; Layton, C.J.; Chang, H.Y.; Snyder, M.P.; Greenleaf, W.J. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat. Biotechnol. 2014, 32, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Pancaldi, V.; Bähler, J. In silico characterization and prediction of global protein-mRNA interactions in yeast. Nucleic Acids Res. 2011, 39, 5826–5836. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, M.; Agostini, F.; Masin, M.; Tartaglia, G.G. Predicting protein associations with long noncoding RNAs. Nat. Methods 2011, 8, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, U.K.; Honavar, V.G.; Dobbs, D. Predicting RNA-Protein Interactions Using Only Sequence Information. BMC Bioinform. 2011, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Liu, Z.P.; Huang, Q.; Wang, Y.; Xu, D.; Zhang, X.S.; Chen, R.; Chen, L. De novo prediction of RNA-protein interactions from sequence information. Mol. Biosyst. 2013, 9, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Ren, S.; Ming, L.; Yong, Z.; Zhu, D.; Zhang, X.; Li, T. Computational prediction of associations between long non-coding RNAs and proteins. BMC Genom. 2013, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Suresh, V.; Liu, L.; Adjeroh, D.; Zhou, X. RPI-Pred: Predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 2015, 43, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fan, Y.X.; Yan, J.; Shen, H.B. IPMiner: Hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genom. 2016, 17, 582. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, R.M.; Lippert, R.A. Notes on Regularized Least Squares. Wseas Org. 2007, 10, 1218–1225. [Google Scholar]
- Cesa-Bianchi, N. Applications of regularized least squares to pattern classification. Theor. Comput. Sci. 2007, 382, 221–231. [Google Scholar] [CrossRef]
- Hermann, T.; Westhof, E. Simulations of the dynamics at an RNA-protein interface. Nat. Struct. Biol. 1999, 6, 540–544. [Google Scholar] [PubMed]
- Han, L.Y.; Cai, C.Z.; Lo, S.L.; Chung, M.C.; Chen, Y.Z. Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA 2004, 10, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Terribilini, M.; Lee, J.H.; Yan, C.; Jernigan, R.L.; Honavar, V.; Dobbs, D. Prediction of RNA binding sites in proteins from amino acid sequence. RNA 2006, 12, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Kohavi, R. A Study of Cross-Validation and Bootstrap for Accuracy Estimation and Model Selection. In Proceedings of the 14th International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; Morgan Kaufmann: San Francisco, CA, USA, 1995; pp. 1137–1143. [Google Scholar]
- Wu, T.; Wang, J.; Liu, C.; Zhang, Y.; Shi, B.; Zhu, X.; Zhang, Z.; Skogerbø, G.; Chen, L.; Lu, H.; et al. NPInter: The noncoding RNAs and protein related biomacromolecules interaction database. Nucleic Acids Res. 2006, 34, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chang, H. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.J.; Fu, H.J.; Wu, Y.G.; Zheng, X.F. Function of lncRNAs and approaches to lncRNA-protein interactions. Sci. China Life Sci. 2013, 56, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.F.; Parker, R. Principles and Properties of Eukaryotic mRNPs. Mol. Cell 2014, 54, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Milek, M.; Wyler, E.; Landthaler, M. Transcriptome-wide analysis of protein–RNA interactions using high-throughput sequencing. Semin. Cell Dev. Biol. 2012, 23, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.; Chung, I.F.; Miyano, S. A neural network method for identification of RNA-interacting residues in protein. Genome Inform. 2004, 15, 105–116. [Google Scholar] [PubMed]
- Murakami, Y.; Spriggs, R.V.; Nakamura, H.; Jones, S. PiRaNhA: A server for the computational prediction of RNA-binding residues in protein sequences. Nucleic Acids Res. 2010, 38, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Wu, L.Y.; Wang, Y.; Zhang, X.S.; Chen, L. Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics 2010, 26, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Roy, N.; Babu, M.M.; Janga, S.C. Dissecting the expression dynamics of RNA-binding proteins in posttranscriptional regulatory networks. Proc. Natl. Acad. Sci. USA 2009, 106, 20300–20305. [Google Scholar] [CrossRef] [PubMed]
