
Targeting Transcription Factors for Cancer Treatment


Abstract
:1. Introduction
- -
- Their inhibition (or activation) at the expression level,
- -
- Their inhibition through physical degradation,
- -
- Their inhibition (or activation) at the protein/protein interaction level,
- -
- Their inhibition (or activation) through the binding of a ligand-based molecule in an activation/inhibition pocket,
- -
- Their inhibition (or activation) at the protein/DNA binding level.
2. Targeting Transcription Factor at the Expression Level
2.1. Example of HOXA Cluster Expression Controlled by MLL Complex
- -
- The macrocyclic peptidomimetic MCP-1 [52],
- -
- The thienopyrimidine MI-2-2 [53] and its derivatives MI-463/503 [54] with MI-2-2 being poorly stable and could not be used in vivo rather than MI-463 and MI-503 (a derivative of MI-463 by the addition of a single methylpyrazole) which both interact with menin at nanomolar range, are more metabolically stable and exert strong cellular and in vivo activity, MI-503 being the most efficient one with deeper contacts with the menin pocket [54],
- -
- The hydroxymethylpiperidines ML227, MIV-6 and cyclopentylphenylpiperidine derivative M-525 [55,56,57] that mimic the interacting MLL peptide and may be used together with DOT1L inhibitors to restore differentiation in MLL-rearranged leukemias [58]. ML227 presents poor metabolic stability as well as off target activities that limited its developement and an IC50 for interation to menin of 390 nM [56]. MIV-6 differs from ML227 by an amine group that substitute to the hydroxyl group of ML227 and is more stable but with similar range of IC50 for menin (185 nM) whereas M-525 is much more efficient on menin interaction with IC50 of 3.3 nM and is 30-fold more potent in cellular activities with a hiogh specificity on mixed lineage leukemia cell models such as MV4;11 [57]. Inhibitors of BRD4 also showed efficiency to target mutated MLL functional complex, based on their interaction to control gene expression [59,60] and to collaborate with DOT1L [61]. This is the case for the thienodiazepine (+)-JQ1, I-BET762 (GSK525762), OTX015, GW841819X, CPI-0610 and RVX-208 that are developed by different companies and entered into clinical trials in various hematological malignancies and solid tumors while other compounds such as, MS436 or the iridium based inhibitor 1a (Figure 1) are in developmental stages (for reviews Huang 2016; Liu 2017; Kharenko 2017) [62,63,64]. Moreover, it is worth noting that both BRD4 and DOT1L inhibitors could synergistically inhibit proliferation of MLL-rearranged leukemic cells [61].
2.2. Example of MYC Transcription Factors Expression Control at the Epigenetic Level
3. Targeting Transcription Factor at the Protein Degradation Level
4. Targeting Transcription Factor at the Protein/Protein Interaction Level
5. Targeting Transcription Factor through a Binding Pocket
5.1. Targeting a Ligand-Binding Pocket
5.2. Targeting a Pocket in the DNA-Binding Domain
6. Targeting Transcription Factor at the Protein/DNA Interaction Level
6.1. DNA Alkylating Drugs for Transcription Factor DNA Binding Modulation
6.2. DNA Intercalating Drugs for Transcription Factor DNA Binding Modulation
6.3. Major Groove DNA Binding Drugs for Transcription Factor DNA Binding Modulation
6.4. Minor Groove DNA Binding Drugs for Transcription Factor DNA Binding Modulation
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Badis, G.; Berger, M.F.; Philippakis, A.A.; Talukder, S.; Gehrke, A.R.; Jaeger, S.A.; Chan, E.T.; Metzler, G.; Vedenko, A.; Chen, X.; et al. Diversity and complexity in DNA recognition by transcription factors. Science 2009, 324, 1720–1723. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.A. Identification of Small Molecules That Disrupt SSB-Protein Interactions Using a High-Throughput Screen. Methods Mol. Biol. 2012, 922, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- An, O.; Dall’Olio, G.M.; Mourikis, T.P.; Ciccarelli, F.D. NCG 5.0: Updates of a manually curated repository of cancer genes and associated properties from cancer mutational screenings. Nucleic Acids Res. 2016, 44, D992–D999. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, T.; Suzuki, H.; Cannistraci, C.V.; Katayama, S.; Bajic, V.B.; Tan, K.; Akalin, A.; Schmeier, S.; Kanamori-Katayama, M.; Bertin, N.; et al. An Atlas of Combinatorial Transcriptional Regulation in Mouse and Man. Cell 2010, 140, 744–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Lahiri, D.K. Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, P.C.; Pal, R. The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues Clin. Neurosci. 2017, 19, 71–80. [Google Scholar] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Bencini, L.; Galli, A. Peroxisome proliferator activated receptors at the crossroad of obesity, diabetes, and pancreatic cancer. World J. Gastroenterol. 2016, 22, 2441–2459. [Google Scholar] [CrossRef] [PubMed]
- Lilly, A.J.; Lacaud, G.; Kouskoff, V. SOXF transcription factors in cardiovascular development. Semin. Cell Dev. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; Lim, J.W.C.; Richards, L.J.; Bunt, J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett. 2017, 410, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.J.; van Riggelen, J. MYC—Master regulator of the cancer epigenome and transcriptome. Genes 2017, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Oh, S.; Yoo, K.H. Functional Enhancers as Master Regulators of Tissue-Specific Gene Regulation and Cancer Development. Mol. Cells 2017, 40, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two sides of the same coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Higgins, P.J. Drugging the undruggable: Transcription therapy for cancer. Biochim. Biophys. Acta Rev. Cancer 2013, 1835, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawagoe, H.; Humphries, R.K.; Blair, A.; Sutherland, H.J.; Hogge, D.D. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 1999, 13, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.; Katryniok, C.; Scholz, B.; Merkens, J.; Löscher, D.; Marschalek, R.; Steinhilber, D. Inhibition of class I HDACs abrogates the dominant effect of MLL-AF4 by activation of wild-type MLL. Oncogenesis 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, G.M.; Mehdipour, P.; Cluse, L.A.; Falkenberg, K.J.; Wang, E.; Roth, M.; Santoro, F.; Vidacs, E.; Stanley, K.; House, C.M.; et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 2015, 126, 2392–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; Taranova, O.; He, J.; Zhang, Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9—Mediated leukemogenesis. Blood 2011, 117, 6912–6922. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Stanojevic, B.; Kanai, A.; Kawamura, T.; Takahashi, S.; Matsui, H.; Takaori-Kondo, A.; Yokoyama, A. Cooperative gene activation by AF4 and DOT1L drives MLL-rearranged leukemia. J. Clin. Investig. 2017, 127, 1918–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.J.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, A.; Cleary, M.L. Menin Critically Links MLL Proteins with LEDGF on Cancer-Associated Target Genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, A.T.; Huang, J.; Lei, M.; Hua, X. Menin as a hub controlling mixed lineage leukemia. BioEssays 2012, 34, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ashkar, S.; Schwaller, J.; Pieters, T.; Goossens, S.; Demeulemeester, J.; Christ, F.; Van Belle, S.; Juge, S.; Boeckx, N.; Engelman, A.; et al. LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 2018, 131, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C.; Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; et al. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7, 37740–37754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W.E. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016, 29, 32–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhilber, D.; Marschalek, R. How to effectively treat acute leukemia patients bearing MLL-rearrangements? Biochem. Pharmacol. 2018, 147, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Gjertsen, B.T.; Bruserud, Ø. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenet. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Morabito, F.; Voso, M.T.; Hohaus, S.; Gentile, M.; Vigna, E.; Recchia, A.G.; Iovino, L.; Benedetti, E.; Lo-Coco, F.; Galimberti, S. Panobinostat for the treatment of acute myelogenous leukemia. Expert Opin. Investig. Drugs 2016, 25, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.H.; Foon, K.A.; Frankel, P.; Ruel, C.; Pulone, B.; Tuscano, J.M.; Newman, E.M. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: A California Cancer Consortium Study. Leuk. Lymphoma 2014, 55, 2301–2304. [Google Scholar] [CrossRef] [PubMed]
- Mihăilă, R.G. From a better understanding of the mechanisms of action of histone deacetylases inhibitors to the progress of the treatment of malignant lymphomas and plasma cell myeloma. Recent Pat. Anticancer Drug Discov. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xia, X.; Reisenauer, M.R.; Hemenway, C.S.; Kone, B.C. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. J. Biol. Chem. 2006, 281, 18059–18068. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Jiang, Q.; Lemieux, M.; Jeannotte, L.; Su, L.; Zhang, Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 2006, 8, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basavapathruni, A.; Jin, L.; Daigle, S.R.; Majer, C.R.A.; Therkelsen, C.A.; Wigle, T.J.; Kuntz, K.W.; Chesworth, R.; Pollock, R.M.; Scott, M.P.; et al. Conformational Adaptation Drives Potent, Selective and Durable Inhibition of the Human Protein Methyltransferase DOT1L. Chem. Biol. Drug Des. 2012, 80, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Deshpande, A.J.; Banka, D.; Bernt, K.M.; Dias, S.; Buske, C.; Olhava, E.J.; Daigle, S.R.; Richon, V.M.; Pollock, R.M.; et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 2013, 27, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Rau, R.E.; Rodriguez, B.