2. Results and Discussion
Compound
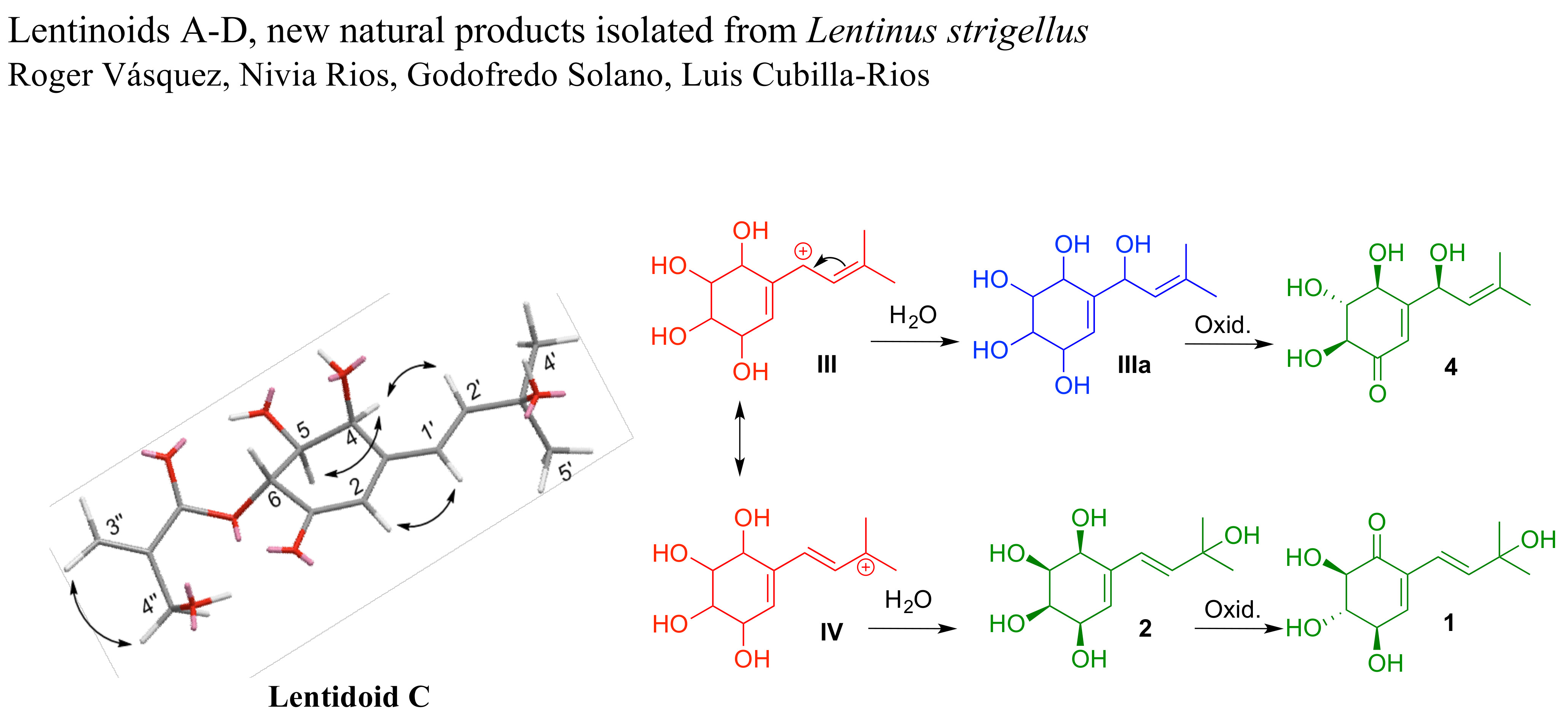
1 was obtained as a brown viscous liquid. Its molecular formula, C
11H
16O
5, was determined by High-resolution electrospray ionisation time-of-flight mass spectrometry (HR-ESI-TOF-MS) analysis on the basis of its cationized molecular peak [M + Na]
+ at
m/
z 251.0891. The ion at
m/
z 269.0568 (100%) corresponded to [M + H
2O + Na
+] and the tandem mass spectrometry (MS
2) spectrum showed that this ion is derived from the cluster [2M + H
2O + Na
+],
m/
z 497.0824. The Infrared (IR) spectrum showed bands at 3350 and 1690 cm
−1 corresponding to a hydroxyl and carbonyl group, respectively. The nuclear magnetic resonance (NMR) data (
Table 1) revealed the existence of an isoprenyl unit: two methyl groups (δ
H 1.30 (s); δ
C 29.7), a quaternary sp
3 oxycarbon (δ
C 71.4), and two methines sp
2 (δ
H 6.36, 6.44 (d); δ
C 120.1, 143.1). These signals have been observed in striguellone A (
5) [
3], a compound previously isolated from a culture of
L.
strigellus. This was confirmed by the correlation spectroscopy (COSY) experiment.
