Liposomal Drug Delivery Systems and Anticancer Drugs

 ,
, 

Abstract
:1. Introduction
2. Cancer
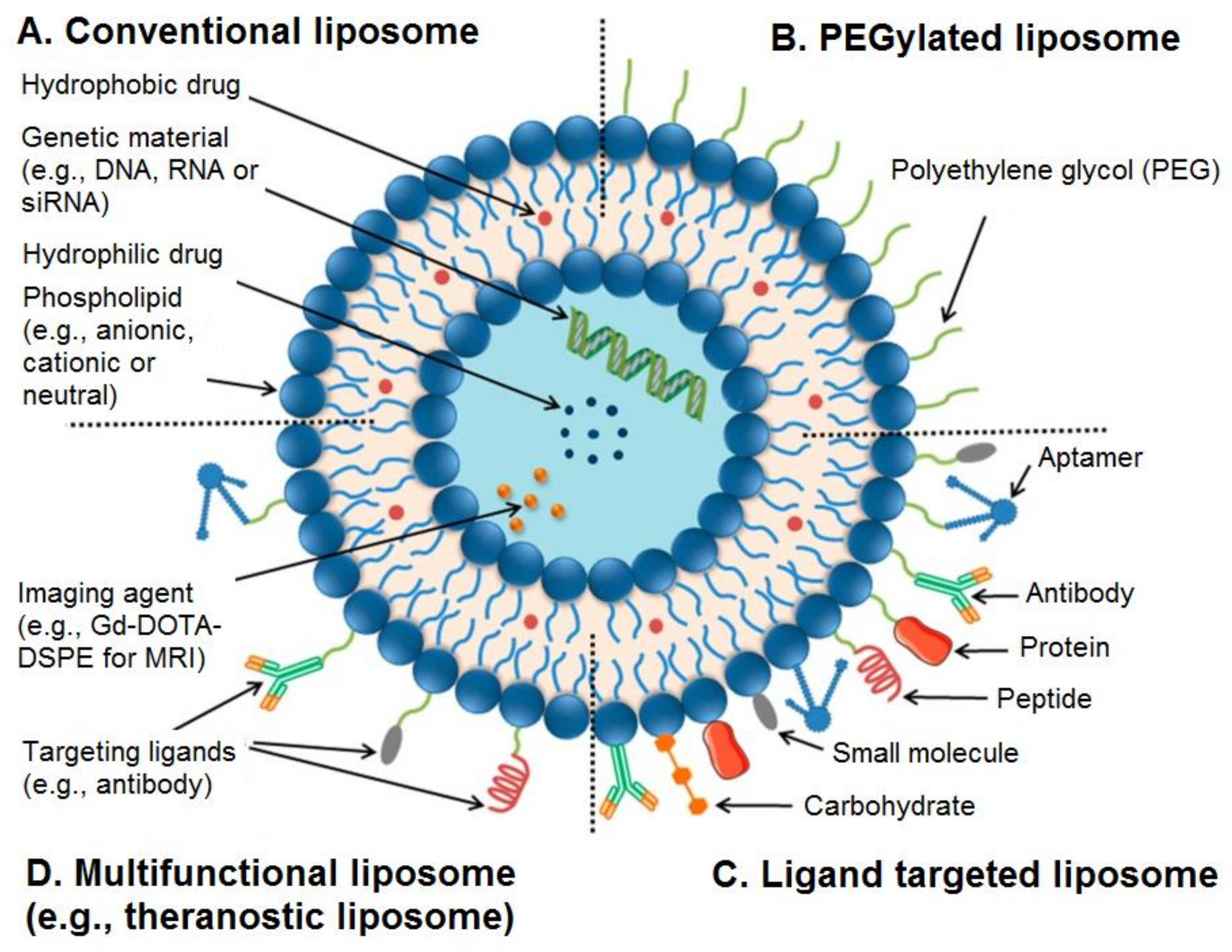
3. Categories of Liposomes
4. Stability of Liposomes
5. Influence of Liposomal Composition in Drug Delivery
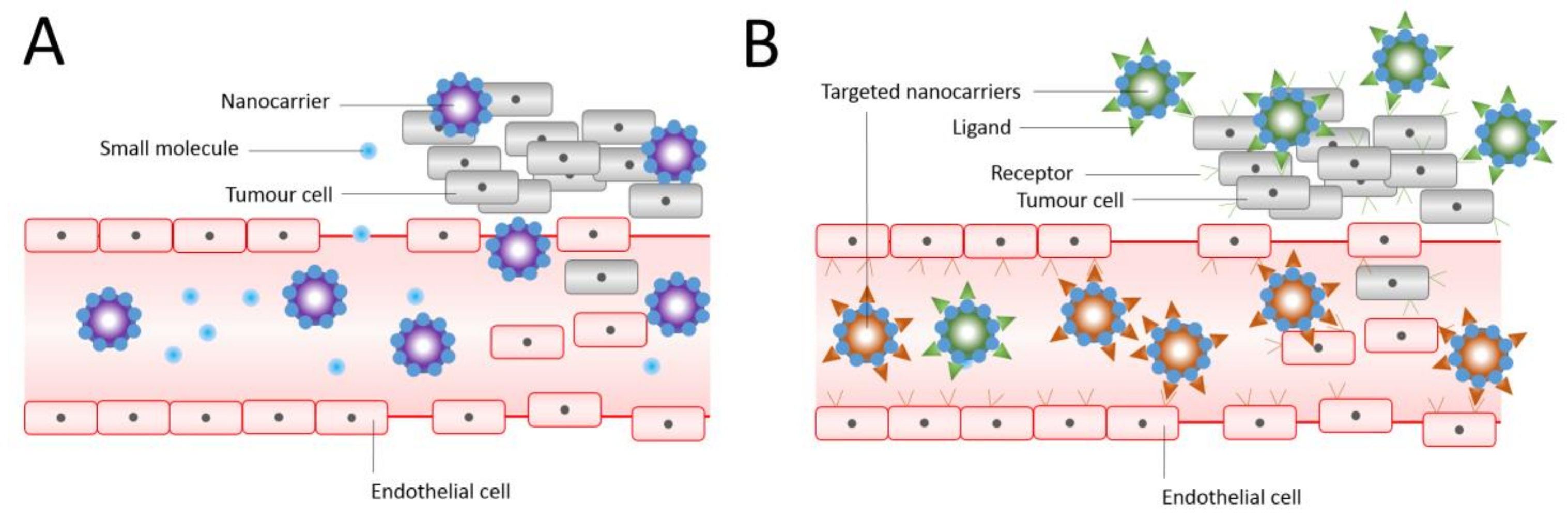
6. Liposomes as Targeted Drug Delivery System for Cancer Treatment
7. Applications of Liposomes in Anticancer Drug Formulations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kumar, A.; Chen, F.; Mozhi, A.; Zhang, X.; Zhao, Y.; Xue, X.; Hao, Y.; Zhang, X.; Wang, P.C.; Liang, X.J. Innovative pharmaceutical development based on unique properties of nanoscale delivery formulation. Nanoscale 2013, 5, 8307–8325. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kieler-Ferguson, H.M.; Chan, D.; Sockolosky, J. Encapsulation, controlled release, and antitumor efficacy of cisplatin delivered in liposomes composed of sterol-modified phospholipids. Eur. J. Pharm. Sci. 2017, 130, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Balzus, B.; Sahle, F.F.; Hönzke, S.; Gerecke, C.; Schumacher, F.; Hedtrich, S.; Kleuser, B.; Bodmeier, R. Formulation and ex vivo evaluation of polymeric nanoparticles for controlled delivery of corticosteroids to the skin and the corneal epithelium. Eur. J. Pharm. Biopharm. 2017, 115, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Al-Kinani, A.A.; Haj-Ahmad, R.; Arshad, M.S.; Chang, M.W.; Alany, R.G.; Ahmad, Z. Electrically atomised formulations of timolol maleate for direct and on-demand ocular lens coatings. Eur. J. Pharm. Biopharm. 2017, 119, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Hui, H.-W.; Lee, T.; Kurtulik, P.; Surapaneni, S. Nanosuspension of a Poorly Soluble Drug via Microfluidization Process. U.S. Patent 9,023,886, 10 November 2009. [Google Scholar]
- Takechi-Haraya, Y.; Goda, Y.; Sakai-Kato, K. Control of liposomal penetration into three-dimensional multicellular tumor spheroids by modulating liposomal membrane rigidity. Mol. Pharm. 2017, 14, 2158–2165. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D. Physical structure and behavior of lipids and lipid enzymes. Adv. Lipid Res. 1963, 1, 65–104. [Google Scholar] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.R.; Weston, N.; Coombes, A.G.A.; Fitzgerald, M.; Perrie, Y. Liposome formulation of poorly water soluble drugs: optimisation of drug loading and ESEM analysis of stability. Int. J. Pharm. 2004, 285, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Patra, B.; Layek, B.; Mukherjee, A. Sustained release of acyclovir from nano-liposomes and nano-niosomes: An in vitro study. Int. J. Nanomed. 2007, 2, 213–225. [Google Scholar]
- Allen, T.M. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol. Sci. 1994, 15, 215–220. [Google Scholar] [CrossRef]
- Allen, T.M.; Martin, F.J. Advantages of liposomal delivery systems for anthracyclines. Semin. Oncol. 2004, 31, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, M.C.; Cosco, D.; Celia, C.; Tudose, A.; Mare, R.; Paolino, D.; Fresta, M. Anticancer activity of all-trans retinoic acid-loaded liposomes on human thyroid carcinoma cells. Colloids Surf. B: Biointerfaces 2017, 150, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cheng, W.W.; Hare, J.I.; Laginha, K.M. Pharmacokinetics and pharmacodynamics of lipidic nano-particles in cancer. Anticancer Agents Med. Chem. 2006, 6, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Wakasugi, M.; Takanaga, H.; Ohtani, H.; Naito, M.; Tsuruo, T.; Sawada, Y. Possibility of the reversal of multidrug resistance and the avoidance of side effects by liposomes modified with MRK-16, a monoclonal antibody to P-glycoprotein. J. Control. Release 2001, 77, 77–86. [Google Scholar] [CrossRef]