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ge, W. The histone methyltransferase DOT1L inhibits osteoclastogenesis and protects against osteoporosis article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-B.; Liu, L.; Huang, J.; Bernard, D.; Karatas, H.; Navarro, A.; Lei, M.; Wang, S. Structure-based Design of High-Affinity Macrocyclic Peptidomimetics to Block the Menin-MLL1 Protein-Protein Interaction. J. Med. Chem. 2013, 56, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Murai, M.J.; He, S.; Lund, G.; Hartley, T.; Purohit, T.; Reddy, G.; Chruszcz, M.; Grembecka, J.; Cierpicki, T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [Google Scholar] [CrossRef] [PubMed]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Senter, T.J.; Pollock, J.; Han, C.; Upadhyay, S.K.; Purohit, T.; Gogliotti, R.D.; Lindsley, C.W.; Cierpicki, T.; Stauffer, S.R.; et al. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J. Med. Chem. 2014, 57, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Senter, T.; Gogliotti, R.D.; Han, C.; Locuson, C.W.; Morrison, R.; Daniels, J.S.; Cierpicki, T.; Grembecka, J.; Lindsley, C.W.; Stauffer, S.R. Progress towards small molecule menin-mixed lineage leukemia (MLL) interaction inhibitors with in vivo utility. Bioorgan. Med. Chem. Lett. 2015, 25, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Aguilar, A.; Xu, T.; Zheng, K.; Huang, L.; Stuckey, J.; Chinnaswamy, K.; Bernard, D.; Fernández-Salas, E.; Liu, L.; et al. Design of the First-in-Class, Highly Potent Irreversible Inhibitor Targeting the Menin-MLL Protein–Protein Interaction. Angew. Chem. Int. Ed. 2018, 57, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Dafflon, C.; Craig, V.J.; Méreau, H.; Gräsel, J.; Schacher Engstler, B.; Hoffman, G.; Nigsch, F.; Gaulis, S.; Barys, L.; Ito, M.; et al. Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 2017, 31, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Flajollet, S.; Rachez, C.; Ploton, M.; Schulz, C.; Gallais, R.; Métivier, R.; Pawlak, M.; Leray, A.; Issulahi, A.A.; Héliot, L.; et al. The Elongation Complex Components BRD4 and MLLT3/AF9 Are Transcriptional Coactivators of Nuclear Retinoid Receptors. PLoS ONE 2013, 8, e64880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carretta, M.; Brouwers-Vos, A.Z.; Bosman, M.; Horton, S.J.; Martens, J.H.A.; Vellenga, E.; Schuringa, J.J. BRD3/4 inhibition and FLT3-ligand deprivation target pathways that are essential for the survival of human MLL-AF9+ leukemic cells. PLoS ONE 2017, 12, e189102. [Google Scholar] [CrossRef] [PubMed]
- Gilan, O.; Lam, E.Y.N.; Becher, I.; Lugo, D.; Cannizzaro, E.; Joberty, G.; Ward, A.; Wiese, M.; Fong, C.Y.; Ftouni, S.; et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat. Struct. Mol. Biol. 2016, 23, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zheng, X.; Yang, Y.; Wang, X.; Shen, Z. An Overview on Small Molecule Inhibitors of BRD4. Mini Rev. Med. Chem. 2016, 16, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. [Google Scholar] [CrossRef] [PubMed]
- Kharenko, O.A.; Hansen, H.C. Novel approaches to targeting BRD4. Drug Discov. Today Technol. 2017, 24, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Townsend, E.C.; Cao, F.; Chen, Y.; Bernard, D.; Liu, L.; Lei, M.; Dou, Y.; Wang, S. High-affinity, small-molecule peptidomimetic inhibitors of mll1/wdr5 protein-protein interaction. J. Am. Chem. Soc. 2013, 135, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Li, Y.; Liu, L.; Ji, J.; Lee, S.; Chen, Y.; Yang, J.; Huang, L.; Bernard, D.; Xu, J.; et al. Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)-Mixed Lineage Leukemia (MLL) Protein-Protein Interaction. J. Med. Chem. 2017, 60, 4818–4839. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Chen, W.L.; Wang, Z.H.; Xie, Y.Y.; Xu, X.L.; Jiang, Z.Y.; Zhang, X.J.; You, Q.D.; Guo, X.K. High-affinity small molecular blockers of mixed lineage leukemia 1 (MLL1)-WDR5 interaction inhibit MLL1 complex H3K4 methyltransferase activity. Eur. J. Med. Chem. 2016, 124, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-L.; Li, D.-D.; Wang, Z.-H.; Xu, X.-L.; Zhang, X.-J.; Jiang, Z.-Y.; Guo, X.-K.; You, Q.-D. Design, synthesis, and initial evaluation of affinity-based small molecular probe for detection of WDR5. Bioorg. Chem. 2018, 76. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.B.C.; Bachovchin, D.A.; Brown, S.J.; Finn, M.G.; Rosen, H.; Cravatt, B.F.; Mowen, K.A. Novel inhibitors for PRMT1 discovered by high-throughput screening using activity-based fluorescence polarization. ACS Chem. Biol. 2012, 7, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Yang, L.; Xie, M.; Lin, C.; Merkurjev, D.; Yang, J.C.; Tanasa, B.; Oh, S.; Zhang, J.; Ohgi, K.A.; et al. Chem-seq permits identification of genomic targets of drugs against androgen receptor regulation selected by functional phenotypic screens. Proc. Natl. Acad. Sci. USA 2014, 111, 9235–9240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Liu, L.; Gao, H.; Pinnamaneni, J.P.; Sanagasetti, D.; Singh, V.P.; Wang, K.; Mathison, M.; Zhang, Q.; Chen, F.; et al. The stem cell factor SALL4 is an essential transcriptional regulator in mixed lineage leukemia-rearranged leukemogenesis. J. Hematol. Oncol. 2017, 10, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, S.; Peirs, S.; Van Loocke, W.; Wang, J.; Takawy, M.; Matthijssens, F.; Sonderegger, S.E.; Haigh, K.; Nguyen, T.; Vandamme, N.; et al. Oncogenic ZEB2 activation drives sensitivity toward KDM1A inhibition in T-cell acute lymphoblastic leukemia. Blood 2017, 129, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Konopleva, M.Y. ORY-1001: Overcoming the Differentiation Block in AML. Cancer Cell 2018, 33, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Pabst, C.; Su, L.; Miller, M.; Liu, F.; Yi, L.; Zhang, R.; Krosl, J.; Yung, E.; Kirschner, J.; et al. The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev. 2014, 28, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; On, D.M.; Ma, A.; Parton, T.; Konze, K.D.; Pattenden, S.G.; Allison, D.F.; Cai, L.; Rockowitz, S.; Liu, S.; et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL- rearranged leukemia. Blood 2015, 125, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, R.G.; Gehling, V.S.; Dakin, L.A.; Cook, A.S.; Nasveschuk, C.G.; Duplessis, M.; Iyer, P.; Balasubramanian, S.; Zhao, F.; Good, A.C.; et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitabl. J. Med. Chem. 2016, 59, 9928–9941. [Google Scholar] [CrossRef] [PubMed]
- Brach, D.; Johnston-Blackwell, D.; Drew, A.; Lingaraj, T.; Motwani, V.; Warholic, N.M.; Feldman, I.; Plescia, C.; Smith, J.J.; Copeland, R.A.; et al. EZH2 inhibition by tazemetostat results in altered dependency on B-cell activation signaling in DLBCL. Mol. Cancer Ther. 2017, 16, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2016, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.; Béguelin, W.; Giulino-Roth, L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk. Lymphoma 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Trop-Steinberg, S.; Azar, Y. Is Myc an Important Biomarker? Myc Expression in Immune Disorders and Cancer. Am. J. Med. Sci. 2018, 355, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma: Basic and clinical updates. Int. J. Hematol. 2013, 97, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; Bettess, M.D.; Oser, G.M.; Pasche, A.-C.; Knabenhans, C.; Macdonald, H.R.; Trumpp, A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004, 18, 2747–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, T.; Ghatak, P.; Price, D.H.; Conaway, R.; Conaway, J.; Chiang, C.M.; Bradner, J.E.; Shilatifard, A.; Roy, A.L. Regulation of MYC expression and differential JQ1 sensitivity in cancer cells. PLoS ONE 2014, 9, e87003. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loosveld, M.; Castellano, R.; Gon, S.; Goubard, A.; Crouzet, T.; Pouyet, L.; Prebet, T.; Vey, N.; Nadel, B.; Collette, Y.; et al. Therapeutic Targeting of c-Myc in T-Cell Acute Lymphoblastic Leukemia (T-ALL). Oncotarget 2014, 5, 3168–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Ciro, A.; Martens, J.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. C-Myc modulation and acetylation is a key HDAC inhibitor target in cancer. Clin. Cancer Res. 2016, 23, 2542–2555. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sánchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delehouzé, C.; Godl, K.; Loaëc, N.; Bruyère, C.; Desban, N.; Oumata, N.; Galons, H.; Roumeliotis, T.I.; Giannopoulou, E.G.; Grenet, J.; et al. CDK/CK1 inhibitors roscovitine and CR8 downregulate amplified MYCN in neuroblastoma cells. Oncogene 2014, 33, 5675–5687. [Google Scholar] [CrossRef] [PubMed]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The expanding world of N-MYC-driven tumors. Cancer Discov. 2018, 8, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. Regulation of transcription factors by protein degradation. Cell. Mol. Life Sci. 2000, 57, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Yokosawa, H. Degradation of transcription factor IRF-1 by the ubiquitin–proteasome pathway. Eur. J. Biochem. 2000, 267, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Muratani, M.; Tansey, W.P. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 2003, 4, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Geng, F.; Daulny, A.; Collins, G.; Guzzardo, P.; Tansey, W.P. Transcriptional control and the ubiquitin-proteasome system. Ernst Scher. Found. Symp. Proc. 2008, 75–97. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.X.; Wen, X.F.; Guo, Y.X.; Zhang, G.J. Targeting Notch degradation system provides promise for breast cancer therapeutics. Crit. Rev. Oncol. Hematol. 2016, 104, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Walf-Vorderwülbecke, V.; Pearce, K.; Brooks, T.; Hubank, M.; van den Heuvel-Eibrink, M.M.; Zwaan, C.M.; Adams, S.; Edwards, D.; Bartram, J.; Samarasinghe, S.; et al. Targeting acute myeloid leukemia by drug-induced c-MYB degradation. Leukemia 2018, 32, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Li, H.; Xing, C.; Ye, H.; Feng, J.; Wu, J.; Lu, Z.; Fang, J.; Gao, S. Honokiol induces proteasomal degradation of AML1-ETO oncoprotein via increasing ubiquitin conjugase UbcH8 expression in leukemia. Biochem. Pharmacol. 2017, 128, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Kerres, N.; Steurer, S.; Schlager, S.; Bader, G.; Berger, H.; Caligiuri, M.; Dank, C.; Engen, J.R.; Ettmayer, P.; Fischerauer, B.; et al. Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep. 2017, 20, 2860–2875. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Misawa, T.; Kurihara, M.; Demizu, Y.; Naito, M. Development of a peptide-based inducer of protein degradation targeting NOTCH1. Bioorgan. Med. Chem. Lett. 2017, 27, 4985–4988. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, S.; Fan, J.; Li, X.; Wen, Q.; Luo, N. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem. Pharmacol. 2016, 116, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von hippel-lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef] [PubMed]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and human malignancies: Expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M.C. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Lemos, A.; Leão, M.; Soares, J.; Palmeira, A.; Pinto, M.; Saraiva, L.; Sousa, M.E. Medicinal Chemistry Strategies to Disrupt the p53–MDM2/MDMX Interaction. Med. Res. Rev. 2016, 36, 789–844. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.K.; Khatik, G.L.; Narang, R.; Monga, V.; Chopra, H.K. p53-Mdm2 interaction inhibitors as novel nongenotoxic anticancer agents. Curr. Cancer Drug Targets 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.Y. Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: Progress and challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McEachern, D.; Li, S.; Ellis, M.J.; Wang, S. Reactivation of p53 by MDM2 Inhibitor MI-77301 for the Treatment of Endocrine-Resistant Breast Cancer. Mol. Cancer Ther. 2016, 15, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Bharadwaj, M.; Kumar, A.; Mehrotra, R. Spiro-oxindoles as a Promising Class of Small Molecule Inhibitors of p53–MDM2 Interaction Useful in Targeted Cancer Therapy. Top. Curr. Chem. 2017, 375. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Zhao, Y.; Wang, S. AM-8553: A novel MDM2 inhibitor with a promising outlook for potential clinical development. J. Med. Chem. 2012, 55, 4934–4935. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Sun, D. Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J. Med. Chem. 2014, 57, 6332–6341. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.Z.; Eksterowicz, J.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Fox, B.M.; Fu, J.; et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J. Med. Chem. 2014, 57, 2472–2488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ding, Q.; Liu, J.J.; Zhang, J.; Jiang, N.; Chu, X.J.; Bartkovitz, D.; Luk, K.C.; Janson, C.; Tovar, C.; et al. Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorgan. Med. Chem. 2014, 22, 4001–4009. [Google Scholar] [CrossRef] [PubMed]
- Dos-Santos, O.; Lagarde, P.; Pérot, G.; Chibon, F.; Ratet, N.; Flamand, O.; Debussche, L. Human Dedifferentiated Liposarcomas Growth Inhibition by SAR299155, a Potent and Selective Disruptor of the MDM2-p53 Interaction. Eur. J. Cancer 2012, 48, S245–S246. [Google Scholar] [CrossRef]
- Soares, J.; Espadinha, M.; Raimundo, L.; Ramos, H.; Gomes, A.S.; Gomes, S.; Loureiro, J.B.; Inga, A.; Reis, F.; Gomes, C.; et al. DIMP53-1: A novel small-molecule dual inhibitor of p53-MDM2/X interactions with multifunctional p53-dependent anticancer properties. Mol. Oncol. 2017, 11, 612–627. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, B.B.; Drew, K.; Guarracino, D.A.; Brewer, T.F.; Heindel, D.W.; Bonneau, R.; Arora, P.S. Rational design of topographical helix mimics as potent inhibitors of protein-protein interactions. J. Am. Chem. Soc. 2014, 136, 7877–7888. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, P.; Berlicki, Ł. Peptide-based inhibitors of protein-protein interactions. Bioorgan. Med. Chem. Lett. 2016, 26, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Wallbrecher, R.; Chène, P.; Ruetz, S.; Stachyra, T.; Vorherr, T.; Brock, R. A critical assessment of the synthesis and biological activity of p53/human double minute 2-stapled peptide inhibitors. Br. J. Pharmacol. 2017, 174, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Sajadimajd, S.; Khazaei, M. Oxidative Stress and Cancer: The role of Nrf2. Curr. Cancer Drug Targets 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, H.M.; Kansanen, E.; Pölönen, P.; Heinäniemi, M.; Levonen, A.L. Role of the keap1-Nrf2 pathway in cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar] [CrossRef] [PubMed]
- Watai, Y.; Kobayashi, A.; Nagase, H.; Mizukami, M.; Mcevoy, J.; Singer, J.D.; Itoh, K.; Yamamoto, M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 2007, 12, 1163–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Lleres, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Nakajima, M.; Yuasa, A.; Obata, R.; Takahashi, K.; Ohe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Imamura, R.; et al. Synthesis of Keap1-phosphorylated p62 and Keap1-Nrf2 protein-protein interaction inhibitors and their inhibitory activity. Bioorgan. Med. Chem. Lett. 2016, 26, 5956–5959. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Yuasa, A.; Obata, R.; Nakajima, M.; Takahashi, K.; Ohe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Imamura, R.; et al. Discovery of benzo[g]indoles as a novel class of non-covalent Keap1-Nrf2 protein-protein interaction inhibitor. Bioorg. Med. Chem. Lett. 2017, 27, 5006–5009. [Google Scholar] [CrossRef] [PubMed]
- Meng, N.; Tang, H.; Zhang, H.; Jiang, C.; Su, L.; Min, X.; Zhang, W.; Zhang, H.; Miao, Z.; Zhang, W.; et al. Fragment-growing guided design of Keap1-Nrf2 protein-protein interaction inhibitors for targeting myocarditis. Free Radic. Biol. Med. 2018, 117, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Jiao, Q.; Liu, T.; Tan, S.J.; Zhou, H.S.; You, Q.D.; Jiang, Z.Y. Discovery of a head-to-tail cyclic peptide as the Keap1-Nrf2 protein-protein interaction inhibitor with high cell potency. Eur. J. Med. Chem. 2018, 143, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, M.J.; Grembecka, J.; Zhou, Y.; Kong, Y.; Roudaia, L.; Douvas, M.G.; Newman, M.; Bielnicka, I.; Baber, G.; Corpora, T.; et al. Allosteric Inhibition of the Protein-Protein Interaction between the Leukemia-Associated Proteins Runx1 and CBFβ. Chem. Biol. 2007, 14, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, L.; Finckbeiner, S.; Hyde, R.K.; Southall, N.; Marugan, J.; Yedavalli, V.R.K.; Dehdashti, S.J.; Reinhold, W.C.; Alemu, L.; Zhao, L.; et al. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBF interaction. Proc. Natl. Acad. Sci. USA 2012, 109, 14592–14597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illendula, A.; Pulikkan, J.A.; Zong, H.; Grembecka, J.; Xue, L.; Sen, S.; Zhou, Y.; Boulton, A.; Kuntimaddi, A.; Gao, Y.; et al. A small-molecule inhibitor of the aberrant transcription factor CBFβ-SMMHC delays leukemia in mice. Science 2015, 347, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Youbi, S.E.; Hong, S.P.; Kallakury, B.; Monroe, P.; Erkizan, H.V.; Barber-Rotenberg, J.S.; Houghton, P.; Üren, A.; Toretsky, J.A. Pharmacokinetic modeling optimizes inhibition of the “undruggable” EWS-FLI1 transcription factor in Ewing Sarcoma. Oncotarget 2014, 5, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Barber-Rotenberg, J.S.; Selvanathan, S.P.; Kong, Y.; Erkizan, H.V.; Snyder, T.M.; Hong, S.P.; Kobs, C.L.; South, N.L.; Summer, S.; Monroe, P.J.; et al. Single enantiomer of YK-4-279 demonstrates specificity in targeting the oncogene EWS-FLI1. Oncotarget 2012, 3, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Minas, T.Z.; Han, J.; Javaheri, T.; Hong, S.-H.; Schlederer, M.; Saygideger-Kont, Y.; Celik, H.; Mueller, K.M.; Temel, I.; Ozdemirli, M.; et al. YK-4-279 effectively antagonizes EWS-FLI1 induced leukemia in a transgenic mouse model. Oncotarget 2015, 6, 37678–37694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Rojas, Y.; Wang, H.; Yu, Y.; Wang, Y.; Chen, Z.; Rajapakshe, K.; Xu, X.; Huang, W.; Agarwal, S.; et al. EWS-FLI1 and RNA helicase A interaction inhibitor YK-4-279 inhibits growth of neuroblastoma. Oncotarget 2017, 8, 94780–94792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahim, S.; Beauchamp, E.M.; Kong, Y.; Brown, M.L.; Toretsky, J.A.; Üren, A. YK-4-279 inhibits ERG and ETV1 mediated prostate cancer cell invasion. PLoS ONE 2011, 6, e19343. [Google Scholar] [CrossRef] [PubMed]
- Winters, B.; Brown, L.; Coleman, I.; Nguyen, H.; Minas, T.Z.; Kollath, L.; Vasioukhin, V.; Nelson, P.; Corey, E.; Üren, A.; et al. Inhibition of ERG Activity in Patient-derived Prostate Cancer Xenografts by YK-4-279. Anticancer Res. 2017, 37, 3385–3396. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.A.Z.; Gunning, P.T.; Glen, M.; Katt, W.P.; Zhang, S.; Schroeck, C.; Sebti, S.M.; Jove, R.; Hamilton, A.D.; Turkson, J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, A.; Xu, Z.; Yu, W.; Wang, H.; Li, C.; Lin, J. XZH-5 inhibits STAT3 phosphorylation and causes apoptosis in human hepatocellular carcinoma cells. Apoptosis 2011, 16, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Daka, P.; Liu, A.; Karunaratne, C.; Csatary, E.; Williams, C.; Xiao, H.; Lin, J.; Xu, Z.; Page, R.C.; Wang, H. Design, synthesis and evaluation of XZH-5 analogues as STAT3 inhibitors. Bioorgan. Med. Chem. 2015, 23, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 2011, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Zuo, M.; Li, C.; Lin, J.; Javle, M. LLL12, a novel small inhibitor targeting STAT3 for hepatocellular carcinoma therapy. Oncotarget 2015, 6, 10940–10949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Luo, W.; Liu, L.; Pang, X.; Zhu, H.; Liu, A.; Lu, J.; Ma, D.L.; Leung, C.H.; Wang, Y.; et al. Isocryptotanshinone, a STAT3 inhibitor, induces apoptosis and pro-death autophagy in A549 lung cancer cells. J. Drug Target. 2016, 24, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Pallandre, J.R.; Borg, C.; Rognan, D.; Boibessot, T.; Luzet, V.; Yesylevskyy, S.; Ramseyer, C.; Pudlo, M. Novel aminotetrazole derivatives as selective STAT3 non-peptide inhibitors. Eur. J. Med. Chem. 2015, 103, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Xu, X.; Ma, S.; Fan, J.; Zhou, Q.; Mao, X.; Qiao, C. Novel 2-Carbonylbenzo[b]thiophene 1,1-Dioxide Derivatives as Potent Inhibitors of STAT3 Signaling Pathway. ACS Med. Chem. Lett. 2015, 6, 1010–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, P.; Yuan, C.; Ma, S.; Fan, J.; Fu, W.; Qiao, C. 4-Carbonyl-2,6-dibenzylidenecyclohexanone derivatives as small molecule inhibitors of STAT3 signaling pathway. Bioorgan. Med. Chem. 2016, 24, 6174–6182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, T.; Li, S.; Yang, Y.; Guo, J.; Yu, W.; Kong, L. Antagonizing STAT3 activation with benzo[b]thiophene 1, 1-dioxide based small molecules. Eur. J. Med. Chem. 2017, 125, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; He, L.; Cao, P.; Yu, Q. Eriocalyxin B inhibits STAT3 signaling by covalently targeting STAT3 and blocking phosphorylation and activation of STAT3. PLoS ONE 2015, 10, e128406. [Google Scholar] [CrossRef] [PubMed]