In the 13C-NMR spectrum, there was a quaternary carbon at δC 191.8 indicative of an α,β-unsaturated ketone. The chemical shift for the olefinic proton at C-3 displayed low-field shift at 6.89 ppm and appeared as a doublet, due to the coupling between H-3 and H-4, while in striguellone A, this appears as a singlet at 6.04 ppm (C-2) due the absence of this coupling.
The quaternary olefinic carbon (δ
C 135.2) at the α-position of the α,β-unsaturated ketone displayed a high-field shift in comparison to C-3 (δ
C 155.7) in striguellone A; additionally, the methine carbon (δ
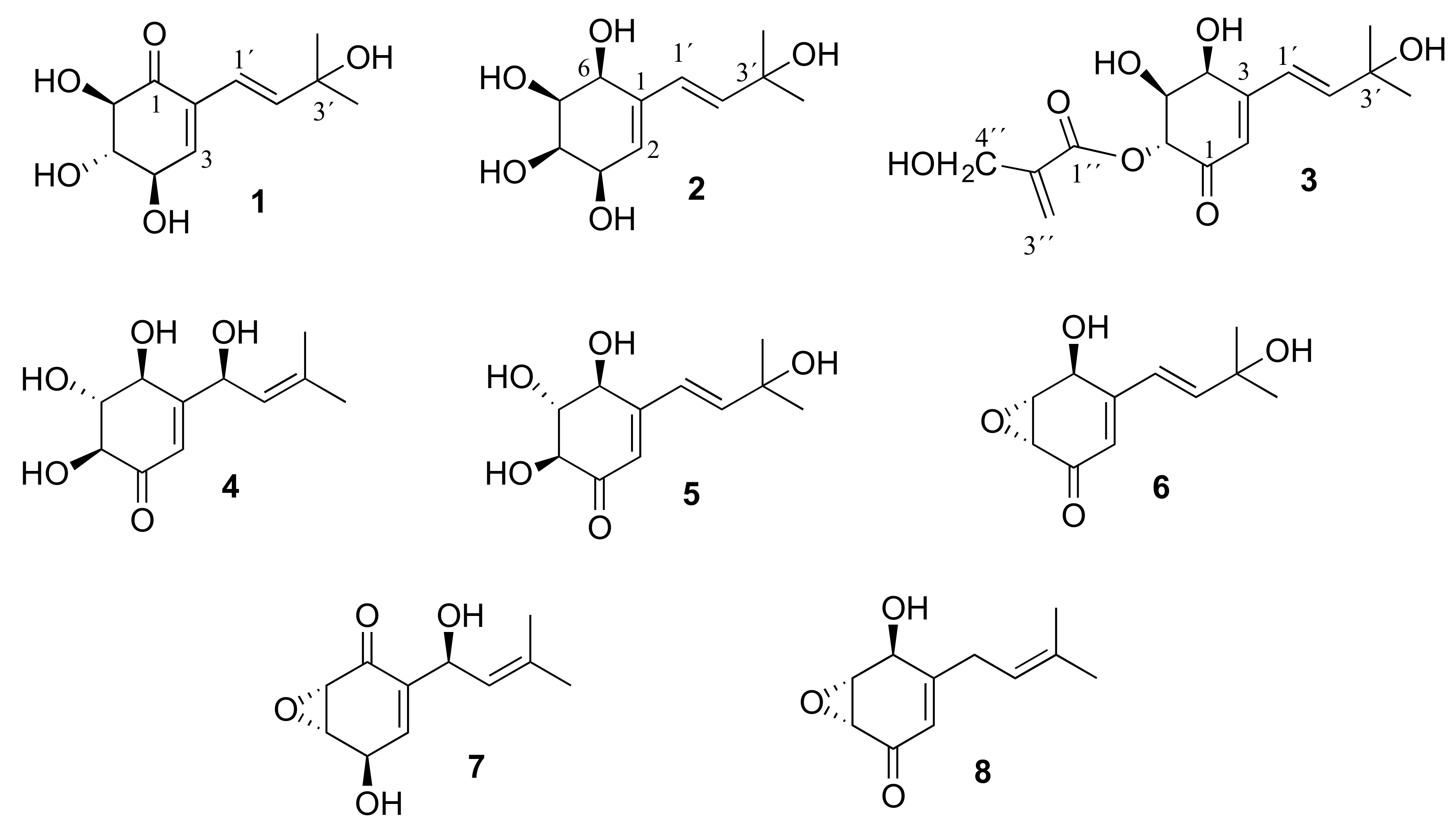
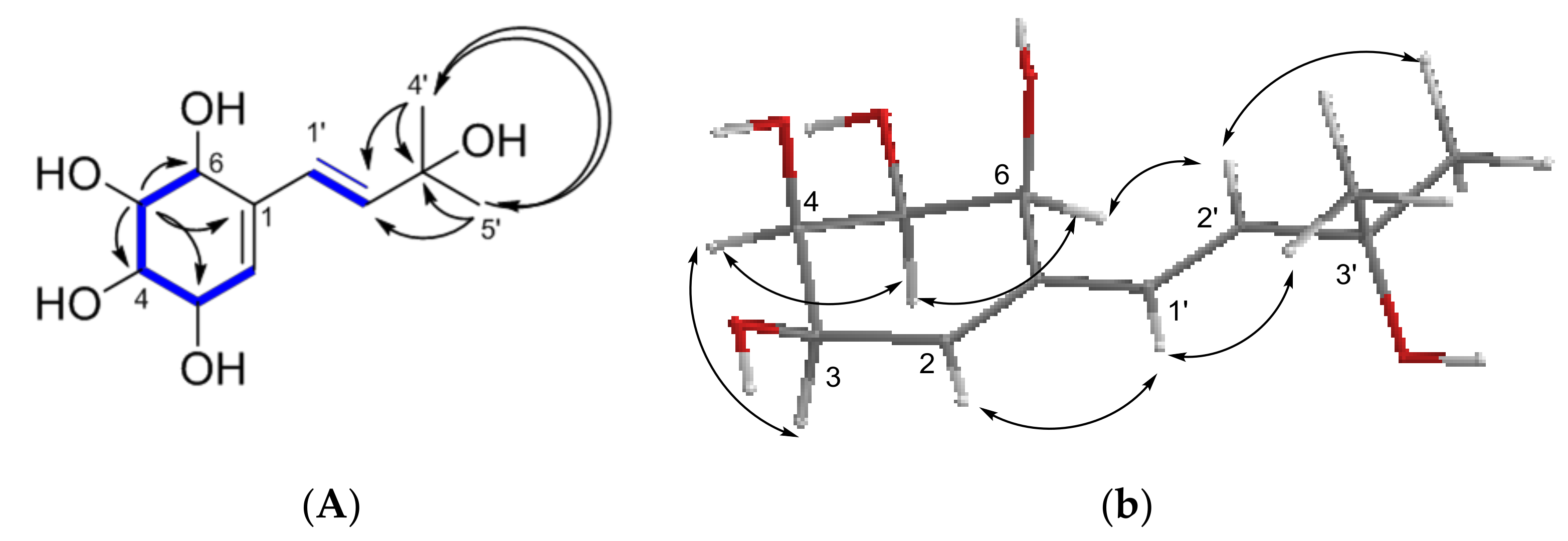
C 145.4) appeared at low-field shift occupying the β-position. The COSY spectrum revealed correlations between H-3/H-4, H-4/H-5, H-5/H-6 and H-1′/H-2′ (
Figure 2A). The position of the carbonyl group was confirmed by the heteronuclear multiple-bond correlation (HMBC) experiment (
Figure 2A). The nuclear Overhauser-effect spectroscopy (NOESY) experiment showed, clearly, correlation of H-1′/H-2′ with H-3 and H-4′/H-5′. On the other hand, a correlation between H-3 and H-4 was observed (