- Wang, C.X.; Li, C.L.; Zhao, X.; Yang, H.Y.; Wei, N.; Li, Y.-H.; Zhang, L.; Zhang, L. Pharmacodynamics, pharmacokinetics and tissue distribution of liposomal mitoxantrone hydrochloride. Yao Xue Xue Bao 2010, 45, 1565–1569. [Google Scholar] [PubMed]
- Park, K.; Kwon, I.C.; Park, K. Oral protein delivery: Current status and future prospect. React. Funct. Polym. 2011, 71, 280–287. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Anderson, A.S.; Birch, J.; Forster, A.S.; Rosenberg, G.; Bauld, L.; Vohra, J. Public awareness and healthcare professional advice for obesity as a risk factor for cancer in the UK: a cross-sectional survey. J. Public Health 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Edmiston, S.N.; Parrish, E.; Bryant, C.; Tse, C.K.; Swift-Scanlan, T.; McCullough, L.E.; Kuan, P.F. Breast tumor DNA methylation patterns associated with smoking in the Carolina Breast Cancer Study. Breast Cancer Res. Treat. 2017, 163, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Licaj, I.; Jacobsen, B.K.; Selmer, R.M.; Maskarinec, G.; Weiderpass, E.; Gram, I.T. Smoking and risk of ovarian cancer by histological subtypes: An analysis among 300,000 Norwegian women. Br. J. Cancer 2017, 116, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Stewart, B.W. How do we judge what causes cancer? The meat controversy. Int. J. Cancer 2016, 138, 2309–2311. [Google Scholar] [CrossRef] [PubMed]
- Revenco, T.; Lapouge, G.; Moers, V.; Brohée, S.; Sotiropoulou, P.A. Low dose radiation causes skin cancer in mice and has a differential effect on distinct epidermal stem cells. Stem Cells 2017, 35, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Dumalaon-Canaria, J.A.; Hutchinson, A.D.; Prichard, I.; Wilson, C. What causes breast cancer? A systematic review of causal attributions among breast cancer survivors and how these compare to expert-endorsed risk factors. Cancer Causes Control 2014, 25, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogoi, M.; Kumar, N.; Patra, S. Multifunctional magnetic liposomes for cancer imaging and therapeutic applications. In Nanoarchitectonics Smart Delivery Drug Targeting; Holban, A.M., Grumezescu, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 743–782. [Google Scholar]
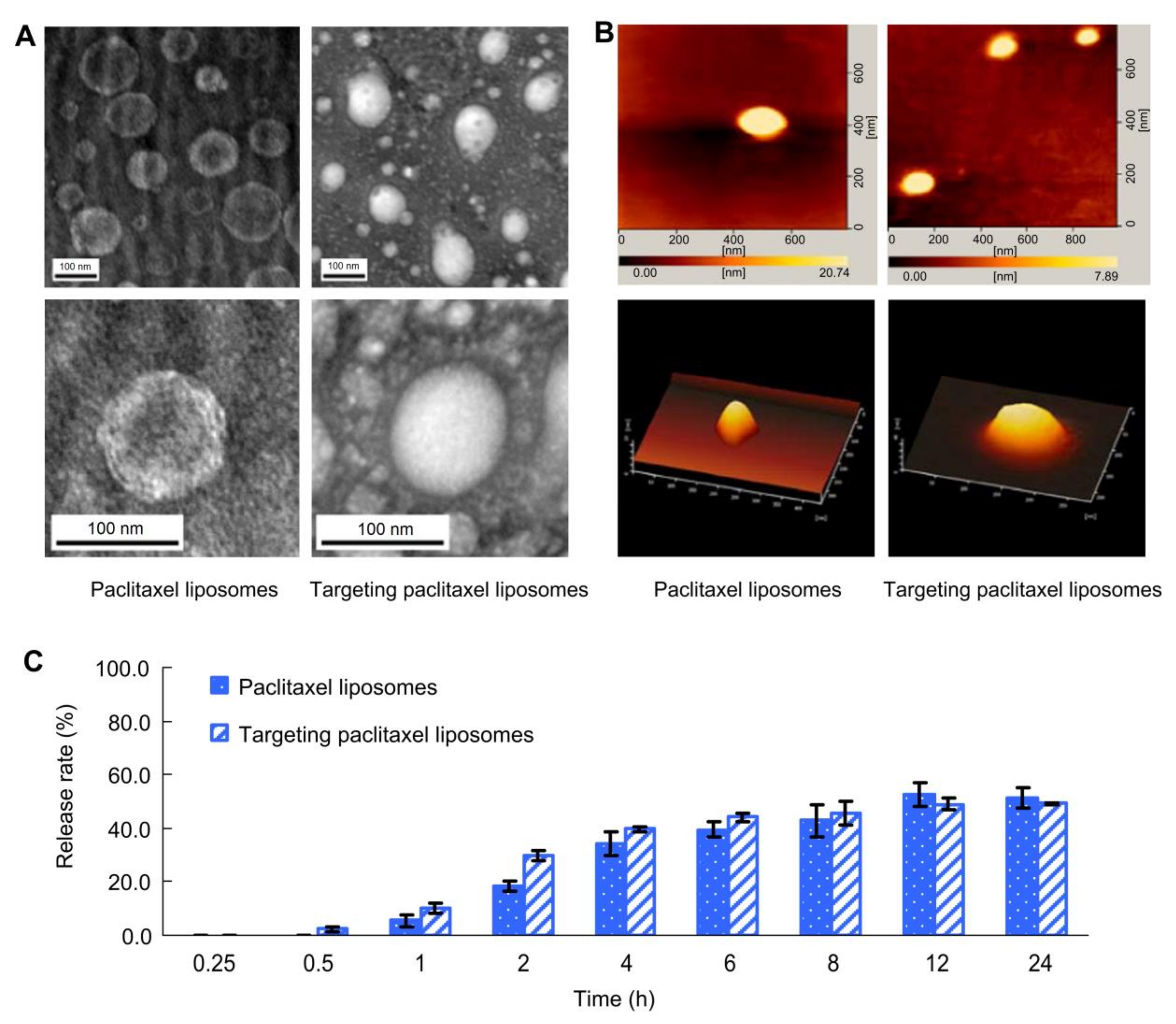
- Zhou, J.; Zhao, W.-Y.; Ma, X.; Ju, R.J.; Li, X.Y.; Li, N.; Sun, M.G.; Shi, J.F.; Zhang, C.X.; Lu, W.L. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials 2013, 34, 3626–3638. [Google Scholar] [CrossRef] [PubMed]
- Forster, J.C.; Harriss-Phillips, W.M.; Douglass, M.J.; Bezak, E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia 2017, 5, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 1999, 51, 691–743. [Google Scholar] [PubMed]