- Don-Doncow, N.; Escobar, Z.; Johansson, M.; Kjellström, S.; Garcia, V.; Munoz, E.; Sterner, O.; Bjartell, A.; Hellsten, R. Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J. Biol. Chem. 2014, 289, 15969–15978. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Evans-Axelsson, S.; Hellsten, R.; Sterner, O.; Krzyzanowska, A.; Andersson, T.; Bjartell, A. The STAT3 Inhibitor Galiellalactone Effectively Reduces Tumor Growth and Metastatic Spread in an Orthotopic Xenograft Mouse Model of Prostate Cancer. Eur. Urol. 2016, 69, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, T.; Ko, H.; Lee, J.; Kim, Y.S.; Suh, Y.G. Identification of galiellalactone-based novel STAT3-selective inhibitors with cytotoxic activities against triple-negative breast cancer cell lines. Bioorgan. Med. Chem. 2017, 25, 5032–5040. [Google Scholar] [CrossRef] [PubMed]
- Escobar, Z.; Bjartell, A.; Canesin, G.; Evans-Axelsson, S.; Sterner, O.; Hellsten, R.; Johansson, M.H. Preclinical Characterization of 3β-(N-Acetyl l-cysteine methyl ester)-2aβ,3-dihydrogaliellalactone (GPA512), a Prodrug of a Direct STAT3 Inhibitor for the Treatment of Prostate Cancer. J. Med. Chem. 2016, 59, 4551–4562. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, J.R.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, M.A.; Lambert, M.; David-Cordonnier, M.-H. Targeting transcription factor binding to DNA by competition using DNA binders as an approach for controlling gene expression. Curr. Top. Med. Chem. 2015, 15, 1323–1358. [Google Scholar] [CrossRef] [PubMed]
- Kerckaert, J.P.; Deweindt, C.; Tilly, H.; Quief, S.; Lecocq, G.; Bastard, C. LAZ3, a novel zinc-finger encoding gene, is disrupted by recurring chromosome 3q27 translocations in human lymphomas. Nat. Genet. 1993, 5, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, Y.; Dutra-Clarke, M.; Mayakonda, A.; Hazawa, M.; Savinoff, S.E.; Doan, N.; Said, J.W.; Yong, W.H.; Watkins, A.; et al. BCL6 promotes glioma and serves as a therapeutic target. Proc. Natl. Acad. Sci. USA 2017, 114, 3981–3986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, J.M.; Dell’Oso, T.; Ranuncolo, S.M.; Cerchietti, L.; Beck, D.; Da Silva, G.F.; Prive, G.G.; Licht, J.D.; Melnick, A. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat. Med. 2004, 10, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Cerchietti, L.C.; Yang, S.N.; Shaknovich, R.; Hatzi, K.; Polo, J.M.; Chadburn, A.; Dowdy, S.F.; Melnick, A. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 2009, 113, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Sogabe, S.; Kamada, Y.; Sakai, N.; Asano, K.; Yoshimatsu, M.; Ida, K.; Imaeda, Y.; Sakamoto, J. ichi Discovery of high-affinity BCL6-binding peptide and its structure-activity relationship. Biochem. Biophys. Res. Commun. 2017, 482, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Osher, E.L.; Castillo, F.; Elumalai, N.; Waring, M.J.; Pairaudeau, G.; Tavassoli, A. A genetically selected cyclic peptide inhibitor of BCL6 homodimerization. Bioorgan. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, M.G.; Yu, W.; Beguelin, W.; Teater, M.R.; Geng, H.; Goldstein, R.L.; Oswald, E.; Hatzi, K.; Yang, S.-N.; Cohen, J.; et al. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J. Clin. Investig. 2016, 126, 3351–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, Y.; Sakai, N.; Sogabe, S.; Ida, K.; Oki, H.; Sakamoto, K.; Lane, W.; Snell, G.; Iida, M.; Imaeda, Y.; et al. Discovery of a B-Cell Lymphoma 6 Protein-Protein Interaction Inhibitor by a Biophysics-Driven Fragment-Based Approach. J. Med. Chem. 2017, 60, 4358–4368. [Google Scholar] [CrossRef] [PubMed]
- Yasui, T.; Yamamoto, T.; Sakai, N.; Asano, K.; Takai, T.; Yoshitomi, Y.; Davis, M.; Takagi, T.; Sakamoto, K.; Sogabe, S.; et al. Discovery of a novel B-cell lymphoma 6 (BCL6)–corepressor interaction inhibitor by utilizing structure-based drug design. Bioorgan. Med. Chem. 2017, 25, 4876–4886. [Google Scholar] [CrossRef] [PubMed]
- McCoull, W.; Abrams, R.D.; Anderson, E.; Blades, K.; Barton, P.; Box, M.; Burgess, J.; Byth, K.; Cao, Q.; Chuaqui, C.; et al. Discovery of Pyrazolo[1,5-a]pyrimidine B-Cell Lymphoma 6 (BCL6) Binders and Optimization to High Affinity Macrocyclic Inhibitors. J. Med. Chem. 2017, 60, 4386–4402. [Google Scholar] [CrossRef] [PubMed]
- Sameshima, T.; Yamamoto, T.; Sano, O.; Sogabe, S.; Igaki, S.; Sakamoto, K.; Ida, K.; Gotou, M.; Imaeda, Y.; Sakamoto, J.; et al. Discovery of an Irreversible and Cell-Active BCL6 Inhibitor Selectively Targeting Cys53 Located at the Protein-Protein Interaction Interface. Biochemistry 2018, 57, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Fancher, A.T.; Hua, Y.; Camarco, D.P.; Close, D.A.; Strock, C.J.; Johnston, P.A. Reconfiguring the AR-TIF2 Protein–Protein Interaction HCS Assay in Prostate Cancer Cells and Characterizing the Hits from a LOPAC Screen. Assay Drug Dev. Technol. 2016, 14, 453–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viziteu, E.; Grandmougin, C.; Goldschmidt, H.; Seckinger, A.; Hose, D.; Klein, B.; Moreaux, J. Chetomin, targeting HIF-1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br. J. Cancer 2016, 114, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.; El-Tanani, M.; Hunter, K.D.; Harrington, K.J.; Pandha, H.S. Targeting HOX/PBX dimers in cancer. Oncotarget 2015, 8, 32322–32331. [Google Scholar] [CrossRef] [PubMed]
- Gibault, F.; Bailly, F.; Corvaisier, M.; Coevoet, M.; Huet, G.; Melnyk, P.; Cotelle, P. Molecular Features of the YAP Inhibitor Verteporfin: Synthesis of Hexasubstituted Dipyrrins as Potential Inhibitors of YAP/TAZ, the Downstream Effectors of the Hippo Pathway. ChemMedChem 2017, 12, 954–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor α gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kitamura, K.; Tanaka, K.; Omura, S.; Miyazaki, T.; Hachiya, T.; Ohno, R.; Naoe, T. Accelerated degradation of PML-retinoic acid receptor alpha (PML-RARA) oncoprotein by all-trans-retinoic acid in acute promyelocytic leukemia: Possible role of the proteasome pathway. Cancer Res. 1996, 56, 2945–2948. [Google Scholar] [PubMed]
- Tang, H.; Chen, F.; Tan, Q.; Tan, S.; Liu, L.; Zhang, F. Regulation of CD11b transcription by decreasing PRC2 and increased acH4 level during ATRA-induced HL-60 differentiation. Acta Biochim. Biophys. Sin. 2009, 41, 588–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimwade, D.; Mistry, A.R.; Solomon, E.; Guidez, F. Acute promyelocytic leukemia: A paradigm for differentiation therapy. Cancer Treat. Res. 2010, 145, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ban, F.; Dalal, K.; Leblanc, E.; Frewin, K.; Ma, D.; Adomat, H.; Rennie, P.S.; Cherkasov, A. Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor. J. Med. Chem. 2014, 57, 6458–6467. [Google Scholar] [CrossRef] [PubMed]
- Dalal, K.; Roshan-Moniri, M.; Sharma, A.; Li, H.; Ban, F.; Hessein, M.; Hsing, M.; Singh, K.; LeBlanc, E.; Dehm, S.; et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J. Biol. Chem. 2014, 289, 26417–26429. [Google Scholar] [CrossRef] [PubMed]
- Poulain, S.; Roumier, C.; Bertrand, E.; Renneville, A.; Caillault-Venet, A.; Doye, E.; Geffroy, S.; Sebda, S.; Nibourel, O.; Nudel, M.; et al. TP53 Mutation and Its Prognostic Significance in Waldenstrom’s Macroglobulinemia. Clin. Cancer Res. 2017, 23, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 in breast cancer: Potential as a therapeutic target and biomarker. Breast Cancer Res. Treat. 2018, 170, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.W.; Mawji, I.A.; Macrae, C.J.; Koch, C.A.; Datti, A.; Wrana, J.L.; Dennis, J.W.; Schimmer, A.D. A high-content chemical screen identifies ellipticine as a modulator of p53 nuclear localization. Apoptosis 2008, 13, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.R.; Rökaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izetti, P.; Hautefeuille, A.; Abujamra, A.L.; De Farias, C.B.; Giacomazzi, J.; Alemar, B.; Lenz, G.; Roesler, R.; Schwartsmann, G.; Osvaldt, A.B.; et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Investig. New Drugs 2014, 32, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Deben, C.; Lardon, F.; Wouters, A.; Op de Beeck, K.; Van den Bossche, J.; Jacobs, J.; Van Der Steen, N.; Peeters, M.; Rolfo, C.; Deschoolmeester, V.; et al. APR-246 (PRIMA-1MET) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016, 375, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; Van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. P53 Reactivation by PRIMA-1Met(APR-246) sensitisesV600E/KBRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.R.; Joerger, A.C.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef] [PubMed]
- Synnott, N.C.; Murray, A.; McGowan, P.M.; Kiely, M.; Kiely, P.A.; O’Donovan, N.; O’Connor, D.P.; Gallagher, W.M.; Crown, J.; Duffy, M.J. Mutant p53: A novel target for the treatment of patients with triple-negative breast cancer? Int. J. Cancer 2017, 140, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Dong, Z.; Wang, F.; Peng, H.; Liu, J.Y.; Zhang, J.T. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem. Biol. 2014, 9, 1188–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Dong, Z.; Chen, Y.; Wang, F.; Wang, C.J.; Peng, H.; He, Y.; Hangoc, G.; Pollok, K.; Sandusky, G.; et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2015, 35, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Corzano, R.; Rashid, R.; Lin, J.; Senthil, M.; Hedvat, M.; Schroeder, A.; Mao, A.; Herrmann, A.; Yim, J.; et al. Alkylation of cysteine 468 in stat3 defines a novel site for therapeutic development. ACS Chem. Biol. 2011, 6, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. The inhibition of Stat5 by a peptide aptamer ligand specific for the DNA binding domain prevents target gene transactivation and the growth of breast and prostate tumor cells. Pharmaceuticals 2013, 6, 960–987. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. Stat5 exerts distinct, vital functions in the cytoplasm and nucleus of Bcr-Abl+K562 and Jak2(V617F)+HEL leukemia cells. Cancers 2015, 7, 503–537. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492–4503. [Google Scholar] [CrossRef] [PubMed]
- Shahi, M.H.; Holt, R.; Rebhun, R.B. Blocking signaling at the level of GLI regulates downstream gene expression and inhibits proliferation of canine osteosarcoma cells. PLoS ONE 2014, 9, e96593. [Google Scholar] [CrossRef] [PubMed]