Figure 2B). No interaction was detected for H-4/H-5 and H-5/H-6, placing them, in both cases, on opposite sides, which was confirmed by the coupling constants (3.6 and 10.8 Hz, respectively). Thereafter, we have assigned the new name lentinoid A to compound
1.
For compound
2, a brown viscous liquid, the molecular formula C
11H
18O
5 was assigned, which has three degrees of unsaturation according to its HR-ESI-TOF-MS data ([M + Na]
+,
m/z 253.1034, calcd. 253.1046 for C
11H
18O
5Na). The ion at
m/z 235.0940 (100%) in the mass spectrum corresponded to the loss of water [M + Na − H
2O]
+, which was confirmed by an MS
2 experiment. The IR spectrum showed a band at 3300 cm
−1 corresponding to hydroxyl groups, although no signal for a carbonyl group was observed. The COSY and HMBC experiments (
Figure 3A) confirmed the presence of an isoprenyl unit as in compound
1 (δ
H 1.30 (s); δ
C 29.9), a quaternary sp
3 oxycarbon (δ
C 71.4) and two sp
2 methines (δ
H 6.15, 6.24 (d); δ
C 126.5, 140.9). Above,
2 showed a quaternary sp
2 carbon (δ
C 136.5), an sp
2 methine (δ
H 5.67 (br t); δ
C 125.9) and four sp
3 oxymethines (δ
H 3.37 (t), 3.49 (br m), 4.35 (d), 4.59 (br d); δ
C 57.1, 56.0, 63.9, 65.3).