- Kishore, S.; Luber, S.; Zavolan, M. Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief. Funct. Genom. 2010, 9, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.A.; Walia, R.R.; Terribilini, M.; Ferguson, J.; Zheng, C.; Honavar, V.; Dobbs, D. PRIDB: A Protein-RNA interface database. Nucl. Acids Res. 2011, 39, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.; Rosasco, L.; Bouvrie, J.; Caponnetto, A.; Poggio, T. Mathematics of the Neural Response. Found. Comput. Math. 2010, 10, 67–91. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.J.; Wong, H.S.; Xiao, Q.W.; Guo, X.; Smale, S. Introduction to the Peptide Binding Problem of Computational Immunology: New Results. Found. Comput. Math. 2013, 14, 951–984. [Google Scholar] [CrossRef]
- Golub, G.H.; Wahba, G. Generalized cross-validation as a method for choosing a good ridge parameter. Technometrics 1977, 21, 215–223. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Template Sizes | l = 1 | l = 2 | l = 3 | l = 4 | l = 5 | l = 6 | l = 7 | l = 8 |
|---|---|---|---|---|---|---|---|---|
| k = 1 | 0.705 | 0.813 | 0.850 | 0.872 | 0.851 | 0.832 | 0.816 | 0.802 |
| k = 2 | 0.375 | 0.767 | 0.853 | 0.911 | 0.926 | 0.920 | 0.915 | 0.910 |
| k = 3 | 0.219 | 0.644 | 0.802 | 0.881 | 0.910 | 0.921 | 0.924 | 0.922 |
| k = 4 | 0.202 | 0.321 | 0.767 | 0.854 | 0.887 | 0.902 | 0.912 | 0.918 |
| Template Sizes | l = 1 | l = 2 | l = 3 | l = 4 | l = 5 | l = 6 | l = 7 | l = 8 |
|---|---|---|---|---|---|---|---|---|
| k = 1 | 0.673 | 0.763 | 0.812 | 0.817 | 0.779 | 0.769 | 0.756 | 0.743 |
| k = 2 | 0.412 | 0.730 | 0.796 | 0.830 | 0.830 | 0.814 | 0.800 | 0.794 |
| k = 3 | 0.261 | 0.646 | 0.731 | 0.784 | 0.811 | 0.823 | 0.821 | 0.815 |
| k = 4 | 0.243 | 0.317 | 0.702 | 0.747 | 0.785 | 0.804 | 0.816 | 0.824 |
| Template Sizes | l = 1 | l = 2 | l = 3 | l = 4 | l = 5 | l = 6 | l = 7 | l = 8 |
|---|---|---|---|---|---|---|---|---|
| k = 1 | 0.663 | 0.737 | 0.776 | 0.766 | 0.733 | 0.656 | 0.605 | 0.578 |
| k = 2 | 0.644 | 0.746 | 0.792 | 0.803 | 0.783 | 0.770 | 0.760 | 0.752 |
| k = 3 | 0.433 | 0.755 | 0.796 | 0.823 | 0.816 | 0.795 | 0.791 | 0.782 |
| k = 4 | 0.347 | 0.673 | 0.764 | 0.805 | 0.822 | 0.816 | 0.803 | 0.794 |
| k = 5 | 0.262 | 0.615 | 0.727 | 0.779 | 0.804 | 0.816 | 0.821 | 0.813 |
| k = 6 | 0.242 | 0.320 | 0.703 | 0.754 | 0.791 | 0.808 | 0.815 | 0.818 |
| Measurements | RPiRLS | RPiRLS-7G | RPI-Pred | IPMiner |
|---|---|---|---|---|
| Accuracy | 0.85 | 0.79 | 0.49 | 0.50 |
| AUC | 0.92 | 0.90 | - | - |
| Specificity | 0.84 | 0.72 | 0.34 | 0.52 |
| Sensitivity | 0.86 | 0.87 | 0.63 | 0.48 |
| Measurements | RPiRLS | RPiRLS-7G | RPI-Pred | IPMiner |
|---|---|---|---|---|
| Accuracy | 0.80 | 0.67 | 0.49 | 0.50 |
| AUC | 0.80 | 0.74 | - | - |
| Specificity | 0.82 | 0.58 | 0.38 | 0.20 |
| Sensitivity | 0.79 | 0.76 | 0.61 | 0.79 |
| Organism | # ncRPIs | RPiRLS | RPiRLS-7G | RPI-Pred |
|---|---|---|---|---|
| Caenorhabditis elegans | 36 | 0.92 | 0.61 | 0.92 |
| Drosophila melanogaster | 95 | 0.80 | 0.52 | 0.88 |
| Escherichia coli | 202 | 0.54 | 0.52 | 0.90 |
| Homo sapiens | 8246 | 0.92 | 0.74 | 0.86 |
| Mus musculus | 3669 | 0.91 | 0.80 | 0.94 |
| Saccharomyces cerevisiae | 905 | 0.91 | 0.83 | 0.80 |
| Weighted average | 13,153 | 0.91 | 0.76 | 0.88 |
| Organism | # lncRPIs | RPiRLS | RPiRLS-7G | RPI-Pred |
|---|---|---|---|---|
| Caenorhabditis elegans | 4 | 1.00 | 0.75 | 0.75 |
| Drosophila melanogaster | 61 | 0.87 | 0.69 | 1.00 |
| Escherichia coli | 78 | 0.45 | 0.45 | 0.86 |
| Homo sapiens | 8039 | 0.93 | 0.74 | 0.86 |
| Mus musculus | 3495 | 0.92 | 0.83 | 0.95 |
| Saccharomyces cerevisiae | 437 | 0.94 | 0.90 | 0.87 |
| Weighted average | 12,114 | 0.92 | 0.77 | 0.89 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, W.-J.; Cui, W.; Chen, D.; Zhang, J.; Xu, J. RPiRLS: Quantitative Predictions of RNA Interacting with Any Protein of Known Sequence. Molecules 2018, 23, 540. https://doi.org/10.3390/molecules23030540
Shen W-J, Cui W, Chen D, Zhang J, Xu J. RPiRLS: Quantitative Predictions of RNA Interacting with Any Protein of Known Sequence. Molecules. 2018; 23(3):540. https://doi.org/10.3390/molecules23030540
Chicago/Turabian StyleShen, Wen-Jun, Wenjuan Cui, Danze Chen, Jieming Zhang, and Jianzhen Xu. 2018. "RPiRLS: Quantitative Predictions of RNA Interacting with Any Protein of Known Sequence" Molecules 23, no. 3: 540. https://doi.org/10.3390/molecules23030540
APA StyleShen, W. -J., Cui, W., Chen, D., Zhang, J., & Xu, J. (2018). RPiRLS: Quantitative Predictions of RNA Interacting with Any Protein of Known Sequence. Molecules, 23(3), 540. https://doi.org/10.3390/molecules23030540