- Maeda, H.; Noguchi, Y.; Sato, K.; Akaike, T. Enhanced vascular permeability in solid tumor is mediated by nitric oxide and inhibited by both new nitric oxide scavenger and nitric oxide synthase inhibitor. Cancer Sci. 1994, 85, 331–334. [Google Scholar] [CrossRef]
- Maeda, H.; Matsumura, Y.; Kato, H. Purification and identification of [hydroxyprolyl3]bradykinin in ascitic fluid from a patient with gastric cancer. J. Biol. Chem. 1988, 263, 16051–16054. [Google Scholar] [PubMed]
- Matsumura, Y.; Kimura, M.; Yamamoto, T.; Maeda, H. Involvement of the kinin-generating cascade in enhanced vascular permeability in tumor tissue. Cancer Sci. 1988, 79, 1327–1334. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Pilat, M.J.; McCormick, J.; LoRusso, P.M. Vascular targeting agents. Curr. Oncol. Rep. 2004, 6, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.M.; Shakil Ahmed, F.R.; Hossen, M.N.; Ahmed, M.; Amran, M.S.; Ul-Islam, M.A. Liposome as a carrier for advanced drug delivery. Pak J. Biol. Sci. 2006, 9, 1181–1191. [Google Scholar]
- Yadav, A.; Murthy, M.S.; Shete, A.S.; Sakhare, S. Stability aspects of liposomes. Indian J. Pharm. Educ. Res. 2011, 45, 402–413. [Google Scholar]
- Briuglia, M.-L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanbarzadeh, S.; Valizadeh, H.; Zakeri-Milani, P. The Effects of Lyophilization on the Physico-Chemical Stability of Sirolimus Liposomes. Adv. Pharm. Bull. 2013, 3, 25–29. [Google Scholar] [PubMed]
- Ball, R.L.; Bajaj, P.; Whitehead, K.A. Achieving long-term stability of lipid nanoparticles: Examining the effect of pH, temperature, and lyophilization. Int. J. Nanomed. 2016, 12, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Patil-Gadhe, A.; Pokharkar, V. Single step spray drying method to develop proliposomes for inhalation: A systematic study based on quality by design approach. Pulm. Pharmacol. Ther. 2014, 27, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Karn, P.R.; Cho, W.; Park, H.-J.; Park, J.-S.; Hwang, S.-J. Characterization and stability studies of a novel liposomal cyclosporin A prepared using the supercritical fluid method: Comparison with the modified conventional Bangham method. Int. J. Nanomed. 2013, 8, 365–377. [Google Scholar]
- Ceh, B.; Lasic, D.D. A rigorous theory of remote loading of drugs into liposomes. Langmuir 1995, 11, 3356–3368. [Google Scholar] [CrossRef]
- Crommelin, D.J. Influence of lipid composition and ionic strength on the physical stability of liposomes. J. Pharm. Sci. 1984, 73, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Frokjaer, S.; Hjorth, E.L.; Worts, O. Stability and storage of liposomes. In Optimization of Drug Delivery; Bundgaard, H., Bagger Hansen, A., Kofod, H., Eds.; Munkgaard: Copenhagen, Denmark, 1982. [Google Scholar]
- Chawdhury, D.F. Pharmaceutical Nanosystems: Manufacture, characterisation and safety. In Pharmaceutical Manufacturing Handbook: Production and Processes; Gad, S.C., Ed.; John Wiley and Sons. Inc.: Hoboken, NJ, USA, 2008; pp. 1286–1326. [Google Scholar]
- Munye, M.M.; Ravi, J.; Tagalakis, A.D.; McCarthy, D.; Ryadnov, M.G.; Hart, S.L. Role of liposome and peptide in the synergistic enhancement of transfection with a lipopolyplex vector. Sci. Rep. 2015, 5, 9292. [Google Scholar] [CrossRef] [PubMed]
- Demel, R.A.; de Kruyff, B. The function of sterols in membranes. Biochim. Biophys. Acta BBA-Biomembr. 1976, 457, 109–132. [Google Scholar] [CrossRef]