The union of the oxymethines was established based on the COSY experiment (
Figure 3A), observing correlations between H-2/H-3, H-3/H-4, H-4/H-5 and H-5/H-6, and the corresponding coupling constants. The relative stereochemistry was determined taking into account the following NOESY correlations: H-3/H-4, H-4/H-5 and H-5/H-6 (
Figure 3B and
Figure S2.6), as well as the values of the coupling constants (all bellow 4.6 Hz) between the hydrogens of the cyclohexyl unit. Therefore, all the hydroxyl groups were determined to be on the same site of the molecule. See
1H- and
13C-NMR data in
Table 1. We have assigned the new name lentinoid B to compound
2.
For lentinoid C (
3), the molecular formula C
15H
20O
7 was assigned with six degrees of unsaturation according to its HR-ESI-TOF-MS data, [M + Na]
+,
m/z 335.1109 (calcd. 335.1101 for C
15H
20O
7Na), with the bimolecular ion presented at
m/z 647.2311. The IR spectrum showed a band at 3400 cm
−1 corresponding to an OH group, and two bands for carbonyl groups: 1632 cm
−1 for an α,β-unsaturated ketone and 1649 cm
−1 for an α,β-unsaturated ester. Compound
3 showed similar NMR data (
Table 2) to striguellone A, including the prenyl unit (two methyls (δ
C 29.6 and 29.7), an oxygenated quaternary carbon (δ
C 71.6), two sp
2 carbons (δ
C 126.6 and 149.5)) and the α,β-unsaturated ketone (carbonyl group (δ
C 194.9), an sp
2 quaternary carbon (δ
C 156.9) and sp
2 methyne (δ
C 125.9)). Moreover, three oxygenated sp
3 carbons were present (δ
C 76.6, 71.8 and 67.9). Altogether, the signals resemble the prenyl group and the cyclohexenyl ketone characteristic of lentinoids.
Additionally, the NMR data revealed the existence of a terminal methylene (δ
C 125.5, δ
H 5.96 and 6.35), a hydroxymethylene (δ
C 61.6, δ
H 4.34) and two quaternary carbons (δ
C 141.6 and δ
C 167.0), with the last one characteristic of a carboxyester group. The 2D NMR data (
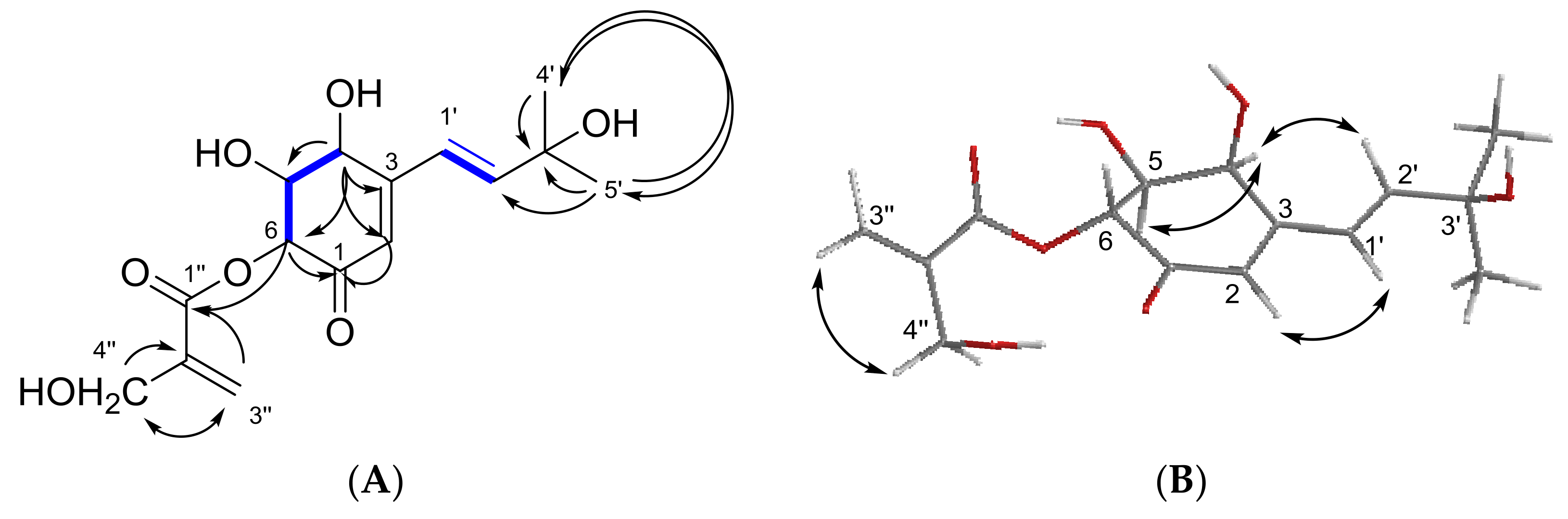
Figure 4A) showed the correlations between all protons and carbons for this fragment of compound
3. Moreover, the connection of the carboxy group and the cyclohexenyl ring was established through the correlation of H-6 (δ
H 5.72, δ
C 76.6) to both carbonyl groups (δ
C 167.0 and 194.9) in equal intensity.