- Ghezali, L.; Liagre, B.; Limami, Y.; Beneytout, J.L.; Leger, D.Y. Sonic hedgehog activation is implicated in diosgenin-induced megakaryocytic differentiation of human erythroleukemia cells. PLoS ONE 2014, 9, e95016. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Peng, X.; Yuan, X.; Huang, D.; Chen, J.; Lu, Q.; Lv, N.; Luo, S. Suppression of growth and migration by blocking the Hedgehog signaling pathway in gastric cancer cells. Cell. Oncol. 2013, 36, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Tabata, K.; Suzuki, T. The GANT61, a GLI Inhibitor, Induces Caspase-Independent Apoptosis of SK-N-LO Cells. Biol. Pharm. Bull. 2014, 37, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Mayr, C.; Wachter, J.; Bach, D.; Fuereder, J.; Wagner, A.; Alinger, B.; Pichler, M.; Di Fazio, P.; Ocker, M.; et al. Activated hedgehog pathway is a potential target for pharmacological intervention in biliary tract cancer. Mol. Cell. Biochem. 2014, 396, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Oladapo, H.O.; Tarpley, M.; Sauer, S.J.; Addo, K.A.; Ingram, S.M.; Strepay, D.; Ehe, B.K.; Chdid, L.; Trinkler, M.; Roques, J.R.; et al. Pharmacological targeting of GLI1 inhibits proliferation, tumor emboli formation and in vivo tumor growth of inflammatory breast cancer cells. Cancer Lett. 2017, 411, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, J.; Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017, 108, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Hu, L.; Liu, Z.; Qin, Y.; Li, R.; Zhang, G.; Zhao, B.; Bi, C.; Lei, Y.; Bai, Y. Inhibition of Gli1-mediated prostate cancer cell proliferation by inhibiting the mTOR/S6K1 signaling pathway. Oncol. Lett. 2017, 14, 7970–7976. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Qiu, L.; Qi, M.; Liu, J.; Hu, K.; Lin, W.; Huang, Y.; Fu, J. GANT-61 and GDC-0449 induce apoptosis of prostate cancer stem cells through a GLI-dependent mechanism. J. Cell. Biochem. 2018, 119, 3641–3652. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Yamagishi, M.; Nakano, K.; Yamochi, T.; Yamochi, T.; Fujikawa, D.; Nakashima, M.; Tanaka, Y.; Uchimaru, K.; Utsunomiya, A.; et al. Epigenetic deregulation of Ellis Van Creveld confers robust Hedgehog signaling in adult T-cell leukemia. Cancer Sci. 2014, 105, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Latuske, E.-M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jücker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao-Kitamoto, H.; Nagata, M.; Nagano, S.; Kitamoto, S.; Ishidou, Y.; Yamamoto, T.; Nakamura, S.; Tsuru, A.; Abematsu, M.; Fujimoto, Y.; et al. GLI2 is a novel therapeutic target for metastasis of osteosarcoma. Int. J. Cancer 2015, 136, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xu, R.; Zeng, C.; Lu, Q.; Huang, D.; Shi, C.; Zhang, W.; Deng, L.; Yan, R.; Rao, H.; et al. Down-regulation of Gli transcription factor leads to the inhibition of migration and invasion of ovarian cancer cells via integrin β4-mediated FAK signaling. PLoS ONE 2014, 9, e88386. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; McKee, C.M.; Dok, R.; Haustermans, K.; Muschel, R.J. The hedgehog inhibitor GANT61 sensitizes prostate cancer cells to ionizing radiation both in vitro and in vivo. Oncotarget 2016, 7, 84286–84298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cai, J.; Zhao, S.; Yao, K.; Sun, Y.; Li, Y.; Chen, L.; Li, R.; Zhai, X.; Zhang, J.; et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 184. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Iwasawa, S.; Ebata, T.; Fan, M.; Tada, Y.; Tatsumi, K.; Takiguchi, Y. Inhibition of Gli leads to antitumor growth and enhancement of cisplatin-induced cytotoxicity in large cell neuroendocrine carcinoma of the lung. Oncol. Rep. 2018, 39, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Pop, M.S.; Stransky, N.; Garvie, C.W.; Theurillat, J.-P.; Hartman, E.C.; Lewis, T.A.; Zhong, C.; Culyba, E.K.; Lin, F.; Daniels, D.S.; et al. A Small Molecule That Binds and Inhibits the ETV1 Transcription Factor Oncoprotein. Mol. Cancer Ther. 2014, 13, 1492–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimley, E.; Liao, C.; Ranghini, E.J.; Nikolovska-Coleska, Z.; Dressler, G.R. Inhibition of Pax2 Transcription Activation with a Small Molecule that Targets the DNA Binding Domain. ACS Chem. Biol. 2017, 12, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Vilaboa, N.; Boré, A.; Martin-Saavedra, F.; Bayford, M.; Winfield, N.; Firth-Clark, S.; Kirton, S.B.; Voellmy, R. New inhibitor targeting human transcription factor HSF1: Effects on the heat shock response and tumor cell survival. Nucleic Acids Res. 2017, 45, 5797–5817. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei-Dakhili, S.A.; Aguayo-Ortiz, R.; Domínguez, L.; Velázquez-Martínez, C.A. Untying the knot of transcription factor druggability: Molecular modeling study of FOXM1 inhibitors. J. Mol. Graph. Model. 2018, 80, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Gormally, M.V.; Dexheimer, T.S.; Marsico, G.; Sanders, D.A.; Lowe, C.; Matak-Vinkoviä, D.; Michael, S.; Jadhav, A.; Rai, G.; Maloney, D.J.; et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Marsico, G.; Gormally, M.V. Small molecule inhibition of FOXM1: How to bring a novel compound into genomic context. Genom. Data 2015, 3, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N. Inhibition of transcription by Pluramycin and Bleomycin. J. Antibiot. 1970, 23, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Nikolov, D.B.; Burley, S.K. Co-crystal structure of TBP recognizing the minor groove of a TATA element. Nature 1993, 365, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Hurley, L.H. TBP binding to the TATA box induces a specific downstream unwinding site that is targeted by pluramycin. Chem. Biol. 1995, 2, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Yun, S.; Hurley, L. Hedamycin intercalates the DNA helix and, through carbohydrate-mediated recognition in the minor groove, directs N7-alkylation of guanine in the major groove in a sequence-specific manner. Chem. Biol. 1995, 2, 229–240. [Google Scholar] [CrossRef]
- Chiang, S.Y.; Welch, J.; Beerman, T.A.; Rauscher, F.J. Effects of Minor Groove Binding Drugs on the Interaction of TATA Box Binding Protein and TFIIA with DNA. Biochemistry 1994, 33, 7033–7040. [Google Scholar] [CrossRef] [PubMed]
- Cairns, M.J.; Murray, V. Detection of protein-DNA interactions at β-globin gene cluster in intact human cells utilizing hedamycin as DNA-damaging agent. DNA Cell Biol. 1998, 17, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Malinge, J.M.; Pérez, C.; Leng, M. Base sequence-independent distorsions induced by interstrand cross-links in cis-diamminedichloroplatinum (II)-modified DNA. Nucleic Acids Res. 1994, 22, 3834–3839. [Google Scholar] [CrossRef] [PubMed]
- Bellon, S.F.; Coleman, J.H.; Lippard, S.J. DNA Unwinding Produced by Site-Specific Intrastrand Cross-Links of the Antitumor Drug cis-Diamminedichloroplatinum (II). Biochemistry 1991, 30, 8026–8035. [Google Scholar] [CrossRef] [PubMed]
- Kasparkova, J.; Marini, V.; Bursova, V.; Brabec, V. Biophysical studies on the stability of DNA intrastrand cross-links of transplatin. Biophys. J. 2008, 95, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L.; Dragan, A.I.; Crane-Robinson, C.; Breslauer, K.J.; Remeta, D.P.; Minetti, C.A.S.A. What Drives Proteins into the Major or Minor Grooves of DNA? J. Mol. Biol. 2007, 365, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Rossetti, G.; Arnesano, F.; Ippoliti, E.; Natile, G.; Carloni, P. Molecular recognition of platinated DNA from chromosomal HMGB1. J. Chem. Theory Comput. 2014, 10, 3578–3584. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ray, R.; Rajeswari, M.R. Overexpression of high mobility group (HMG) B1 and B2 proteins directly correlates with the progression of squamous cell carcinoma in skin. Cancer Investig. 2008, 26, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ramanjaneyulu, A.; Ray, R.; Rajeswari, M.R. Involvement of High Mobility Group B Proteins in Cisplatin-Induced Cytotoxicity in Squamous Cell Carcinoma of Skin. DNA Cell Biol. 2009, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lippard, S.J. Binding Interaction of HMGB4 with Cisplatin-Modified DNA. Biochemistry 2012, 51, 6728–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimmer, E.E.; Zamble, D.B.; Lippard, S.J.; Essigmann, J.M. Human testis-determining factor SRY binds to the major DNA adduct of cisplatin and a putative target sequence with comparable affinities. Biochemistry 1998, 37, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Chválová, K.; Sari, M.A.; Bombard, S.; Kozelka, J. LEF-1 recognition of platinated GG sequences within double-stranded DNA. Influence of flanking bases. J. Inorg. Biochem. 2008, 102, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Bounaix Morand du Puch, C.; Barbier, E.; Kraut, A.; Couté, Y.; Fuchs, J.; Buhot, A.; Livache, T.; Sève, M.; Favier, A.; Douki, T.; et al. TOX4 and its binding partners recognize DNA adducts generated by platinum anticancer drugs. Arch. Biochem. Biophys. 2011, 507, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Ninomiya, W.; Sakamoto, E.; Shibata, T.; Akiyama, H.; Tashiro, F. SRY and OCT4 Are Required for the Acquisition of Cancer Stem Cell-Like Properties and Are Potential Differentiation Therapy Targets. Stem Cells 2015, 33, 2652–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389–1406. [Google Scholar] [PubMed]
- Cohen, S.M.; Jamieson, E.R.; Lippard, S.J. Enhanced binding of the TATA-binding protein to TATA boxes containing flanking cisplatin 1,2-cross-links. Biochemistry 2000, 39, 8259–8265. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Aiba, I.; Chen, H.H.W.; Kuo, M.T. Effects of Cu(II) and cisplatin on the stability of Specific protein 1 (Sp1)-DNA binding: Insights into the regulation of copper homeostasis and platinum drug transport. J. Inorg. Biochem. 2016, 161, 37–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasparkova, J.; Thibault, T.; Kostrhunova, H.; Stepankova, J.; Vojtiskova, M.; Muchova, T.; Midoux, P.; Malinge, J.M.; Brabec, V. Different affinity of nuclear factor-kappa B proteins to DNA modified by antitumor cisplatin and its clinically ineffective trans isomer. FEBS J. 2014, 281, 1393–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasparkova, J.; Fojta, M.; Farrell, N.; Brabec, V. Differential recognition by the tumor suppressor protein p53 of DNA modified by the novel antitumor trinuclear platinum drug BBR3464 and cisplatin. Nucleic Acids Res. 2004, 32, 5546–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzel, C.C.; Berberich, S.J. p53 binds to cisplatin-damaged DNA. Biochim. Biophys. Acta Gene Struct. Expr. 2001, 1517, 392–397. [Google Scholar] [CrossRef]
