
Design, Synthesis, and In Vitro Evaluation of Novel Histone H3 Peptide-Based LSD1 Inactivators Incorporating α,α-Disubstituted Amino Acids with γ-Turn-Inducing Structures


Abstract
:1. Introduction
2. Results
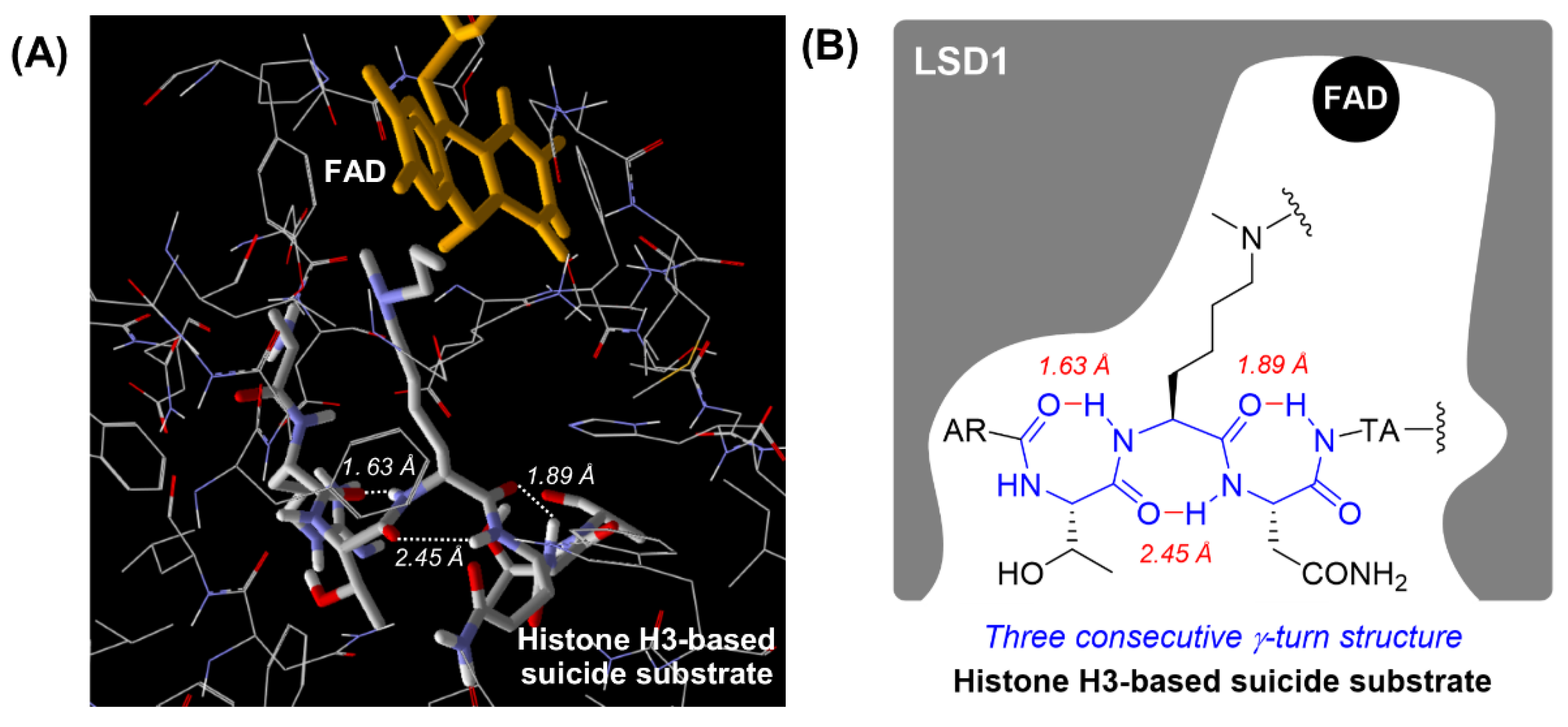
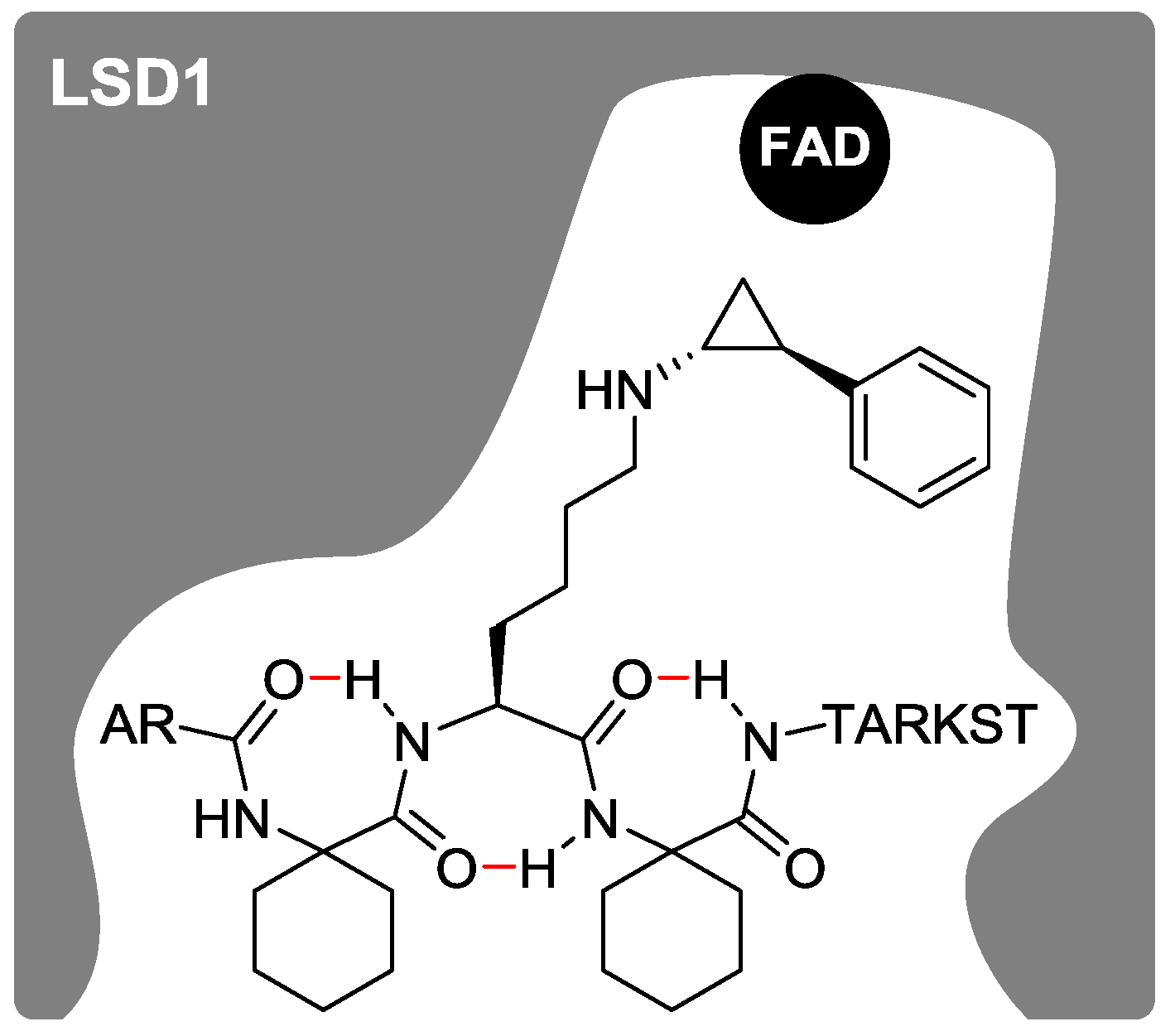
2.1. Design
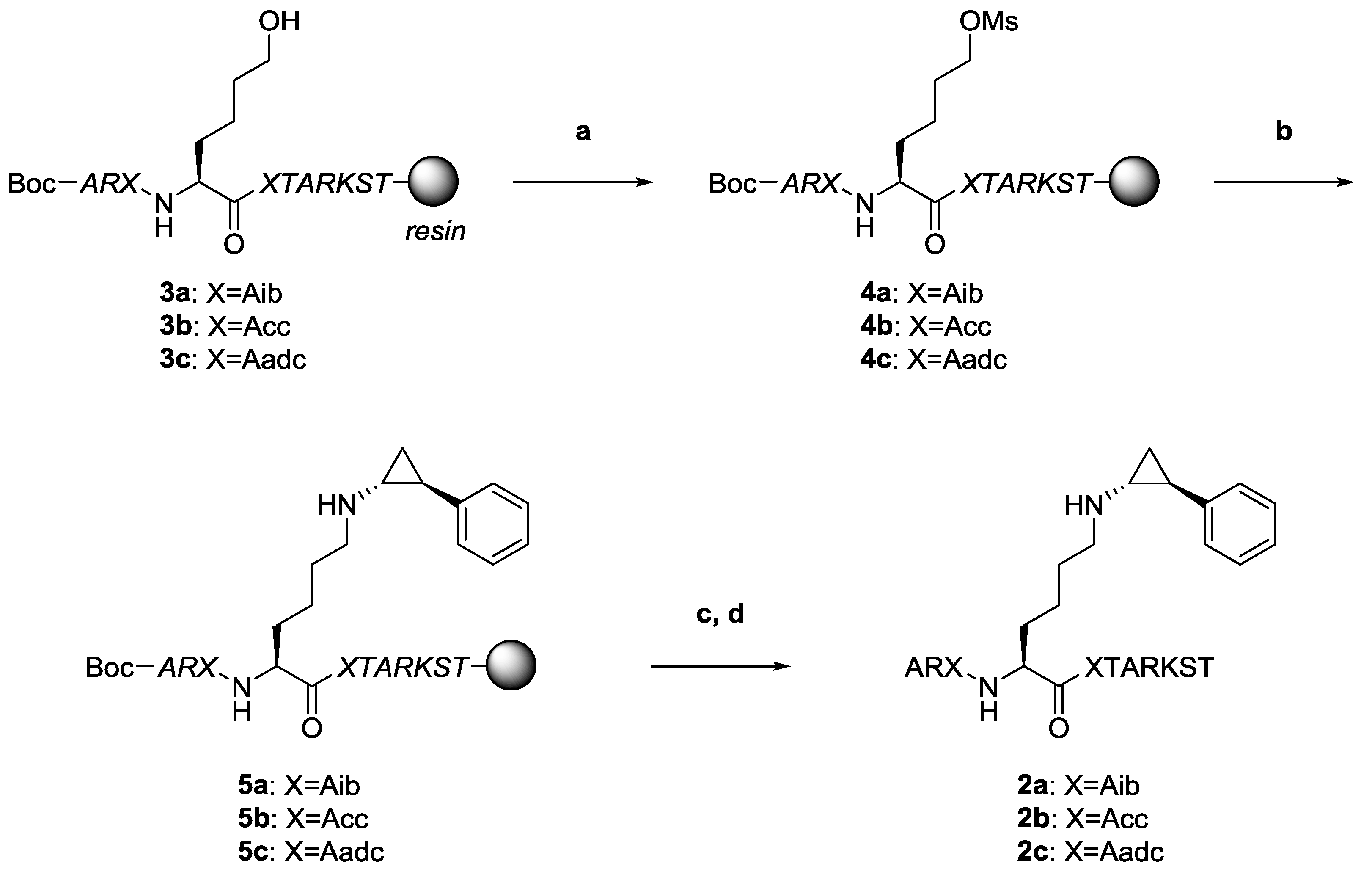
2.2. Synthesis
2.3. In Vitro Evaluation of LSD1 Inhibitory Activity
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. Synthesis of Peptides with γ-Turn Inducers
4.3. Assay for LSD1 Inhibitory Activity
4.4. Assay for MAO Inhibitory Activity
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Greer:, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Kontaki, H.; Talianidis, I. Lysine Methylation Regulates E2F1-Induced Cell Death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Takezawa, S.; Schüle, R.; Kitagawa, H.; Kato, S. Transrepressive function of TLX requires the histone demethylase LSD1. Mol. Cell. Biol. 2008, 28, 3995–4003. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, M.; Yin, N.; Müller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Günther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Højfeldt, J.W.; Agger, K.; Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 2013, 12, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Cui, S.; Engel, J.D.; Tanabe, O. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat. Med. 2013, 19, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase LSD1 blocks α-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Miyata, N. Lysine Demethylases Inhibitors. J. Med. Chem. 2011, 54, 8236–8250. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, B.; Suzuki, T.; Liu, X.; Zhan, P. Medicinal chemistry insights in the discovery of novel LSD1 inhibitors. Epigenomics 2015, 7, 1379–1396. [Google Scholar] [CrossRef] [PubMed]
- McAllister, T.E.; England, K.S.; Hopkinson, R.J.; Brennan, P.E.; Kawamura, A.; Schofield, C.J. Recent Progress in Histone Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1308–1329. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Mascaró, C.; Ortega, A.; Lunardi, S.; Ciceri, F.; Somervaille, T.C.P.; Buesa, C. KDM1 histone lysine demethylases as targets for treatments of oncological and neurodegenerative disease. Epigenomics 2015, 7, 609–626. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Ueda, R.; Suzuki, T.; Mino, K.; Tsumoto, H.; Nakagawa, H.; Hasegawa, M.; Sasaki, R.; Mizukami, T.; Miyata, N. Identification of Cell-Active Lysine Specific Demethylase 1-Selective Inhibitors. J. Am. Chem. Soc. 2009, 131, 17536–17537. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, D.; Suzuki, T.; Mino, K.; Ueda, R.; Khan, M.N.A.; Matsubara, T.; Koseki, K.; Hasegawa, M.; Sasaki, R.; Nakagawa, H.; et al. Synthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitor. Bioorg. Med. Chem. 2011, 19, 3702–3708. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, D.; Itoh, Y.; Tsumoto, H.; Kakizawa, T.; Mino, K.; Fukuhara, K.; Nakagawa, H.; Hasegawa, M.; Sasaki, R.; Mizukami, T.; et al. Lysine-Specific Demethylase 1-Selective Inactivators: Protein-Targeted Drug Delivery Mechanism. Angew. Chem. Int. Ed. 2013, 52, 8620–8624. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Ogasawara, D.; Ota, Y.; Mizukami, T.; Suzuki, T. Synthesis, LSD1 Inhibitory Activity, and LSD1 Binding Model of Optically Pure Lysine-PCPA Conjugates. Comput. Struct. Biotechnol. J. 2014, 9, e201402002. [Google Scholar] [CrossRef] [PubMed]
- Ahmed Khan, M.N.; Tsumoto, H.; Itoh, Y.; Ota, Y.; Suzuki, M.; Ogasawara, D.; Nakagawa, H.; Mizukami, T.; Miyata, N.; Suzuki, T. Design, synthesis, and biological activity of N-alkylated analogue of NCL1, a selective inhibitor of lysine-specific demethylase 1. Medchemcomm 2015, 6, 407–412. [Google Scholar] [CrossRef]
- Kakizawa, T.; Ota, Y.; Itoh, Y.; Tsumoto, H.; Suzuki, T. Histone H3 peptide based LSD1-selective inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Aihara, K.; Mellini, P.; Tojo, T.; Ota, Y.; Tsumoto, H.; Solomon, V.R.; Zhan, P.; Suzuki, M.; Ogasawara, D.; et al. Identification of SNAIL1 Peptide-Based Irreversible Lysine-Specific Demethylase 1-Selective Inactivators. J. Med. Chem. 2016, 59, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, S.; Araki, M.; Ota, Y.; Itoh, Y.; Yasuda, S.; Masuda, M.; Taniguchi, T.; Sowa, Y.; Sakai, T.; Suzuki, T.; et al. C–H activation enables a rapid structure–activity relationship study of arylcyclopropyl amines for potent and selective LSD1 inhibitors. Org. Biomol. Chem. 2016, 14, 8576–8585. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, T.; Ota, Y.; Itoh, Y.; Suzuki, T. Histone H3 peptides incorporating modified lysine residues as lysine-specific demethylase 1 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Miyamura, S.; Araki, M.; Itoh, Y.; Yasuda, S.; Masuda, M.; Taniguchi, T.; Sowa, Y.; Sakai, T.; Itami, K.; et al. Design, synthesis and evaluation of γ-turn mimetics as LSD1-selective inhibitors. Bioorg. Med. Chem. 2018, 26, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.M.Z.; McCafferty, D.G. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Culhane, J.C.; Szewczuk, L.M.; Jalili, P.; Ball, H.L.; Machius, M.; Cole, P.A.; Yu, H. Structural Basis for the Inhibition of the LSD1 Histone Demethylase by the Antidepressant trans-2-Phenylcyclopropylamine. Biochemistry 2007, 46, 8058–8065. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Viswanadhapalli, S.; Surapaneni, P.; Suzuki, T.; Brenner, A.; Vadlamudi, R.K. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene 2017, 36, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Sugino, N.; Kawahara, M.; Tatsumi, G.; Kanai, A.; Matsui, H.; Yamamoto, R.; Nagai, Y.; Fujii, S.; Shimazu, Y.; Hishizawa, M.; et al. A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia 2017, 31, 2302–2314. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Culhane, J.C.; Szewczuk, L.M.; Gocke, C.B.; Brautigam, C.A; Tomchick, D.R.; Machius, M.; Cole, P.A.; Yu, H. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat. Struct. Mol. Biol. 2007, 14, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Ueda, H.; Nozawa, H.; Ogoshi, H. Adamantyl amino acid as γ-turn inducer for peptide. Tetrahedron Lett. 1997, 38, 7901–7904. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of Histone Lysine Trimethylation by the JMJD2 Family of Histone Demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Christophersen, P.C.; Fano, M.; Saaby, L.; Yang, M.; Nielsen, H.M.; Mu, H. Characterization of particulate drug delivery systems for oral delivery of Peptide and protein drugs. Curr. Pharm. Des. 2015, 21, 2611–2628. [Google Scholar] [CrossRef] [PubMed]
- Kumarasinghe, I.R.; Woster, P.M. Synthesis and Evaluation of Novel Cyclic Peptide Inhibitors of Lysine-Specific Demethylase 1. ACS Med. Chem. Lett. 2014, 5, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Kumarasinghe, I.R.; Woster, P.M. Cyclic by peptide inhibitors of lysine-specific demethylase 1 with improved potency identified by alanine scanning mutagenesis. Eur. J. Med. Chem. 2018, 148, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Li, J.; Zheng, M.; Zuo, L.; Sun, C.; Zhu, Y.; Fang, L.; Liu, L.; Zhou, X. Stable H3 peptide was delivered by gold nanorods to inhibit LSD1 activation and induce human mesenchymal stem cells differentiation. Oncotarget 2017, 8, 23110–23119. [Google Scholar] [CrossRef] [PubMed]
- Karstad, R.; Isaksen, G.; Brandsdal, B.O.; Svendsen, J.S.; Svenson, J. Unnatural amino acid side chains as S1, S1’, and S2’ probes yield cationic antimicrobial peptides with stability toward chymotryptic degradation. J. Med. Chem. 2010, 53, 5558–5566. [Google Scholar] [CrossRef] [PubMed]
- Brimble, M.A.; Kowalczyk, R.; Harris, P.W.R.; Dunbar, P.R.; Muir, V.J. Synthesis of fluorescein-labelled O-mannosylated peptides as components for synthetic vaccines: Comparison of two synthetic strategies. Org. Biomol. Chem. 2008, 6, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Culhane, J.C.; Wang, D.; Yen, P.M.; Cole, P.A. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J. Am. Chem. Soc. 2010, 132, 3164–3176. [Google Scholar] [CrossRef] [PubMed]