- Liu, W.; Wei, F.; Ye, A.; Tian, M.; Han, J. Kinetic stability and membrane structure of liposomes during in vitro infant intestinal digestion: Effect of cholesterol and lactoferrin. Food Chem. 2017, 230, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Garg, T.; Goyal, A.K. Liposomes: Targeted and Controlled Delivery System. Drug Del. Lett. 2014, 4, 62–71. [Google Scholar] [CrossRef]
- Cogan, U.; Shinitzky, M.; Weber, G.; Nishida, T. Microviscosity and order in the hydrocarbon region of phospholipid and phospholipid-cholesterol dispersions determined with fluorescent probes. Biochemistry. 1973, 12, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Kaddah, S.; Khreich, N.; Kaddah, F.; Charcosset, C.; Greige-Gerges, H. Cholesterol modulates the liposome membrane fluidity and permeability for a hydrophilic molecule. Food Chem. Toxicol. 2018, 113, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface functionalization and targeting strategies of liposomes in solid tumor therapy: A review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef] [PubMed]
- Motamarry, A.; Asemani, D.; Haemmerich, D. Thermosensitive Liposomes. In Liposomes; Catala, A., Ed.; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul. 2001, 41, 189–207. [Google Scholar] [CrossRef]
- Kunjachan, S.; Ehling, J.; Storm, G.; Kiessling, F.; Lammers, T. Noninvasive imaging of nanomedicines and nanotheranostics: Principles, progress, and prospects. Chem. Rev. 2015, 115, 10907–10937. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.l.; Ishida, O.; Takizawa, T.; Moribe, K. Possibility of active targeting to tumor tissues with liposomes. Adv. Drug Deliv. Rev. 1999, 40, 89–102. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Magin, R.L.; Yatvin, M.B.; Zaharko, D.S. Liposomes and local hyperthermia: Selective delivery of methotrexate to heated tumors. Science 1979, 204, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Yatvin, M.B.; Weinstein, J.N.; Dennis, W.H.; Blumenthal, R. Design of liposomes for enhanced local release of drugs by hyperthermia. Science 1978, 202, 1290–1293. [Google Scholar] [CrossRef] [PubMed]
- Senior, J.H. Fate and behavior of liposomes in vivo: A review of controlling factors. Crit. Rev. Ther. Drug Carrier Syst. 1987, 3, 123–193. [Google Scholar] [PubMed]
- Cullis, P.R.; Chonn, A.; Semple, S.C. Interactions of liposomes and lipid-based carrier systems with blood proteins: Relation to clearance behaviour in vivo. Adv. Drug Deliv. Rev. 1998, 32, 3–17. [Google Scholar] [PubMed]
- Kong, G.; Anyarambhatla, G.; Petros, W.P.; Braun, R.D.; Colvin, O.M.; Needham, D.; Dewhirst, M.W. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: importance of triggered drug release. Cancer Res. 2000, 60, 6950–6957. [Google Scholar] [PubMed]
- Gaber, M.H.; Hong, K.; Huang, S.K.; Papahadjopoulos, D. Thermosensitive sterically stabilized liposomes: formulation and in vitro studies on mechanism of doxorubicin release by bovine serum and human plasma. Pharm. Res. 1995, 12, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Zenitani, K.; Takagishi, T. Novel pH-sensitive liposomes: liposomes bearing a poly(ethylene glycol) derivative with carboxyl groups. Biochim. Biophys. Acta. 1994, 1193, 1–9. [Google Scholar] [CrossRef]