- Wei, Y.D.; Tepperman, K.; Huang, M.Y.; Sartor, M.A.; Puga, A. Chromium Inhibits Transcription from Polycyclic Aromatic Hydrocarbon-inducible Promoters by Blocking the Release of Histone Deacetylase and Preventing the Binding of p300 to Chromatin. J. Biol. Chem. 2004, 279, 4110–4119. [Google Scholar] [CrossRef] [PubMed]
- VonHandorf, A.; Sánchez-Martín, F.J.; Biesiada, J.; Zhang, H.; Zhang, X.; Medvedovic, M.; Puga, A. Chromium disrupts chromatin organization and CTCF access to its cognate sites in promoters of differentially expressed genes. Epigenetics 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.; Hurley, L.H. Specific targeting of protein-DNA complexes by DNA-reactive drugs (+)- CC-1065 and pluramycins. J. Mol. Recognit. 1996, 9, 75–87. [Google Scholar] [CrossRef]
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K.W. DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the caribbean tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309. [Google Scholar] [CrossRef] [PubMed]
- Zewail-Foote, M.; Hurley, L.H. Ecteinascidin 743: A minor groove alkylator that bends DNA toward the major groove. J. Med. Chem. 1999, 42, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- David-Cordonnier, M.-H.; Gajate, C.; Olmea, O.; Laine, W.; De La Iglesia-Vicente, J.; Perez, C.; Cuevas, C.; Otero, G.; Manzanares, I.; Bailly, C.; et al. DNA and non-DNA targets in the mechanism of action of the antitumor drug trabectedin. Chem. Biol. 2005, 12, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc. Natl. Acad. Sci. USA. 2000, 97, 6775–6779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minuzzo, M.; Marchini, S.; Broggini, M.; Faircloth, G.; D’Incalci, M.; Mantovani, R. Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proc. Natl. Acad. Sci. USA 2000, 97, 6780–6784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, D.; Borbone, E.; Palmieri, D.; Uboldi, S.; Esposito, F.; Frapolli, R.; Pacelli, R.; D’Incalci, M.; Fusco, A. The impairment of the High Mobility Group A (HMGA) protein function contributes to the anticancer activity of trabectedin. Eur. J. Cancer 2013, 49, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Forni, C.; Minuzzo, M.; Virdis, E.; Tamborini, E.; Simone, M.; Tavecchio, M.; Erba, E.; Grosso, F.; Gronchi, A.; Aman, P.; et al. Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol. Cancer Ther. 2009, 8, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uboldi, S.; Bernasconi, S.; Romano, M.; Marchini, S.; Fuso Nerini, I.; Damia, G.; Ganzinelli, M.; Marangon, E.; Sala, F.; Clivio, L.; et al. Characterization of a new trabectedin-resistant myxoid liposarcoma cell line that shows collateral sensitivity to methylating agents. Int. J. Cancer 2012, 131, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giandomenico, S.; Frapolli, R.; Bello, E.; Uboldi, S.; Licandro, S.A.; Marchini, S.; Beltrame, L.; Brich, S.; Mauro, V.; Tamborini, E.; et al. Mode of action of trabectedin in myxoid liposarcomas. Oncogene 2013, 33, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.-R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 Interferes with the Activity of EWS-FLI1 in Ewing Sarcoma Cells. Neoplasia 2011, 13, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Harlow, M.L.; Maloney, N.; Roland, J.; Guillen Navarro, M.J.; Easton, M.K.; Kitchen-Goosen, S.M.; Boguslawski, E.A.; Madaj, Z.B.; Johnson, B.K.; Bowman, M.J.; et al. Lurbinectedin inactivates the Ewing sarcoma oncoprotein EWS-FLI1 by redistributing it within the nucleus. Cancer Res. 2016, 76, 6657–6668. [Google Scholar] [CrossRef] [PubMed]
- Uboldi, S.; Craparotta, I.; Colella, G.; Ronchetti, E.; Beltrame, L.; Vicario, S.; Marchini, S.; Panini, N.; Dagrada, G.; Bozzi, F.; et al. Mechanism of action of trabectedin in desmoplastic small round cell tumor cells. BMC Cancer 2017, 17, 107. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.; Bailly, C.; David-Cordonnier, M.-H. Topoisomerase I-mediated DNA relaxation as a tool to study intercalation of small molecules into supercoiled DNA. Methods Mol. Biol. 2010, 613, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, H.; Triebel, H.; Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on Interaction of Anthracycline Antibiotics and Deoxyribonucleic Acid: Geometry of Intercalation of Iremycin and Daunomycin. Biochemistry 1982, 21, 3940–3946. [Google Scholar] [CrossRef] [PubMed]
- Tichadou, J.L.; Genest, D.; Wahl, P.; Aubel-Sadron, G. The use of fluorescence anisotropy decay of poly d(A-T) ethidium bromide complex to estimate the unwinding angle of the double helix. Biophys. Chem. 1975, 3, 142–146. [Google Scholar] [CrossRef]
- Cons, B.M.G.; Fox, K.R. Effects of sequence selective drugs on the gel mobility of a bent DNA fragment. Biochem. Biophys. Res. Commun. 1990, 171, 1064–1070. [Google Scholar] [CrossRef]
- Leoni, L.; Morosetti, S.; Palermo, C.; Sampaolese, B.; Savino, M. Specific interactions between DNA left-handed supercoils and actinomycin D. Biophys. Chem. 1989, 33, 11–17. [Google Scholar] [CrossRef]
- Waring, M.J.; Wakelin, L.P.G. Echinomycin: A bifunctional intercalating antibiotic. Nature 1974, 252, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Aishima, J.; Gitti, R.K.; Noah, J.E.; Gan, H.H.; Schlick, T.; Wolberger, C. A Hoogsteen base pair embedded in undistorted B-DNA. Nucleic Acids Res. 2002, 30, 5244–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; McSally, J.; Andricioaei, I.; Al-Hashimi, H.M. Modulation of Hoogsteen dynamics on DNA recognition. Nat. Commun. 2018, 9, 1473. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef] [PubMed]
- Regazzetti, C.; Bost, F.; Le Marchand-Brustel, Y.; Tanti, J.F.; Giorgetti-Peraldi, S. Insulin induces REDD1 expression through hypoxia-inducible factor 1 activation in adipocytes. J. Biol. Chem. 2010, 285, 5157–5164. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Z.; Wu, Y.; Chen, L.; Luo, Q.; Zhang, J.; Chen, J.; Luo, Z.; Huang, X.; Cheng, Y. Effects of echinomycin on endothelin-2 expression and ovulation in immature rats primed with gonadotropins. Exp. Mol. Med. 2012, 44, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, J.; Zhang, S.; Xu, X.; Zheng, M.; Jiang, G.; Li, F. IGF-1 induces hypoxia-inducible factor 1α-mediated GLUT3 expression through PI3K/Akt/mTOR dependent pathways in PC12 cells. Brain Res. 2012, 1430, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Yonekura, S.; Itoh, M.; Okuhashi, Y.; Takahashi, Y.; Ono, A.; Nara, N.; Tohda, S. Effects of the HIF1 inhibitor, echinomycin, on growth and NOTCH signalling in leukaemia cells. Anticancer Res. 2013, 33, 3099–3104. [Google Scholar] [PubMed]
- Vakili, H.; Jin, Y.; Cattini, P.A. Negative regulation of human growth hormone gene expression by insulin is dependent on hypoxia-inducible factor binding in primary non-tumor pituitary cells. J. Biol. Chem. 2012, 287, 33282–33292. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Okada, H.; Shindoh, H.; Shimoi, K.; Nishigaki, A.; Kanzaki, H. Effects of the hypoxia-inducible factor-1 inhibitor echinomycin on vascular endothelial growth factor production and apoptosis in human ectopic endometriotic stromal cells. Gynecol. Endocrinol. 2016, 32, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Koike, K.; Okuda, K.; Hirayama, T.; Ebihara, M.; Takenaka, M.; Nagasawa, H. Solution-phase synthesis and biological evaluation of triostin A and its analogues. Org. Biomol. Chem. 2016, 14, 2090–2111. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Punchihewa, C.; Mistry, P.; Ooi, A.T.; Yang, D. Novel DNA bis-intercalation by MLN944, a potent clinical bisphenazine anticancer drug. J. Biol. Chem. 2004, 279, 46096–46103. [Google Scholar] [CrossRef] [PubMed]
- Sidell, N.; Mathad, R.I.; Shu, F.J.; Zhang, Z.; Kallen, C.B.; Yang, D. Intercalation of XR5944 with the estrogen response element is modulated by the tri-nucleotide spacer sequence between half-sites. J. Steroid Biochem. Mol. Biol. 2011, 124, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Mathad, R.I.; Zhang, Z.; Sidell, N.; Yang, D. Solution structure of a 2:1 complex of anticancer drug XR5944 with TFF1 estrogen response element: Insights into DNA recognition by a bis-intercalator. Nucleic Acids Res. 2014, 42, 6012–6024. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Bible, R.H.; Kottke, T.J.; Svingen, P.A.; Xu, K.; Pang, Y.; Hajdu, E.; Kaufmann, S.H. Flavopiridol Binds to Duplex DNA Flavopiridol Binds to Duplex DNA 1. Cancer Res. 2000, 60, 2419–2428. [Google Scholar] [PubMed]
- Lee, Y.K.; Isham, C.R.; Kaufman, S.H.; Bible, K.C. Flavopiridol disrupts STAT3/DNA interactions, attenuates STAT3-directed transcription, and combines with the Jak kinase inhibitor AG490 to achieve cytotoxic synergy. Mol. Cancer Ther. 2006, 5, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathi, A.T.; Karp, J.E. New agents in acute myeloid leukemia: Beyond cytarabine and anthracyclines. Curr. Oncol. Rep. 2009, 11, 346–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talib, J.; Beck, J.L.; Urathamakul, T.; Nguyen, C.D.; Aldrich-Wright, J.R.; Mackay, J.P.; Ralph, S.F. A mass spectrometric investigation of the ability of metal complexes to modulate transcription factor activity. Chem. Commun. 2009, 5546–5548. [Google Scholar] [CrossRef] [PubMed]
- Chakree, K.; Ovatlarnporn, C.; Dyson, P.J.; Ratanaphan, A. Altered DNA binding and amplification of human breast cancer suppressor gene BRCA1 induced by a novel antitumor compound, [Ru(η(6)-p-phenylethacrynate)Cl(2)(pta)]. Int. J. Mol. Sci. 2012, 13, 13183–13202. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.J.; Rauscher, F.J.; Beerman, T.A. Targeting DNA-binding drugs to sequence-specific transcription factor-DNA complexes. Differential effects of intercalating and minor groove binding drugs. J. Biol. Chem. 1994, 269, 31051–31058. [Google Scholar] [PubMed]
- Liaw, Y.C.; Gao, Y.G.; Robinson, H.; van der Marel, G.A.; van Boom, J.H.; Wang, A.H.J. Antitumor Drug Nogalamycin Binds DNA in Both Grooves Simultaneously: Molecular Structure of Nogalamycin-DNA Complex. Biochemistry 1989, 28, 9913–9918. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.D.S.; Egli, M.; Gao, Q.; Rich, A. DNA intercalation: Helix unwinding and neighbor exclusion. Struct. Funct. 1992, 1, 107–125. [Google Scholar]
- Odom, D.T.; Parker, C.S.; Barton, J.K. Site-specific inhibition of transcription factor binding to DNA by a metallointercalator. Biochemistry 1999, 38, 5155–5163. [Google Scholar] [CrossRef] [PubMed]
- Hélène, C. The anti-gene strategy: Control of gene expression by triplex-forming-oligonucleotides. Anticancer Drug Des. 1991, 6, 569–584. [Google Scholar] [PubMed]
- Giovannangeli, C.; Helene, C. Triplex-forming molecules for modulation of DNA information processing. Curr. Opin. Mol. Ther. 2000, 2, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008, 36, 5123–5138. [Google Scholar] [CrossRef] [PubMed]
- Vijayalakshmi, R.; Kanthimathi, M.; Subramanian, V.; Nair, B.U. Interaction of DNA with [Cr(Schiff base)(H2O)2]ClO4. Biochim. Biophys. Acta Gen. Subj. 2000, 1475, 157–162. [Google Scholar] [CrossRef]
- Fu, P.K.-L.; Turro, C. Transcription inhibition by Rh(phi)2(phen)3+. Chem. Commun. 2001, 2, 279–280. [Google Scholar] [CrossRef]
- Wang, P.; Leung, C.H.; Ma, D.L.; Sun, R.W.Y.; Yan, S.C.; Chen, Q.S.; Che, C.M. Specific blocking of CREB/DNA binding by cyclometalated platinum(II) complexes. Angew. Chem. Int. Ed. 2011, 50, 2554–2558. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Munde, M.; Wang, S.; Wilson, W.D. Minor groove to major groove, an unusual DNA sequence-dependent change in bend directionality by a distamycin dimer. Biochemistry 2011, 50, 7674–7683. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C.; Reinert, K.E.; Luck, G.; Wähnert, U.; Löber, G.; Thrum, H. Interaction of the oligopeptide antibiotics netropsin and distamycin a with nucleic acids. J. Mol. Biol. 1971, 58, 329–348. [Google Scholar] [CrossRef]
- Melnikova, A.F.; Zasedatelev, A.S.; Kolchinsky, A.M.; Gursky, G.V.; Zhuze, A.L.; Grochovsky, S.L.; Mirzabekov, A.D. Accessibility of the minor groove of DNA in chromatin to the binding of antibiotics netropsin and distamycin A. Mol. Biol. Rep. 1975, 2, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.A.; Baron, R.M.; Macias, A.A.; Layne, M.D.; Perrella, M.A.; Rigby, A.C. Netropsin improves survival from endotoxaemia by disrupting HMGA1 binding to the NOS2 promoter. Biochem. J. 2009, 418, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Baliga, R.; Crothers, D.M. On the kinetics of distamycin binding to its target sites on duplex DNA. Proc. Natl. Acad. Sci. USA 2000, 97, 7814–7818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broggini, M.; Ponti, M.; Ottolenghi, S.; D’incalci, M.; Mongelli, N.; Mantovani, R. Distamycins inhibit the binding of OTF-1 and NFE-1 transfactors to their conserved DNA elements. Nucleic Acids Res. 1989, 17, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciucci, A.; Feriotto, G.; Mischiati, C.; Gambari, R.; Animati, F.; Lombardi, P.; Giorgio Natali, P.; Arcamone, F.; Giacomini, P. Distamycin analogues with improved sequence-specific DNA binding activities. Biochem. Pharmacol. 1994, 48, 1583–1591. [Google Scholar] [CrossRef]
- Feriotto, G.; Ciucci, A.; Mischiati, C.; Animati, F.; Lombardi, P.; Giacomini, P.; Arcamone, F.; Gambari, R. Binding of Epstein-Barr virus nuclear antigen 1 to DNA: Inhibition by distamycin and two novel distamycin analogues. Eur. J. Pharmacol. Mol. Pharmacol. 1994, 267, 143–149. [Google Scholar] [CrossRef]
- Chiang, S.Y.; Azizkhan, J.C.; Beerman, T.A. A comparison DNA-binding drug as inhibitors of E2F1- and Sp1-DNA complexes and associated gene expression. Biochemistry 1998, 37, 3109–3115. [Google Scholar] [CrossRef] [PubMed]
- Massaad-Massade, L.; Navarro, S.; Krummrei, U.; Reeves, R.; Beaune, P.; Barouki, R. HMGA1 enhances the transcriptional activity and binding of the estrogen receptor to its responsive element. Biochemistry 2002, 41, 2760–2768. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.M.; Lopez-Guzman, S.; Riascos, D.F.; Macias, A.A.; Layne, M.D.; Cheng, G.; Harris, C.; Chung, S.W.; Reeves, R.; von Andrian, U.H.; et al. Distamycin A inhibits HMGA1-binding to the P-selectin promoter and attenuates lung and liver inflammation during murine endotoxemia. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munde, M.; Poon, G.M.K.; Wilson, W.D. Probing the electrostatics and pharmacological modulation of sequence-specific binding by the dna-binding domain of the ETS family transcription factor Pu.1: A binding affinity and kinetics investigation. J. Mol. Biol. 2013, 425, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Bazhulina, N.P.; Nikitin, A.M.; Rodin, S.A.; Surovaya, A.N.; Kravatsky, Y.V.; Pismensky, V.F.; Archipova, V.S.; Martin, R.; Gursky, G.V. Binding of Hoechst 33258 and its derivatives to DNA. J. Biomol. Struct. Dyn. 2009, 26, 701–718. [Google Scholar] [CrossRef] [PubMed]
- White, C.M.; Heidenreich, O.; Nordheim, A.; Beerman, T.A. Evaluation of the effectiveness of DNA-binding drugs to inhibit transcription using the c-fos serum response element as a target. Biochemistry 2000, 39, 12262–12273. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Apontes, P.; Song, L.; Liang, P.; Yang, L.; Li, F. Molecular mechanism of upregulation of survivin transcription by the AT-rich DNA-binding ligand, Hoechst33342: Evidence for survivin involvement in drug resistance. Nucleic Acids Res. 2007, 35, 2390–2402. [Google Scholar] [CrossRef] [PubMed]
- Bruice, T.C.; Sengupta, D.; Blaskó, A.; Chiang, S.Y.; Beerman, T.A. A microgonotropen branched decaaza decabutylamine and its DNA and DNA/transcription factor interactions. Bioorgan. Med. Chem. 1997, 5, 685–692. [Google Scholar] [CrossRef]
- White, C.M.; Satz, A.L.; Gawron, L.S.; Bruice, T.C.; Beerman, T.A. Inhibiting transcription factor/DNA complexes using fluorescent microgonotropens (FMGTs). Biochim. Biophys. Acta Gene Struct. Expr. 2002, 1574, 100–108. [Google Scholar] [CrossRef]
- Dervan, P.B.; Doss, R.M.; Marques, M. a Programmable DNA binding oligomers for control of transcription. Curr. Med. Chem. Anticancer Agents 2005, 5, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, Y.; Bando, T.; Sugiyama, H. Sequence-specific DNA binding Pyrrole–imidazole polyamides and their applications. Bioorgan. Med. Chem. 2018, 26, 1393–1411. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Nickols, N.G.; Li, B.C.; Marinov, G.K.; Said, J.W.; Dervan, P.B. Antitumor activity of a pyrrole-imidazole polyamide. Proc. Natl. Acad. Sci. USA 2013, 110, 1863–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehley, J.A.; Melander, C.; Herman, D.; Baird, E.E.; Ferguson, H.A.; Goodrich, J.A.; Dervan, P.B.; Gottesfeld, J.M. Promoter scanning for transcription inhibition with DNA-binding polyamides. Mol. Cell. Biol. 2002, 22, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Supekova, L.; Pezacki, J.P.; Su, A.I.; Loweth, C.J.; Riedl, R.; Geierstanger, B.; Schultz, P.G.; Wemmer, D.E. Genomic effects of polyamide/DNA Interactions on mRNA expression. Chem. Biol. 2002, 9, 821–827. [Google Scholar] [CrossRef]
- Gearhart, M.D.; Dickinson, L.; Ehley, J.; Melander, C.; Dervan, P.B.; Wright, P.E.; Gottesfeld, J.M. Inhibition of DNA binding by human estrogen-related receptor 2 and estrogen receptor alpha with minor groove binding polyamides. Biochemistry 2005, 44, 4196–4203. [Google Scholar] [CrossRef] [PubMed]
- Nickols, N.G.; Dervan, P.B. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc. Natl. Acad. Sci. USA 2007, 104, 10418–10423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenoweth, D.M.; Dervan, P.B. Structural basis for cyclic Py-Im polyamide allosteric inhibition of nuclear receptor binding. J. Am. Chem. Soc. 2010, 132, 14521–14529. [Google Scholar] [CrossRef] [PubMed]
- Fechter, E.J.; Dervan, P.B. Allosteric inhibition of protein-DNA complexes by polyamide-intercalator conjugates. J. Am. Chem. Soc. 2003, 125, 8476–8485. [Google Scholar] [CrossRef] [PubMed]
- Bremer, R.E.; Baird, E.E.; Dervan, P.B. Inhibition of major-groove-binding proteins by pyrrole-imidazole polyamides with an Arg-Pro-Arg positive patch. Chem. Biol. 1998, 5, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Fukuda, N.; Ueno, T.; Tahira, Y.; Ayame, H.; Zhang, W.; Bando, T.; Sugiyama, H.; Saito, S.; Matsumoto, K.; et al. Development of gene silencing pyrrole-imidazole polyamide targeting the TGF-β1 promoter for treatment of progressive renal diseases. J. Am. Soc. Nephrol. 2006, 17. [Google Scholar] [CrossRef] [PubMed]
- Inami, M.; Fukushima, A.; Ueno, T.; Yamada, T.; Tsunemi, A.; Matsumoto, Y.; Fukuda, N.; Soma, M.; Moriyama, M. Reduction of Dimethylnitrosamine-Induced Liver Fibrosis by the Novel Gene Regulator PI Polyamide Targeting Transforming Growth Factor β1 Gene. Biol. Pharm. Bull. 2015, 38, 1836–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Fukuda, N.; Tsunemi, A.; Yao, E.H.; Matsuda, H.; Tahira, K.; Matsumoto, T.; Matsumoto, K.; Matsumoto, Y.; Nagase, H.; et al. A novel gene silencer, pyrrole-imidazole polyamide targeting human lectin-like oxidized low-density lipoprotein receptor-1 gene improves endothelial cell function. J. Hypertens. 2009, 27, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, L.A.; Trauger, J.W.; Baird, E.E.; Dervan, P.B.; Graves, B.J.; Gottesfeld, J.M. Inhibition of Ets-1 DNA binding and ternary complex formation between Ets-1, NF-κB, and DNA by a designed DNA-binding ligand. J. Biol. Chem. 1999, 274, 12765–12773. [Google Scholar] [CrossRef] [PubMed]
- Bashkin, J.K.; Aston, K.; Ramos, J.P.; Koeller, K.J.; Nanjunda, R.; He, G.; Dupureur, C.M.; David Wilson, W. Promoter scanning of the human COX-2 gene with 8-ring polyamides: Unexpected weakening of polyamide-DNA binding and selectivity by replacing an internal N-Me-pyrrole with β-alanine. Biochimie 2013, 95, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sicot, G.; Cui, X.; Vogel, M.; Wuertzer, C.A.; Lezon-Geyda, K.; Wheeler, J.; Harki, D.A.; Muzikar, K.A.; Stolper, D.A.; et al. Targeting a DNA binding motif of the EVI1 protein by a pyrrole-imidazole polyamide. Biochemistry 2011, 50, 10431–10441. [Google Scholar] [CrossRef] [PubMed]
- Syed, J.; Pandian, G.N.; Sato, S.; Taniguchi, J.; Chandran, A.; Hashiya, K.; Bando, T.; Sugiyama, H. Targeted suppression of EVI1 oncogene expression by sequence-specific Pyrrole-imidazole polyamide. Chem. Biol. 2014, 21, 1370–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Wang, S.; Aston, K.; Koeller, K.J.; Kermani, S.F.H.; Castañeda, C.H.; Scuderi, M.J.; Luo, R.; Bashkin, J.K.; Wilson, W.D. β-Alanine and N-terminal cationic substituents affect polyamide-DNA binding. Org. Biomol. Chem. 2017, 15, 9880–9888. [Google Scholar] [CrossRef] [PubMed]
- Gottesfeld, J.M.; Melander, C.; Suto, R.K.; Raviol, H.; Luger, K.; Dervan, P.B. Sequence-specific recognition of DNA in the nucleosome by pyrrole-imidazole polyamides. J. Mol. Biol. 2001, 309, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Neely, L.; Trauger, J.W.; Baird, E.E.; Dervan, P.B.; Gottesfeld, J.M. Importance of minor groove binding zinc fingers within the transcription factor IIIA-DNA complex. J. Mol. Biol. 1997, 274, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Hackley, D.H.; Ramm, E.; Taylor, C.M.; Joung, J.K.; Dervan, P.B.; Pabo, C.O. Allosteric Inhibition of Zinc-Finger Binding in the Major Groove of DNA by Minor-Groove Binding Ligands. Biochemistry 2004, 43, 3880–3890. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.A.; Le, N.M.; Nguyen, B.; Howard, C.M.; Bailey, S.L.; Horick, S.M.; Buchmueller, K.L.; Kotecha, M.; Hochhauser, D.; Hartley, J.A.; et al. Targeting the inverted CCAAT box 2 in the topoisomerase IIalpha promoter by JH-37, an imidazole-pyrrole polyamide hairpin: Design, synthesis, molecular biology, and biophysical studies. Biochemistry 2004, 43, 12249–12257. [Google Scholar] [CrossRef] [PubMed]
- Hochhauser, D.; Kotecha, M.; O’hare, C.; Morris, P.J.; Hartley, J.A.J.M.; Taherbhai, Z.; Harris, D.; Forni, C.; Mantovani, R.; Lee, M. Modulation of topoisomerase IIalpha expression by a DNA sequence-specific polyamide. Mol. Cancer Ther. 2007, 6, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Kiakos, K.; Pett, L.; Satam, V.; Patil, P.; Hochhauser, D.; Lee, M.; Hartley, J.A. Nuclear Localization and Gene Expression Modulation by a Fluorescent Sequence-Selective p-Anisyl-benzimidazolecarboxamido Imidazole-Pyrrole Polyamide. Chem. Biol. 2015, 22, 862–875. [Google Scholar] [CrossRef] [PubMed]
- Brucoli, F.; Hawkins, R.M.; James, C.H.; Jackson, P.J.M.; Wells, G.; Jenkins, T.C.; Ellis, T.; Kotecha, M.; Hochhauser, D.; Hartley, J.A.; et al. An extended pyrrolobenzodiazepine-polyamide conjugate with selectivity for a DNA sequence containing the ICB2 transcription factor binding site. J. Med. Chem. 2013, 56, 6339–6351. [Google Scholar] [CrossRef] [PubMed]
- Pett, L.; Kiakos, K.; Satam, V.; Patil, P.; Laughlin-Toth, S.; Gregory, M.; Bowerman, M.; Olson, K.; Savagian, M.; Lee, M.; et al. Modulation of topoisomerase IIα expression and chemosensitivity through targeted inhibition of NF-Y:DNA binding by a diamino p-anisyl-benzimidazole (Hx) polyamide. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Raskatov, J.A.; Meier, J.L.; Puckett, J.W.; Yang, F.; Ramakrishnan, P.; Dervan, P.B. Modulation of NF-κB-dependent gene transcription using programmable DNA minor groove binders. Proc. Natl. Acad. Sci. USA 2012, 109, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Wang, X.; Fujiwara, K.; Osaka, S.; Yoshida, Y.; Osaka, E.; Taniguchi, M.; Ueno, T.; Fukuda, N.; Soma, M.; et al. Inhibition of Human Osteosarcoma Cell Migration and Invasion by a Gene Silencer, Pyrrole-Imidazole Polyamide, Targeted at the Human MMP9 NF-κB Binding Site. Biol. Pharm. Bull. 2014, 37, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Olenyuk, B.Z.; Zhang, G.-J.; Klco, J.M.; Nickols, N.G.; Kaelin, W.G.; Dervan, P.B. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc. Natl. Acad. Sci. USA 2004, 101, 16768–16773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageyama, Y.; Sugiyama, H.; Ayame, H.; Iwai, A.; Fujii, Y.; Huang, L.E.; Kizaka-Kondoh, S.; Hiraoka, M.; Kihara, K. Suppression of VEGF transcription in renal cell carcinoma cells by pyrrole-imidazole hairpin polyamides targeting the hypoxia responsive element. Acta Oncol. 2006, 45, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szablowski, J.O.; Raskatov, J.A.; Dervan, P.B. An HRE-Binding Py-Im Polyamide Impairs Hypoxic Signaling in Tumors. Mol. Cancer Ther. 2016, 15, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Mysore, V.S.; Szablowski, J.; Dervan, P.B.; Frost, P.J. A DNA-binding Molecule Targeting the Adaptive Hypoxic Response in Multiple Myeloma Has Potent Antitumor Activity. Mol. Cancer Res. 2016, 14, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Fujiwara, K.; Nakai, Y.; Ozaki, T.; Koshikawa, N.; Toshio, K.; Kataba, M.; Oguni, A.; Matsuda, H.; Yoshida, Y.; et al. Inhibition of malignant phenotypes of human osteosarcoma cells by a gene silencer, a pyrrole-imidazole polyamide, which targets an E-box motif. FEBS Open Bio 2014, 4, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Watanabe, T.; Kimura, M.T.; Koshikawa, N.; Ikeda, M.; Uekusa, S.; Kawashima, H.; Wang, X.; Igarashi, J.; Choudhury, D.; et al. Identification of a novel E-box binding pyrrole-imidazole polyamide inhibiting MYC-driven cell proliferation. Cancer Sci. 2015, 106, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obinata, D.; Takayama, K.; Fujiwara, K.; Suzuki, T.; Tsutsumi, S.; Fukuda, N.; Nagase, H.; Fujimura, T.; Urano, T.; Homma, Y.; et al. Targeting Oct1 genomic function inhibits androgen receptor signaling and castration-resistant prostate cancer growth. Oncogene 2016, 35, 6350–6358. [Google Scholar] [CrossRef] [PubMed]
- Hayatigolkhatmi, K.; Padroni, G.; Su, W.; Fang, L.; Gómez-Castañeda, E.; Hsieh, Y.C.; Jackson, L.; Holyoake, T.L.; Pellicano, F.; Burley, G.A.; et al. Investigation of a minor groove-binding polyamide targeted to E2F1 transcription factor in chronic myeloid leukaemia (CML) cells. Blood Cells Mol. Dis. 2018, 69, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.D.; Nguyen, B.; Tanious, F.A.; Mathis, A.; Hall, J.E.; Stephens, C.E.; Boykin, D.W. Dications that target the DNA minor groove: Compound design and preparation, DNA interactions, cellular distribution and biological activity. Curr. Med. Chem. Anticancer Agents 2005, 5, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Yeates, C. DB-289 Immtech International. IDrugs 2003, 6, 1086–1093. [Google Scholar] [PubMed]
- Bailly, C.; Tardy, C.; Wang, L.; Armitage, B.; Hopkins, K.; Kumar, A.; Schuster, G.B.; Boykin, D.W.; Wilson, W.D. Recognition of ATGA sequences by the unfused aromatic dication DB293 forming stacked dimers in the DNA minor groove. Biochemistry 2001, 40, 9770–9779. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, P.; Liu, Y.; Depauw, S.; Hildebrand, M.P.; Boykin, D.W.; Bailly, C.; Wilson, W.D.; David-Cordonnier, M.-H. Direct inhibition of the DNA-binding activity of POU transcription factors Pit-1 and Brn-3 by selective binding of a phenyl-furan-benzimidazole dication. Nucleic Acids Res. 2008, 36, 3341–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.B.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Ichikawa, H.; Tojo, A.; Kaneko, Y.; Maseki, N.; Hayashi, Y.; Ohira, M.; Asano, S.; Ohki, M. An ets-related gene, ERG, is rearranged in human myeloid leukemia with t(16;21) chromosomal translocation. Proc. Natl. Acad. Sci. USA 1993, 90, 10280–10284. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.D.P.; Offor, O.; Ferry, J.A.; Amrein, P.C.; Morton, C.C.; Dal Cin, P. ELF4 is fused to ERG in a case of acute myeloid leukemia with a t(X;21)(q25-26;q22). Leuk. Res. 2006, 30, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Baldus, C.D.; Ruppert, A.S.; Radmacher, M.D.; Mrózek, K.; Whitman, S.P.; Kolitz, J.E.; Edwards, C.G.; Vardiman, J.W.; Powell, B.L.; et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2005, 23, 9234–9242. [Google Scholar] [CrossRef] [PubMed]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013, 41, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Munde, M.; Kumar, A.; Peixoto, P.; Depauw, S.; Ismail, M.A.; Farahat, A.A.; Paul, A.; Say, M.V.; David-Cordonnier, M.-H.; Boykin, D.W.; et al. The unusual monomer recognition of guanine-containing mixed sequence DNA by a dithiophene heterocyclic diamidine. Biochemistry 2014, 53, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Flajollet, S.; Tian, T.V.; Flourens, A.; Tomavo, N.; Villers, A.; Bonnelye, E.; Aubert, S.; Leroy, X.; Duterque-Coquillaud, M. Abnormal Expression of the ERG Transcription Factor in Prostate Cancer Cells Activates Osteopontin. Mol. Cancer Res. 2011, 9, 914–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munde, M.; Wang, S.; Kumar, A.; Stephens, C.E.; Farahat, A.A.; Boykin, D.W.; Wilson, W.D.; Poon, G.M.K. Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines. Nucleic Acids Res. 2014, 42, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debré, I.; Paul, A.; Leite, J.; Mitchell, K.; Kim, H.M.; Carvajal, L.A.; Todorova, T.I.; Huang, K.; Kumar, A.; Farahat, A.A.; et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J. Clin. Investig. 2017, 127, 4297–4313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, L.A.; Li, S.K.H.; Piskorz, J.; Xu, L.S.; DeKoter, R.P. Genome-wide comparison of PU.1 and Spi-B binding sites in a mouse B lymphoma cell line. BMC Genom. 2015, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Rekhtman, N.; Cheng, G.; Krasikov, T.; Skoultchi, A.I. Deregulated expression of the PU.1 transcription factor blocks murine erythroleukemia cell terminal differentiation. Oncogene 1997, 14, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbauer, F.; Wagner, K.; Kutok, J.L.; Iwasaki, H.; Le Beau, M.M.; Okuno, Y.; Akashi, K.; Fiering, S.; Tenen, D.G. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 2004, 36, 624–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonadies, N.; Pabst, T.; Mueller, B.U. Heterozygous deletion of the PU.1 locus in human AML. Blood 2010, 115, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Will, B.; Vogler, T.O.; Narayanagari, S.; Bartholdy, B.; Todorova, T.I.; Da Silva Ferreira, M.; Chen, J.; Yu, Y.; Mayer, J.; Barreyro, L.; et al. Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat. Med. 2015, 21, 1172–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budiman, M.E.; Bierbach, U.; Alexander, R.W. DNA minor groove adducts formed by a platinum-acridine conjugate inhibit association of TATA-binding protein with its cognate sequence. Biochemistry 2005, 44, 11262–11268. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.Y.; Tang, Y.; Knosp, W.M.; Stadler, H.S.; Shaw, J.T. Synthesis of diverse lactam carboxamides leading to the discovery of a new transcription-factor inhibitor. Angew. Chem. Int. Ed. 2007, 46, 5352–5355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, J.; Jin, L.; Song, T.; