
Porphyrin Co(III)-Nitrene Radical Mediated Pathway for Synthesis of o-Aminoazobenzenes


Abstract
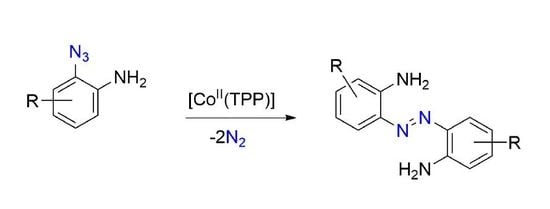
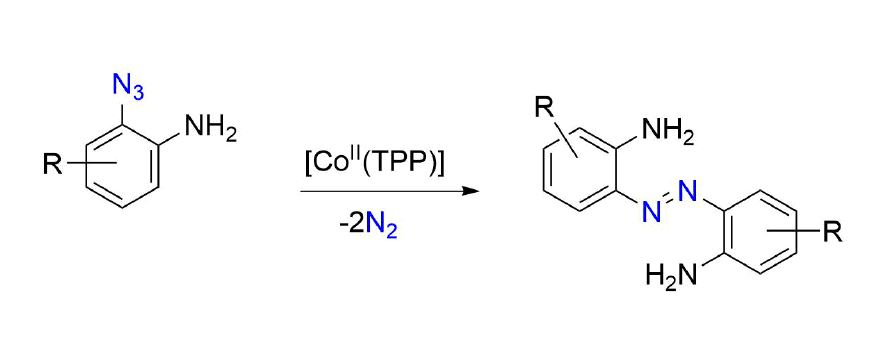
:
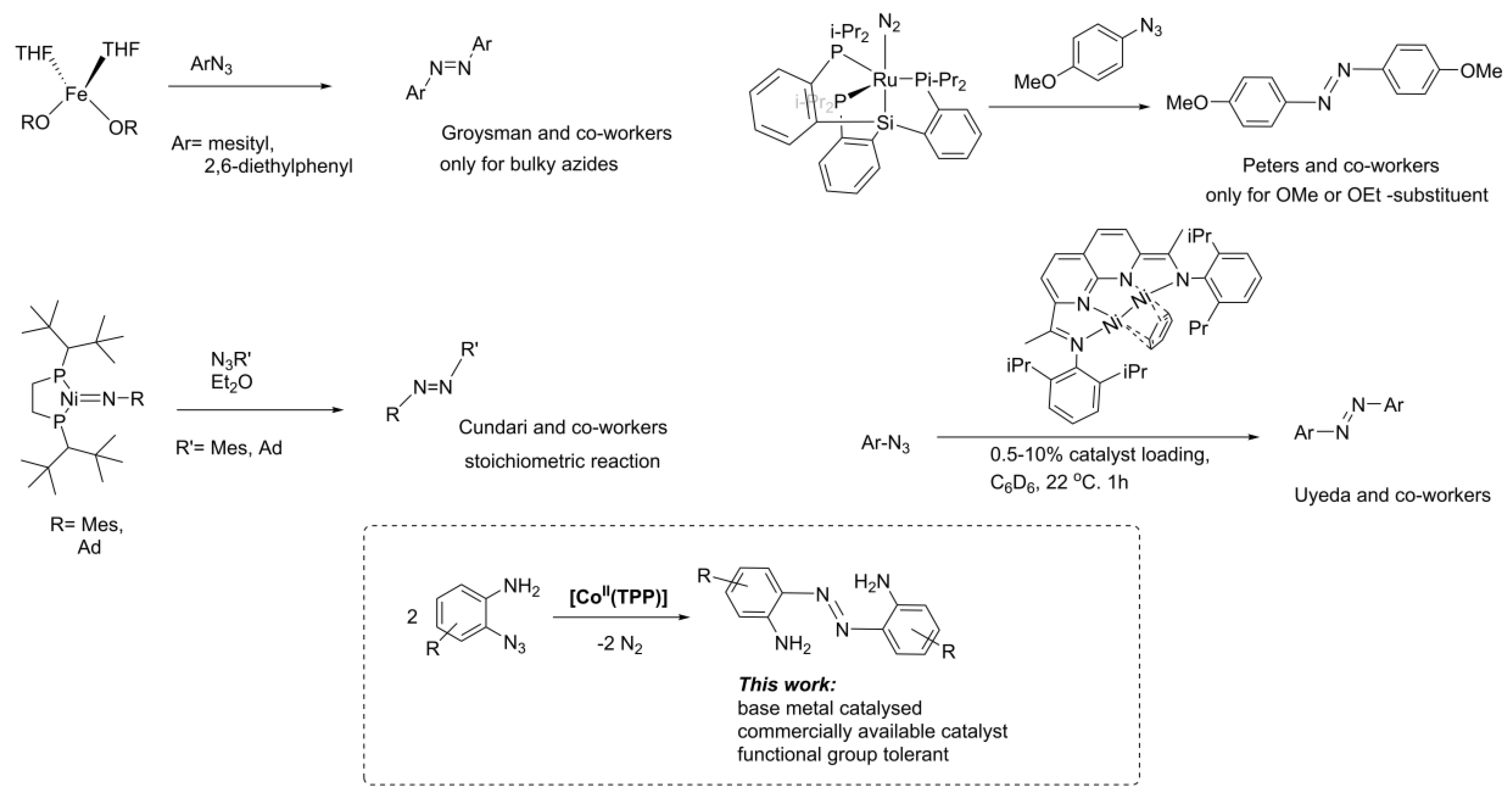
1. Introduction
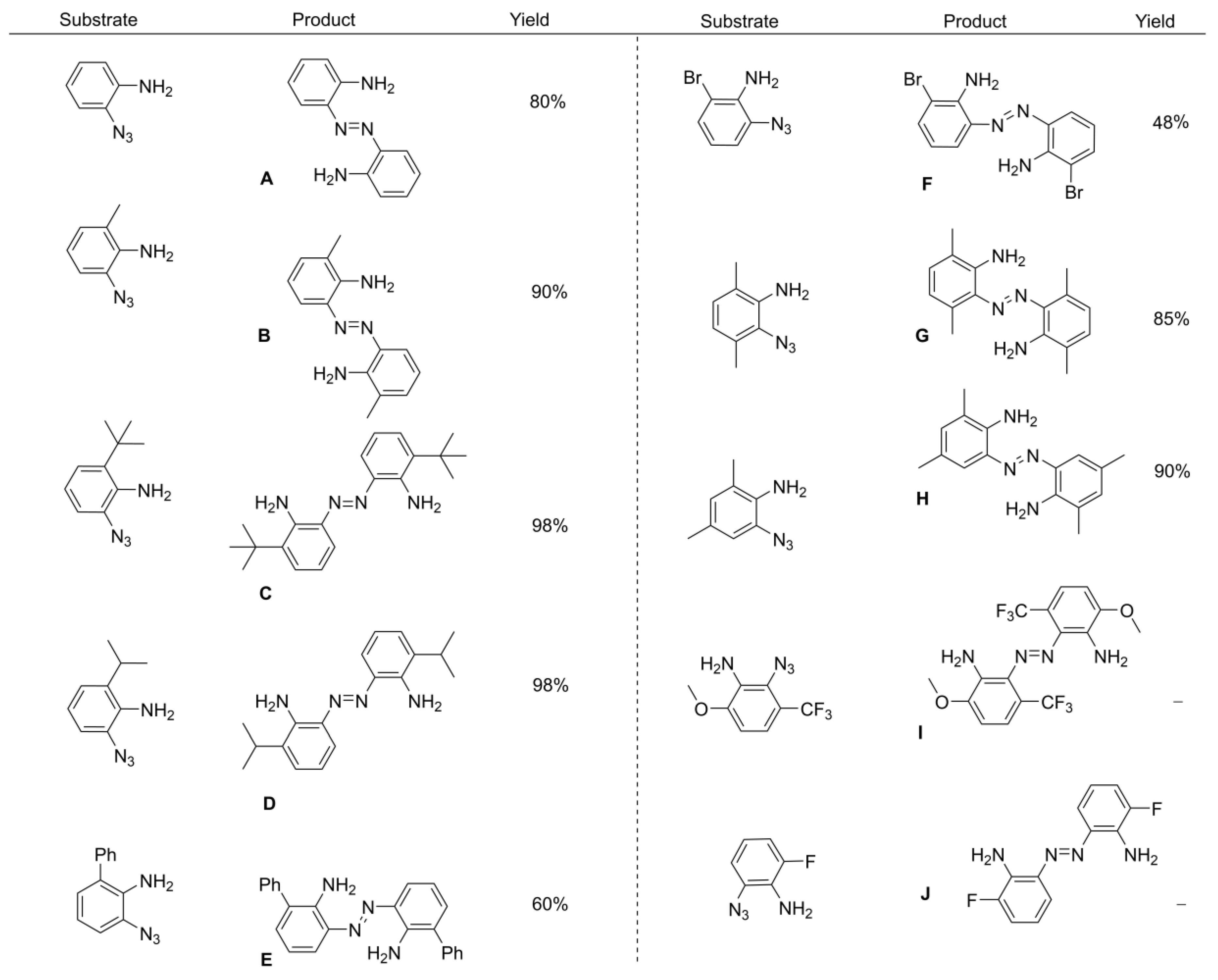
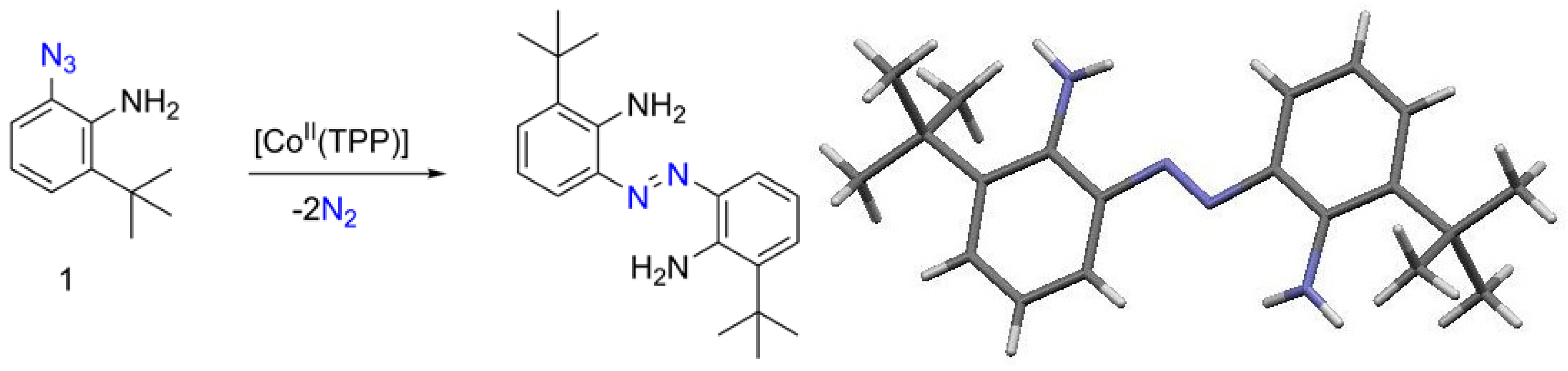
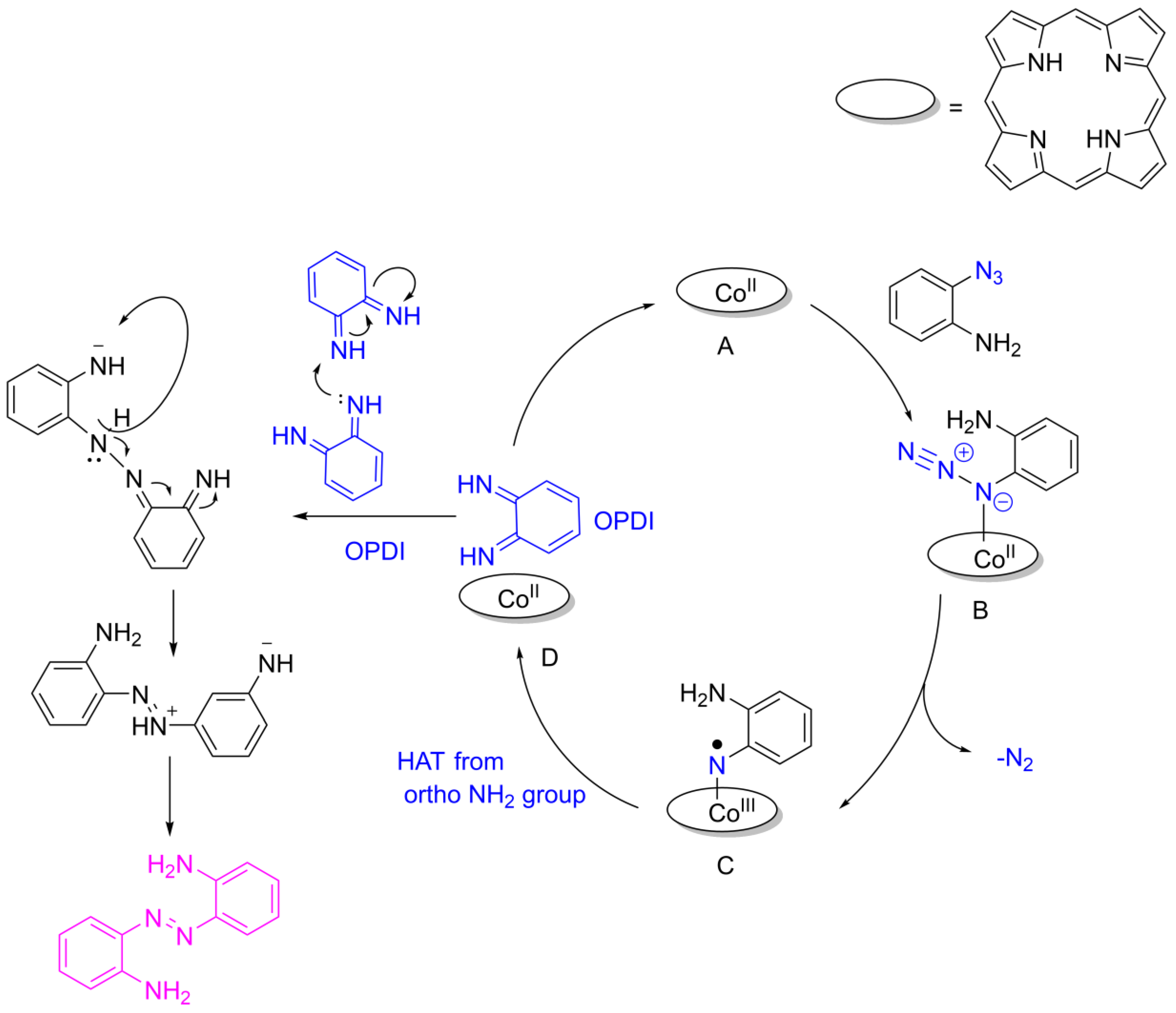
2. Results
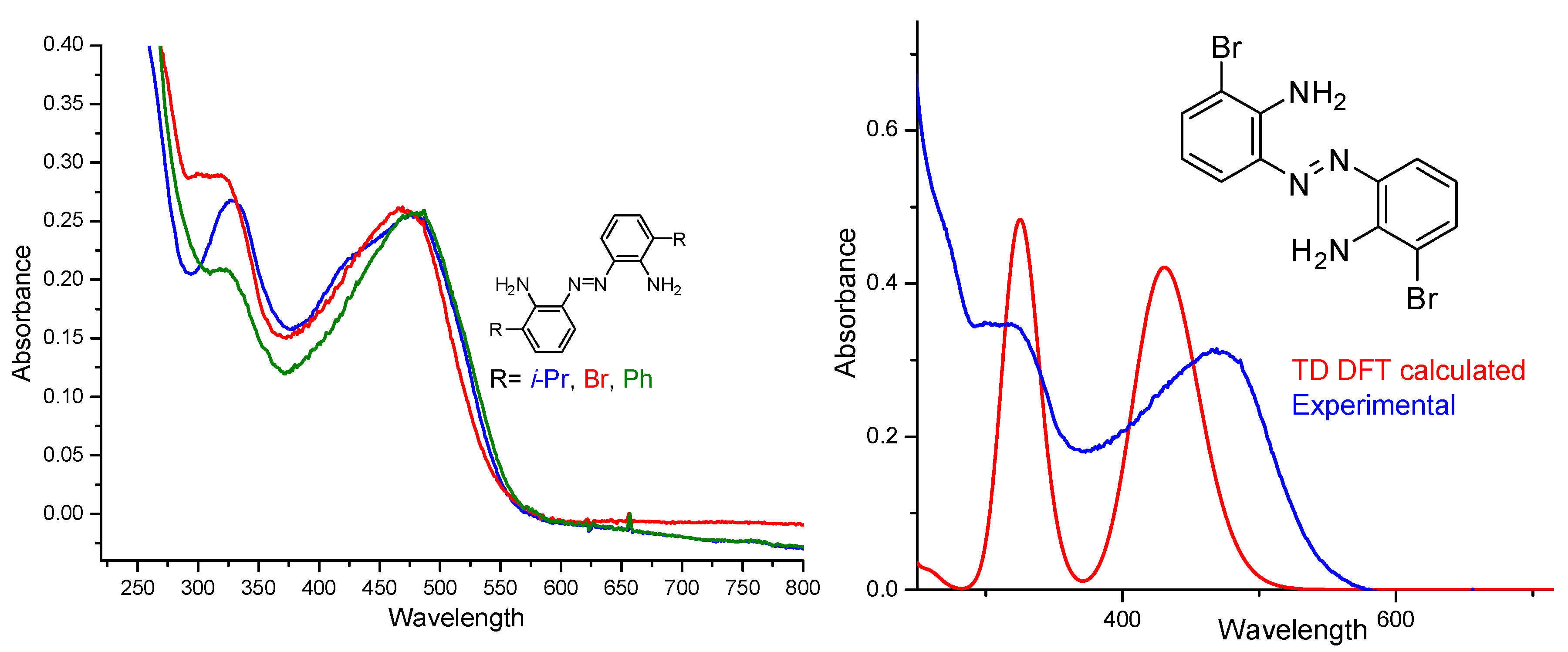
3. Electronic Properties of the Synthesized o-Amino-Substituted-Azobenzenes
4. Materials and Methods
4.1. Synthesis of the Azides








4.2. Catalytic Reactions to Give Azobenzenes
TD-DFT Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Hunger, K. Industrial Dyes: Chemistry, Properties, Applications; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Zollinger, H. Color Chemistry: Syntheses, Properties and Applications of Organic Dyes and Pigments; John Wiley & Sons: New York, NY, USA, 1987; Volume 85. [Google Scholar]
- Gordon, P.F.; Gregory, P. Organic Chemistry in Colour; Springer: New York, NY, USA, 1983; Volume 95. [Google Scholar]
- Ashutosh, P.N.D.; Mehrotra, J.K. Azo dyes as metallochromic indicators. Colourage 1979, 26, 25–36. [Google Scholar]
- Athey, R.D. Free radical initiator basics. Eur. Coat. J. 1998, 3, 146–149. [Google Scholar]
- Sandborn, W.J. Rational selection of oral 5-aminosalicylate formulations and prodrugs for the treatment of ulcerative colitis. Am. J. Gastroenterol. 2002, 97, 2939–2941. [Google Scholar] [CrossRef] [PubMed]
- Cisnetti, F.; Ballardini, R.; Credi, A.; Gandolfi, M.T.; Masiero, S.; Negri, F.; Pieraccini, S.; Spada, G.P. Photochemical and Electronic Properties of Conjugated Bis(azo) Compounds: An Experimental and Computational Study. Chem. Eur. J. 2004, 10, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Gupta, Y.; Jain, S.K. Azo chemistry and its potential for colonic delivery. Crit. Rev. Ther. Drug Carrier Syst. 2006, 23, 349–400. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Tsutsumi, O. Optical switching and image storage by means of azobenzene liquid-crystal films. Science 1995, 268, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Feringa, B.L.; van Delden, R.A.; Koumura, N.; Geertsema, E.M. Chiroptical Molecular Switches. Chem. Rev. 2000, 100, 1789–1816. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.A.; Woolley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef] [PubMed]
- Merino, E. Synthesis of azobenzenes: The coloured pieces of molecular materials. Chem. Soc. Rev. 2011, 40, 3835–3853. [Google Scholar] [CrossRef] [PubMed]