- Gogoi, M.; Jaiswal, M.K.; Sarma, H.D.; Bahadur, D.; Banerjee, R. Biocompatibility and therapeutic evaluation of magnetic liposomes designed for self-controlled cancer hyperthermia and chemotherapy. Integr. Biol. 2017, 9, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Hardiansyah, A.; Huang, L.-Y.; Yang, M.-C.; Liu, T.Y.; Tsai, S.C.; Yang, C.Y.; Kuo, C.Y.; Chan, T.Y.; Zou, H.M.; Lian, W.N.; et al. Magnetic liposomes for colorectal cancer cells therapy by high-frequency magnetic field treatment. Nanoscale Res. Lett. 2014, 9, 497. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Konicek, B.W.; Deddens, J.A.; Chedid, M.; Hurst, B.M.; Colligan, B.; Neubauer, B.L.; Carter, H.W.; Carter, J.H. Expression of group IIa secretory phospholipase A2 increases with prostate tumor grade. Clin. Cancer Res. 2001, 7, 3857–3861. [Google Scholar] [PubMed]
- Yamashita, S.; Yamashita, J.; Sakamoto, K.; Inada, K.; Nakashima, Y.; Murata, K.; Saishoji, T.; Nomura, K.; Ogawa, M. Increased expression of membrane-associated phospholipase A2 shows malignant potential of human breast cancer cells. Cancer 1993, 71, 3058–3064. [Google Scholar] [CrossRef]
- Kiyohara, H.; Egami, H.; Kako, H.; Shibata, Y.; Murata, K.; Ohshima, S.; Sei, K.; Suko, S.; Kurano, R.; Ogawa, M. Immunohistochemical localization of group II phospholipase A2 in human pancreatic carcinomas. Int. J. Pancreatol. 1993, 13, 49–57. [Google Scholar] [PubMed]
- La Rocca, G.; Pucci-Minafra, I.; Marrazzo, A.; Taormina, P.; Minafra, S. Zymographic detection and clinical correlations of MMP-2 and MMP-9 in breast cancer sera. Br. J. Cancer 2004, 90, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Mook, O.R.F.; Frederiks, W.M.; van Noorden, C.J.F. The role of gelatinases in colorectal cancer progression and metastasis. Biochim. Biophys. Acta BBA-Rev. Cancer 2004, 1705, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Keleg, S.; Büchler, P.; Ludwig, R.; Büchler, M.W.; Friess, H. Invasion and metastasis in pancreatic cancer. Mol. Cancer 2003, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osinsky, S.P.; Ganusevich, I.I.; Bubnovskaya, L.N.; Valkovskaya, N.V.; Kovelskaya, A.V.; Sergienko, T.K.; Zimina, S.V. Hypoxia level and matrix metalloproteinases-2 and -9 activity in Lewis lung carcinoma: correlation with metastasis. Exp. Oncol. 2005, 27, 202–205. [Google Scholar] [PubMed]
- Liu, S.; Bugge, T.H.; Leppla, S.H. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem. 2001, 276, 17976–17984. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Alix, A.J.P.; Sapi, J.; Hornebeck, W.; Bourguet, E. Neutrophil elastase as a target in lung cancer. Anticancer Agents Med. Chem. 2012, 12, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Ogawa, M.; Shirakusa, T. Free-form neutrophil elastase is an independent marker predicting recurrence in primary breast cancer. J. Leukoc. Biol. 1995, 57, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Starcher, B.; O’Neal, P.; Granstein, R.D.; Beissert, S. Inhibition of neutrophil elastase suppresses the development of skin tumors in hairless mice. J. Invest. Dermatol. 