- Szewczuk, L.M.; Culhane, J.C.; Yang, M.; Majumdar, A.; Yu, H.; Cole, P.A. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry 2007, 46, 6892–6902. [Google Scholar] [CrossRef] [PubMed]
- Culhane, J.C.; Szewczuk, L.M.; Liu, X.; Da, G.; Marmorstein, R.; Cole, P.A. A mechanism-based inactivator for histone demethylase LSD1. J. Am. Chem. Soc. 2006, 128, 4536–4537. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) 1 | ||
|---|---|---|---|
| LSD1 | MAO-A | MAO-B | |
| PCPA | 16.5 ± 1.9 | 6.00 ± 1.38 | 6.54 ± 0.51 |
| 1 | 0.126 ± 0.002 | >10 | >10 |
| 2a | 0.0891 ± 0.0053 | >10 | >10 |
| 2b | 0.0584 ± 0.0025 | >10 | >10 |
| 2c | 0.0724 ± 0.0040 | >10 | >10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ota, Y.; Kakizawa, T.; Itoh, Y.; Suzuki, T. Design, Synthesis, and In Vitro Evaluation of Novel Histone H3 Peptide-Based LSD1 Inactivators Incorporating α,α-Disubstituted Amino Acids with γ-Turn-Inducing Structures. Molecules 2018, 23, 1099. https://doi.org/10.3390/molecules23051099
Ota Y, Kakizawa T, Itoh Y, Suzuki T. Design, Synthesis, and In Vitro Evaluation of Novel Histone H3 Peptide-Based LSD1 Inactivators Incorporating α,α-Disubstituted Amino Acids with γ-Turn-Inducing Structures. Molecules. 2018; 23(5):1099. https://doi.org/10.3390/molecules23051099
Chicago/Turabian StyleOta, Yosuke, Taeko Kakizawa, Yukihiro Itoh, and Takayoshi Suzuki. 2018. "Design, Synthesis, and In Vitro Evaluation of Novel Histone H3 Peptide-Based LSD1 Inactivators Incorporating α,α-Disubstituted Amino Acids with γ-Turn-Inducing Structures" Molecules 23, no. 5: 1099. https://doi.org/10.3390/molecules23051099
APA StyleOta, Y., Kakizawa, T., Itoh, Y., & Suzuki, T. (2018). Design, Synthesis, and In Vitro Evaluation of Novel Histone H3 Peptide-Based LSD1 Inactivators Incorporating α,α-Disubstituted Amino Acids with γ-Turn-Inducing Structures. Molecules, 23(5), 1099. https://doi.org/10.3390/molecules23051099