- Dhammika Bandara, H.M.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef] [PubMed]
- Bleger, D.; Schwarz, J.; Brouwer, A.M.; Hecht, S. o-Fluoroazobenzenes as Readily Synthesized Photoswitches Offering Nearly Quantitative Two-Way Isomerization with Visible Light. J. Am. Chem. Soc. 2012, 134, 20597–20600. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.A.; Sadovski, O.; Woolley, G.A. Azobenzene Photoswitching without Ultraviolet Light. J. Am. Chem. Soc. 2011, 133, 19684–19687. [Google Scholar] [CrossRef] [PubMed]
- Sadovski, O.; Beharry, A.A.; Zhang, F.; Woolley, G.A. Spectral tuning of azobenzene photoswitches for biological applications. Angew. Chem. Int. Ed. 2009, 48, 1484–1486. [Google Scholar] [CrossRef] [PubMed]
- Bellow, J.A.; Yousif, M.; Cabelof, A.C.; Lord, R.L.; Groysman, S. Reactivity Modes of an Iron Bis(alkoxide) Complex with Aryl Azides: Catalytic Nitrene Coupling vs. Formation of Iron(III) Imido Dimers. Organometallics 2015, 34, 2917–2923. [Google Scholar] [CrossRef]
- Harrold, N.D.; Waterman, R.; Hillhouse, G.L.; Cundari, T.R. Group-Transfer Reactions of Nickel−Carbene and −Nitrene Complexes with Organoazides and Nitrous Oxide that Form New C=N, C=O, and N=N Bonds. J. Am. Chem. Soc. 2009, 131, 12872–12873. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Moret, M.; Peters, J.C. A Ru(I) Metalloradical That Catalyzes Nitrene Coupling to Azoarenes from Arylazides. J. Am. Chem. Soc. 2012, 134, 6695–6706. [Google Scholar] [CrossRef] [PubMed]
- Powers, I.G.; Andjaba, J.M.; Luo, X.; Mei, J.; Uyeda, C. Catalytic Azoarene Synthesis from Aryl Azides Enabled by a dinuclear Ni Complex. J. Am. Chem. Soc. 2018, 140, 4110–4118. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Rebreyend, C.; de Bruin, B. Porphyrin Cobalt(III) “Nitrene Radical” Reactivity; Hydrogen Atom Transfer from Ortho-YH Substituents to the Nitrene Moiety of Cobalt-Bound Aryl Nitrene Intermediates (Y = O, NH). Molecules 2016, 21, 242. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Jiao, N. Copper-Catalyzed C–H Azidation of Anilines under Mild Conditions. J. Am. Chem. Soc. 2012, 134, 18924–18927. [Google Scholar] [CrossRef] [PubMed]
- Dzik, W.I.; Xu, X.; Zhang, X.P.; Reek, J.N.H.; de Bruin, B. ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc. 2010, 132, 10891–10902. [Google Scholar] [CrossRef] [PubMed]
- Suarez, A.I.O.; Jiang, H.; Zhang, X.P.; de Bruin, B. The radical mechanism of cobalt(II) porphyrin-catalyzed olefin aziridination and the importance of cooperative H-bonding. Dalton Trans. 2011, 40, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Lyaskovskyy, V.; Suarez, A.I.O.; Lu, H.; Jiang, H.; Zhang, X.P.; de Bruin, B. Mechanism of Cobalt(II) Porphyrin-Catalyzed C–H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)–Nitrene Intermediates. J. Am. Chem. Soc. 2011, 133, 12264–12273. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Lyaskovskyy, V.; Domingos, S.R.; Buma, W.J.; Woutersen, S.; Troeppner, O.; Ivanović-Burmazović, I.; Lu, H.; Cui, X.; Zhang, X.P.; et al. Characterization of Porphyrin-Co(III)-‘Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. [Google Scholar] [CrossRef] [PubMed]
- Emond, M.; le Saux, T.; Maurin, S.; Baudin, J.B.; Plasson, R.; Jullien, L. 2-Hydroxyazobenzenes to Tailor pH Pulses and Oscillations with Light. Chem. Eur. J. 2010, 16, 8822–8831. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. ORCA—An Ab Initio, Density Functional and Semiempirical Program Package; Version 3.0.3; Max-Planck-Institut für Bioanorganische Chemie: Mülheim an der Ruhr, Germany, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Calculations were performed using the “Turbomole functional b3-lyp”, which is not fully identical to the “Gaussian B3LYP” functional.