1996, 107, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Sokoll, L.J.; Chan, D.W.; Khan, S.R.; Isaacs, J.T. Concentration of enzymatically active prostate-specific antigen (PSA) in the extracellular fluid of primary human prostate cancers and human prostate cancer xenograft models. Prostate 2001, 48, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, F.; Steffen, K.J.; Mallik, S. Enzyme-responsive liposomes for the delivery of anticancer drugs. Bioconjug. Chem. 2017, 28, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.D.; Mager, D.E.; Slack, J.E.; Straubinger, R.M. Effect of repetitive administration of Doxorubicin-containing liposomes on plasma pharmacokinetics and drug biodistribution in a rat brain tumor model. Clin. Cancer Res. 2005, 11, 8856–8865. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.F.; Mason, A.R. European perspective on the costs and cost-effectiveness of cancer therapies. J. Clin. Oncol. 2007, 25, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Yamashita, J.; Ogawa, M. Overexpression of group II phospholipase A2 in human breast cancer tissues is closely associated with their malignant potency. Br. J. Cancer 1994, 69, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Mock, J.N.; Costyn, L.J.; Wilding, S.L.; Arnold, R.D.; Cummings, B.S. Evidence for distinct mechanisms of uptake and antitumor activity of secretory phospholipase A2 responsive liposome in prostate cancer. Integr. Biol. 2013, 5, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Gregoriadis, G. Liposomes as immunological adjuvants. Nature 1974, 252, 252. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Gregoriadis, G. Liposomes as Immunological Adjuvants. In Lymphocytes, Macrophages, and Cancer; Recent Results in Cancer Research/Fortschritte der Krebsforschung/Progrès dans les recherches sur le cancer; Mathe, G., Florentin, I., Simmler, M.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1976; pp. 58–64. [Google Scholar]
- Schwendener, R.A. Liposomes as vaccine delivery systems: A review of the recent advances. Ther. Adv. Vaccines 2014, 2, 159–182. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, G.; Finolezzi, E.; Luciani, S.; Ranucci, E.; Federico, M.; Di Nicola, M.; Zecca, I.A.L.; Angrilli, F. Combination of rituximab and nonpegylated liposomal doxorubicin (R-NPLD) as front-line therapy for aggressive non-Hodgkin lymphoma (NHL) in patients 80 years of age or older: a single-center retrospective study. Hematol. Oncol. 2018, 36, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Sun, Y.; Zhu, J.; Li, W.; Zhang, A.; Kuang, T.; Xie, J.; Yang, Z. Delivery of nanoparticles for treatment of brain tumor. Curr. Drug Metab. 2016, 17, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Sun, Y.; Wang, M.; Cheng, X. Nanosized camptothecin conjugates for single and combined drug delivery. Eur. J. Biomed. Res. 2016, 2, 8–14. [Google Scholar] [CrossRef]
- Tahover, E.; Patil, Y.P.; Gabizon, A.A. Emerging delivery systems to reduce doxorubicin cardiotoxicity and improve therapeutic index: focus on liposomes. Anticancer Drugs 2015, 26, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, M.G.; Mima, T.; Ohnishi, S.T.; Mori, K. S-allylcysteine ameliorates doxorubicin toxicity in the heart and liver in mice. Planta Med. 2000, 66, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Safra, T. Cardiac Safety of Liposomal Anthracyclines. Oncologist 2003, 8, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Petre, C.E.; Dittmer, D.P. Liposomal daunorubicin as treatment for Kaposi’s sarcoma. Int. J. Nanomed. 2007, 2, 277–288. [Google Scholar]