The rotating frame overhauser effect spectroscopy (ROESY) experiment (
Figure 4B) showed correlation between H-4 and H-5, indicating that they are on the same site of the molecule, and this was confirmed by the coupling constant of 3.6 Hz. No correlation between H-5 and H-6 was observed, indicating that they are in axial positions with a coupling constant of 10.8 Hz. Thus, the relative stereochemistry of compound
3 was established.
Due to possible instability and the small amount of compound 4 obtained, the structure was characterized by its 1H-NMR spectrum. Compound 4 showed signals for the isoprenyl group as in panepoxydone (7) characterized by two methyl groups attached to an sp2 carbon (δH 1.69 and 1.70), a methyne (δH 5.29, d, J = 8.8 Hz) and a hydroxymethyne (δH 5.00, d, J = 8.8 Hz). Based on the chemical shift for the other three hydroxymethynes, the coupling constants (H-6: δH 3.96, d, 12.0; H-5: δH 3.63, dd, 8.4 and 12.0; H-4: δH 4.36, d, 8.4), and the olefin proton (δH 6.89) with a chemical shift characteristic of an α-proton in an α,β-unsaturated ketone, we proposed a structure for the newly discovered lentinoid D (4).
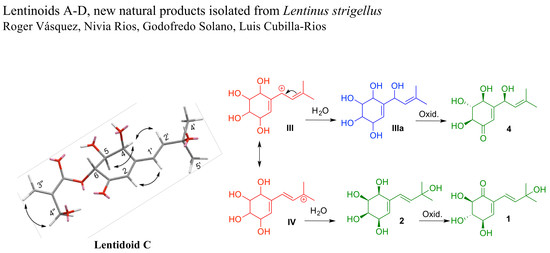
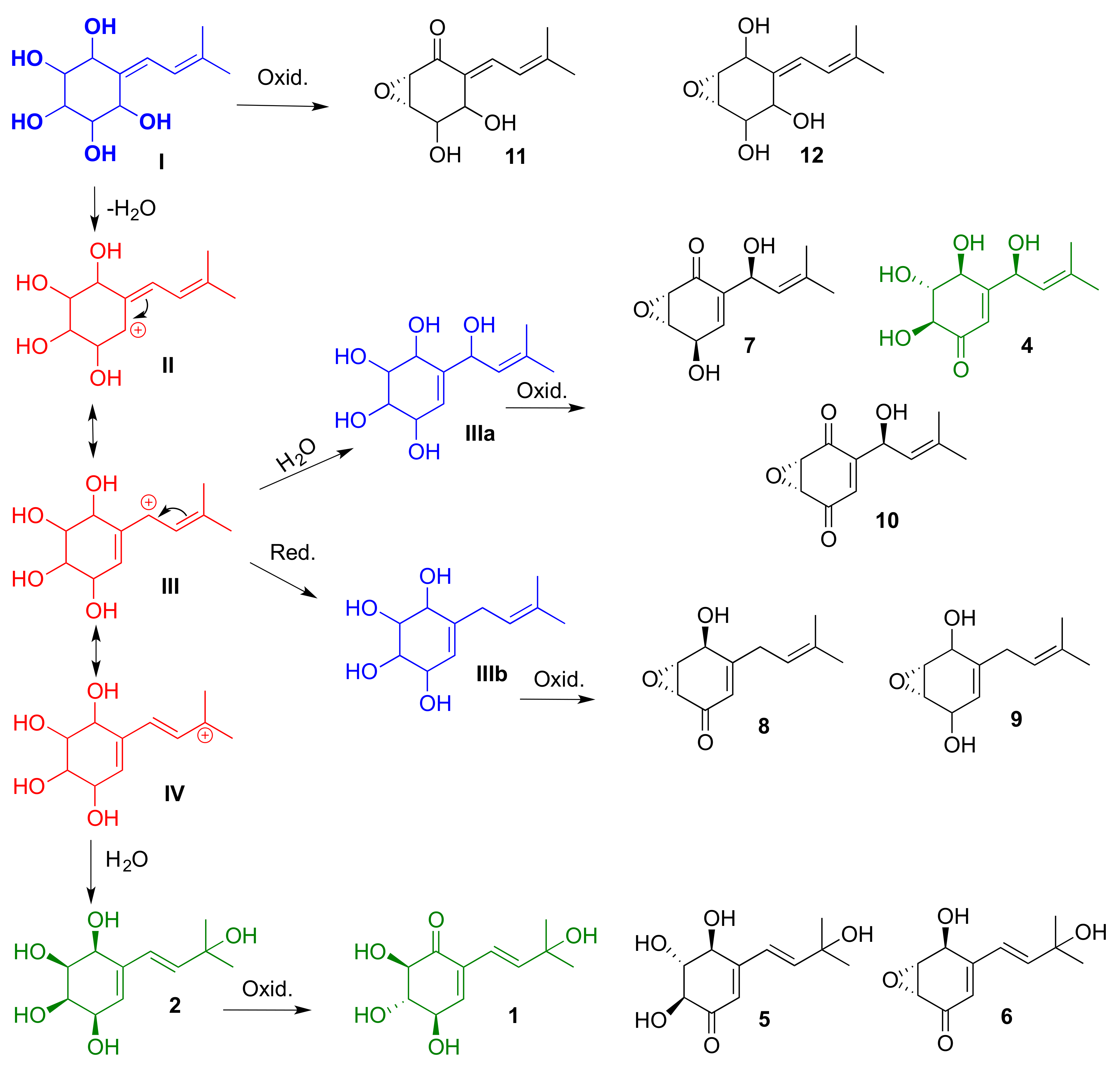
This group of compounds, isolated from lentinoid species (
Lentinus and
Panus), is characterized by an isoprenyl residue and an oxygenated cyclohexenyl ring, and could have similar biogenetic precursors. In
Scheme 1, we proposed a not-yet-isolated precursor (
I), and the structure of three possible intermediates (
II–
IV, in red) for the generation of this series of molecules. The biosynthesis may include two subsequent oxidation reactions of the not-yet-isolated compounds
I,
IIIa,
IIIb and the newly reported lentinoid B (
2). Compounds
I,
IIIa and
IIIb (in blue) could be produced by some species of the genus
Lentinus under different culture conditions.
Antibacterial Activity
The bacterial growth inhibition was evaluated in vitro by the disc diffusion method [
11]. Compounds
1–
3 and
5–
7 were tested at a concentration of 6.25 µg/µL against
Listeria monocytogenes, Enterococcus feacalis, Pseudomonas aeruginosa and