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Temperature (°C) | Catalyst Loading | Yield |
|---|---|---|---|---|
| 1 | Toluene | 90 | 5 mol% | 98% |
| 2 | Benzene | 60 | 5 mol% | - |
| 3 | Toluene | 60 | 5 mol% | - |
| 4 | Toluene | 90 | 1 mol% | 60% |
| 5 | THF | 60 | 5 mol% | - |
| 6 | Toluene | 90 | No catalyst | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goswami, M.; De Bruin, B. Porphyrin Co(III)-Nitrene Radical Mediated Pathway for Synthesis of o-Aminoazobenzenes. Molecules 2018, 23, 1052. https://doi.org/10.3390/molecules23051052
Goswami M, De Bruin B. Porphyrin Co(III)-Nitrene Radical Mediated Pathway for Synthesis of o-Aminoazobenzenes. Molecules. 2018; 23(5):1052. https://doi.org/10.3390/molecules23051052
Chicago/Turabian StyleGoswami, Monalisa, and Bas De Bruin. 2018. "Porphyrin Co(III)-Nitrene Radical Mediated Pathway for Synthesis of o-Aminoazobenzenes" Molecules 23, no. 5: 1052. https://doi.org/10.3390/molecules23051052
APA StyleGoswami, M., & De Bruin, B. (2018). Porphyrin Co(III)-Nitrene Radical Mediated Pathway for Synthesis of o-Aminoazobenzenes. Molecules, 23(5), 1052. https://doi.org/10.3390/molecules23051052