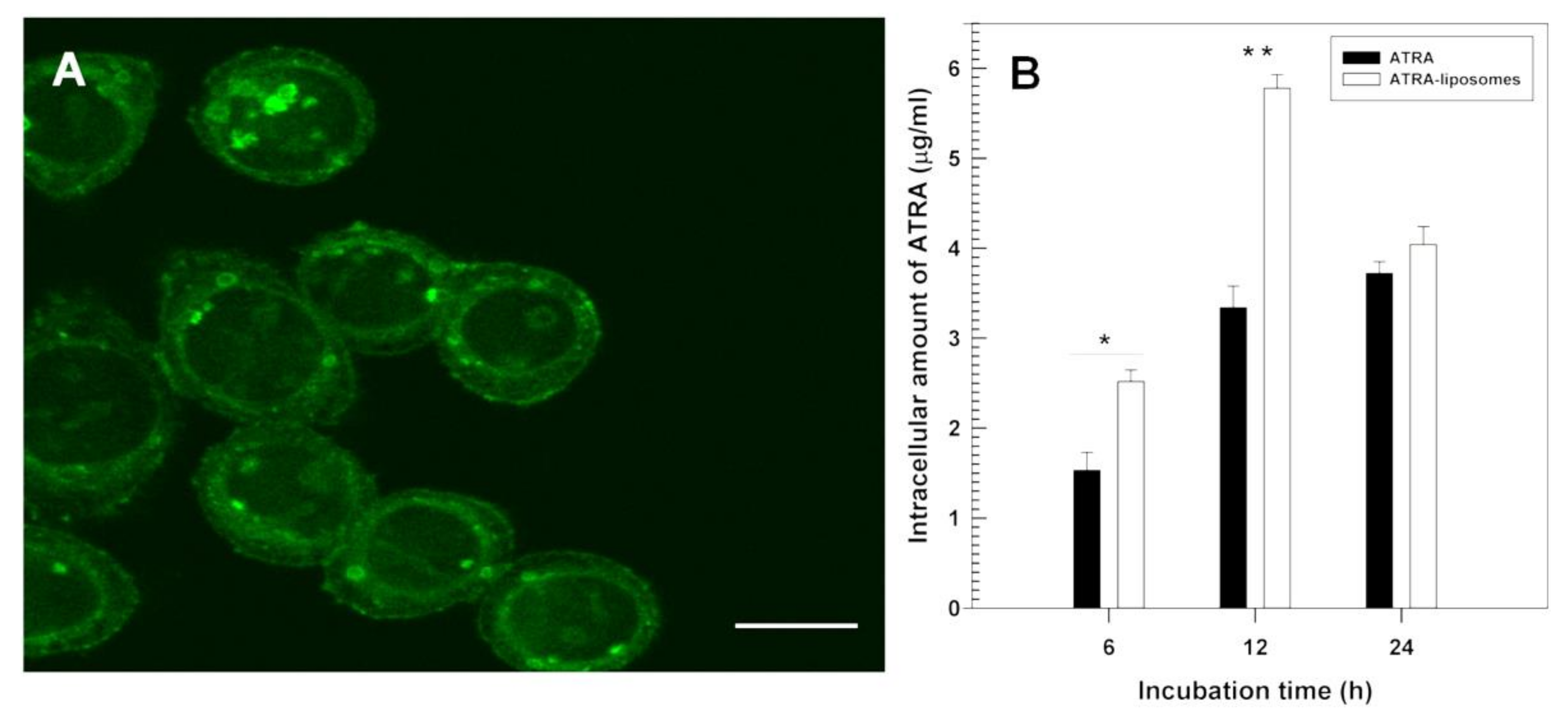
- Berlin Grace, V.M.; Viswanathan, S. Pharmacokinetics and therapeutic efficiency of a novel cationic liposome nano-formulated all trans retinoic acid in lung cancer mice model. J. Drug Deliv. Sci. Technol. 2017, 39, 223–236. [Google Scholar] [CrossRef]
- Legut, M.; Lipka, D.; Filipczak, N.; Piwoni, A.; Kozubek, A.; Gubernator, J. Anacardic acid enhances the anticancer activity of liposomal mitoxantrone towards melanoma cell lines—in vitro studies. Int. J. Nanomed. 2014, 9, 653–668. [Google Scholar]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J. Control Release 2014, 187, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Pouli, A.; Zervas, K.; Grigoraki, V.; Symeonidis, A.; Repoussis, P.; Mitsouli, C.; Papanastasiou, C.; Margaritis, D.; Tokmaktsis, A.; et al. Prospective randomized comparison of vincristine, doxorubicin and dexamethasone (VAD) administered as intravenous bolus injection and VAD with liposomal doxorubicin as first-line treatment in multiple myeloma. Ann. Oncol. 2003, 14, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®--the first FDA-approved nano-drug: Lessons learned. J. Control Release. 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.M.; Sheikh, S.; Ahmad, A.; Ahmad, M.U.; Chen, P.; Paithankar, M. Bioequivalence study of pegylated doxorubicin hydrochloride liposome (PEGADRIA) and DOXIL® in ovarian cancer patients: physicochemical characterization and pre-clinical studies. J. Nanomed. Nanotechnol. 2016, 7, 361. [Google Scholar]
- Gill, P.S.; Wernz, J.; Scadden, D.T.; Cohen, P.; Mukwaya, G.M.; von Roenn, J.H.; Jacobs, M.; Kempin, S.; Silverberg, I.; Gonzales, G.; et al. Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 1996, 14, 2353–2364. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, L.; Zucchetti, M.; Parisi, I.; Viganò, M.G.; Zecca, B.; Careddu, A.; D’Incalci, M.; Lazzarin, A. The pharmacokinetics of liposomal encapsulated daunorubicin are not modified by HAART in patients with HIV-associated Kaposi’s sarcoma. Cancer Chemother. Pharmacol. 2000, 45, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Tulpule, A.; Yung, R.C.; Wernz, J.; Espina, B.M.; Myers, A.; Scadden, D.T.; Cabriales, S.; Ilaw, M.; Boswell, W.; Gill, P.S. Phase II trial of liposomal daunorubicin in the treatment of AIDS-related pulmonary Kaposi’s sarcoma. J. Clin. Oncol. 1998, 16, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Creutzig, U.; Zimmermann, M.; Bourquin, J.-P.; Dworzak, M.N.; Fleischhack, G.; Graf, N.; Klingebiel, T.; Kremens, B.; Lehrnbecher, T.; von Neuhoff, C.; et al. Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: Results from Study AML-BFM 2004. Blood 2013, 122, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucherov, F.A.; Egorova, K.S.; Posvyatenko, A.V.; Eremin, D.B.; Ananikov, V.P. Investigation of cytotoxic activity of mitoxantrone at the individual cell level by using ionic-liquid-tag-enhanced mass spectrometry. Anal. Chem. 2017, 89, 13374–13381. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, G.; Schuler, U.; Proksch, B.; Zeller, K.-P.; Blanz, J. Pharmacokinetics and metabolism of mitoxantrone A Review. Clin. Pharmacokinet. 1990, 18, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Alderton, P.M.; Gross, J.; Green, M.D. Comparative study of doxorubicin, mitoxantrone, and epirubicin in combination with ICRF-187 (ADR-529) in a chronic cardiotoxicity animal model. Cancer Res. 1992, 52, 194–201. [Google Scholar] [PubMed]