Klebsiella pneumoniae. The first three bacteria were inhibited by at least one of the compounds with an inhibition diameter (ID) ranging from 7.5–9.5 mm. No antibacterial activity was detected against
K. pneumoniae. The positive control used was gentamycin sulfate (ID, 15–25 mm) at a concentration of 1.25 µg/µL (
Table 3).
The minimum inhibitory concentration (MIC) was determined for compounds
1,
2 and
3 using
E. faecalis and
P. aeruginosa (
Table 4); nevertheless, only compound
1 resulted in a significant MIC; to inhibit
E. faecalis, twice the amount of lentinoid A will be required. This could indicate that the presence of the carbonyl group is necessary for the antibacterial activity of lentinoid A compared to lentinoid B.
3. Materials and Methods
3.1. General
General experimental procedures. Optical rotations were measured in methanol on an Autopol V Plus Automatic Polarimeter (Whashington, NJ, USA) using a 10 cm cell. Ultra violet spectra were determined on a Waters 2996 PDA detector (Waters, Milford, MA, USA). IR spectra were determined using an Agilent Cary 630 FTIR spectrophotometer (film) (Agilent, Santa Clara, CA, USA). NMR spectra including 1D and 2D experiments were recorded at 400 MHz (JEOL, Ltd. Tokyo, Japan) and 600 MHz (Bruker, Billerica, MA, USA) using CD3OD and CDCl3 as solvents with tetramethylsilane (TMS) as internal standard. HRMS spectra were acquired on a Bruker micrOTOF-QIII (ESI-TOF), direct injection (Bruker, Billerica, MA, USA). The flash columns were made using silica gel 100 (70–230 mesh ASTM, Merck, Darmstadt, Germany) and silica gel (200–400 mesh, 60 Å, Sigma-Aldrich, St. Louis, MO, USA), the fractions collected were monitored by Sigma-Aldrich TLC plates 3110 (Sigma-Aldrich, St. Louis, MO, USA) and spots were detected using p-anisaldehyde spray reagent after heating. HPLC separations (Waters Delta 600) were carried out using a normal-phase semi-preparative column (YMC-Pack Sil, S-5 μm, 12 nm, 150 × 10 mm ID), a chiral column (QuiralPack IA, 5 μm 150 × 4.6 mm ID, Daicel Corporation, Tokyo, Japan) and a photodiode array detector Waters 2996. The injector was equipped with a 200 μL loop. Chemical structures (1D and 3D (force field MM2)) were drawn using Chemdraw (Perkin Elmer, Waltham, USA), version: 16.0.1.4.
3.2. Fungal Material
Fruiting bodies of
Lentinus strigellus (collection number LC46) [
7] were collected on burned wood near La Nevera highway in the Boquete, a region of Chiriquí Province in Panamá. The specimen was identified as
Lentinus strigellus using molecular phylogenetic analyses. The air-dried specimens were deposited at the National Herbarium of the University of Panamá (PMA115711) and in the Herbarium of the Autonomous University of Chiriquí (UCH7481), Panamá.
3.3. Cultivation and Crude Extract
Initially,
L. strigellus was cultivated in potato dextrose agar (PDA), malt extract agar (MEA) and Sabouraud Dextrose Agar (SDA) using six petri dishes each for 15 days. The chemical compositions of these three extracts were analyzed by thin-layer chromatography and NMR spectroscopy. A greater chemical diversity in the SDA extract using this analysis was detected. Hence, SDA cultivation was evaluated further, including the addition of the elicitors: arginine, glutamic acid, CaCl
2, CuSO
4 and FeSO
4, and at three different pH values: 4.6, 5.6 and 6.6. The period of 15 days and temperature at 26 °C were the same for all experiments. The biomass yield (221 mg) was the highest for SDA with CaCl
2 and a pH of 4.6 (calcium in low concentration tends to increase the yield of biomass produced [
11]; in some cases this could bind proteins causing conformational changes and activating or inactivating mechanisms of catalysis [
12]).
Culturing involving 67 petri dishes and the optimal conditions resulted in 3.10 g of crude ethyl acetate extract. To prepare the extract, the content of 15 petri dishes was transferred into one 1 L glass flask, with 400 mL of ethyl acetate, and extracted by sonicating for 20 min. This was repeated four times. The extract was recovered by drying under reduced pressure at 30 °C [
7].
3.4. Compound Isolation
3.0 g of crude extract was fractionated by flash chromatography (4 cm ID) using silica gel (200–400 mesh, 10 g) and 1 L each of the following solvents: n-hexane, CH2Cl2, EtOAc, acetonitrile and water. All fractions (A–E) were analyzed by TLC. Fraction C (EtOAc), having a mass of 1561.4 mg (50.4%), was subjected to flash chromatography (2.5 cm ID) using silica gel (200–400 mesh, 70 g), and eluted with a gradient system from 100% CH2Cl2 to 100% EtOAc followed by a gradient from 100% EtOAc to 100% MeOH to afford 8 fractions (C1 to C8).
Fraction C6 (74.1 mg, 4.7%) was purified by normal-phase HPLC using a linear gradient (from 75:25 n-hexane–isopropanol to 40:60 n-hexane–isopropanol at 1.00 mL/min) in 55 min, to give 4 fractions, where the peak at tR 17 min corresponded to panepoxydone (7) (8.1 mg, 10.9%) and the peak at tR 21.5 min corresponded to isopanepoxydone (6) (12.3 mg, 16.6%).
Fraction C7 (235.0 mg, 15.1%) was flash chromatographed (2 cm ID) with silica gel (200–400 mesh, 22 g), and n-hexane/EtOAc (v/v = 100:0–0:100) and EtOAc/MeOH (v/v = 100:0–0:100) as eluents, to give 14 fractions (C7.1 to C7.14). C7.9 (76.2 mg, 32.4%) was subjected to normal-phase HPLC using a linear gradient (from 75:25 n-hexane–isopropanol to 40:60 n-hexane–isopropanol at 1.00 mL/min) in 55 min, to give 3 fractions, where the peak at tR 18 min corresponds to striguellone A (5) and the peak at tR 23 min corresponds to lentinoid A (1).
Fraction C7.10 (55.0 mg, 23.4%) was purified by normal-phase HPLC using a linear gradient (of 75:25 n-hexane–isopropanol to 40:60 n-hexane–isopropanol at 1.00 mL/min) in 55 min, to give 6 fractions (I–VI), where the peak at tR 33 min (fraction VI) corresponds to lentinoid B (2) 7.4 mg (13.5%). Fraction V (26.0 mg, 47.3%) was purified by normal-phase HPLC using a linear gradient (85:15 to 30:70 n-hexane–isopropanol at 1.00 mL/min) in 55 min, to give 3 fractions (V.1 to V.3). V.1 (7.3 mg, 28.1%) was purified by HPLC (Chiral Pack IA, 5 µm, 150 × 4.6 mm ID) using the isocratic 85:15 n-hexane–isopropanol at 0.50 mL/min to give lentinoid C (3) (tR 17 min; 2.6 mg, 35.6%). V.2 (5.4 mg, 20.8%) was purified by HPLC (Chiral Pack IA, 5 µm, 150 × 4.6 mm ID) using the isocratic 85:15 n-hexane–isopropanol at 0.50 mL/min to give lentinoid D (4) (tR 12 min; 1 mg, 13.7%).
3.5. Lentinoid A (1)
Brown viscous liquid;
−64.5 (c 0.0029, MeOH); UV (MeOH) λ
max 273 and 212 nm; IR mmax: 3350, 2948, 2876, 1690, 1390 and 1040 cm
−1;
1H- and
13C-NMR data, see
Table 1; (+)-HR-ESI-TOF-MS
m/
z 251.0891 (calcd. 251.0890 for C
11H
16O
5Na).
3.6. Lentinoid B (2)
Brown viscous liquid;
+3.3 (c 0.0024, MeOH); UV (Hx/IP) λ
max 233 nm; IR mmax: 3300, 2942, 2831 and 1021 cm
−1;
1H- and
13C-NMR data, see
Table 1; (+)-HR-ESI-TOF-MS
m/
z 253.1034 [M + Na]
+ (calcd. 253.1046 for C
11H
18O
5Na).
3.7. Lentinoid C (3)
Brown viscous liquid;
−75.0 (c 0.0005, MeOH); UV (MeOH) λ
max 278 and 208 nm; IR mmax: 3280, 2951, 2840, 1649, 1632 and 1013 cm
−1;
1H- and
13C-NMR data, see
Table 2; (+)-HR-ESI-TOF-MS
m/
z 335.1109 (calcd. 335.1101 for C
15H
20O
7Na).
3.8. Antibacterial Activities [13,14]
The antibacterial activity was determined through the susceptibility test of the British Society for Antimicrobial Chemotherapy (BSAC).
3.8.1. Preparation of 0.5 McFarland Turbidity Standard
The turbidity standard was prepared using 0.5 mL of solution of BaCl2 (BaCl2.2H2O; 0.048 M) in a tube containing 99.5 mL of H2SO4 (0.18 M). The absorbance of the McFarland standard (between 0.08 and 0.10 at 625 nm) was verified using a spectrophotometer. The solution was stored in the dark at 24 ± 2 °C.
3.8.2. Preparation of Bacterial Inoculum
Each bacterium was cultivated in Tripticase Soy Agar (TSA) for 24 h; thereafter, five colonies were chosen and transferred into a tube containing saline and isotonic solution and then visually compared with the 0.5 McFarland standard.
3.8.3. Bacterial Growth Inhibition in Vitro
The bacterial growth inhibition was evaluated in vitro by the disc diffusion method using Trypticase Soy Agar (TSA) in Petri dishes (145 mm) and placing a disc (6 mm diameter) with 50 µg of each tested compound dissolved in DMSO. The plate was then incubated for 18 h at 37 °C, and the inhibition area were measured. For the positive control, gentamycin sulfate (10 μg/mL) was used and the negative control was DMSO. The diameter of the inhibition around each disk was measured, providing a measure of zone–of-inhibition (ZOI) in mm (including the diameter of the disc).
3.8.4. Minimum Inhibitory Concentration (MIC)
The minimum inhibitory concentration (MIC) was determined using a stock solution prepared by adding 300 µg of each compound in 3 mL of Trypticase Soy Broth (TSB). Serial dilutions of the evaluated compounds and control were performed to determine the MIC. The evaluated concentrations of each compound were 100.0, 40.0, 33.3, 11.1 and 5.6 μg/mL. Each solution was thereafter inoculated with 50 μL (0.5 McFarland) of a culture of E. faecalis and P. aeruginosa, and incubated at 37 °C for 18 h. The positive control (gentamycin sulfate 10 mg/mL) concentrations were similar to those of the compounds. Additionally, two blanks were used, one with culture medium alone and one with DMSO. Each assay was performed in duplicate.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}