- Xu, X.; Wang, L.; Xu, H.-Q.; Huang, X.-E.; Qian, Y.-D.; Xiang, J. Clinical comparison between paclitaxel liposome (Lipusu®) and paclitaxel for treatment of patients with metastatic gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2591–2594. [Google Scholar] [CrossRef] [PubMed]
- Surapaneni, M.S.; Das, S.K.; Das, N.G. Designing paclitaxel drug delivery systems aimed at improved patient outcomes: Current status and challenges. ISRN Pharmacol. 2012, 2012, 623139. [Google Scholar] [CrossRef] [PubMed]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Magin, R.L.; Hunter, J.M.; Niesman, M.R.; Bark, G.A. Effect of vesicle size on the clearance, distribution, and tumor uptake of temperature-sensitive liposomes. Cancer Drug Deliv. 1986, 3, 223–237. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Ingredient | Liposome Composition | Size (nm) | Cancer Type Being Targeted | Reference | |
|---|---|---|---|---|---|
| DOX | HSPC/DSPE/cholesterol (12.5:1:8.25 molar ratio) | 130 | Colorectal (in-vitro) | [68] | |
| DOX | Cholesterol, DSPC, DSPE and DSPE-PEG2000 (10 µmol total phospholipid). | 100 | Prostate cancer (in-vivo/in-vitro | [86] | |
| DOX | HSPC: cholesterol: lipid with a PEG head group (DSPE-PEG2000) (molar ratio 56.4:38.3:5.3) | 100 | Colorectal (in-vitro) | [68] | |
| DOX | 1-Palmitoyl-2-oleoylphosphatidylcholine: cholesterol (molar ratio 55.8:44.2) | 180 | Metastatic (clinical trial & in clinic) | [15,96] | |
| DNR | DSPC:cholesterol (molar ratio 2:1) | 50 | Kaposi’s sarcoma | [97] | |
| ATRA | DPPC:cholesterol:1,2-distearoyl-sn-glycero-3-phosphoethanolamine - Methoxy PEG2000 (molar ratio 6:3:1) | 200 | Human Thyroid carcinoma (in-vitro) | [16] | |
| ATRA | DOTAP, cholesterol and ATRA (molar ratio 70:20:10) | 263 | Lung cancer (in-vivo in animal) | [98] | |
| MXT | HSPC: DSPE-PEG2000: cholesterol: anacardic acid (molar ratio 0.55:0.05:0.35:0.05) | 112 | Melanoma cell lines (in-vitro) | [99] | |
| PCX | Egg phosphatidylcholine: cholesterol: TPGS1000-TPP (molar ratio 88:3.5:8.5) | 80 | Lung cancer cell lines (in-vivo & in-vitro) | [30] | |
| Irinotecan | - | - | Pancreatic ductal adenocarcinoma | [100] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907. https://doi.org/10.3390/molecules23040907
Olusanya TOB, Haj Ahmad RR, Ibegbu DM, Smith JR, Elkordy AA. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules. 2018; 23(4):907. https://doi.org/10.3390/molecules23040907
Chicago/Turabian StyleOlusanya, Temidayo O. B., Rita Rushdi Haj Ahmad, Daniel M. Ibegbu, James R. Smith, and Amal Ali Elkordy. 2018. "Liposomal Drug Delivery Systems and Anticancer Drugs" Molecules 23, no. 4: 907. https://doi.org/10.3390/molecules23040907
APA StyleOlusanya, T. O. B., Haj Ahmad, R. R., Ibegbu, D. M., Smith, J. R., & Elkordy, A. A. (2018). Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules, 23(4), 907. https://doi.org/10.3390/molecules23040907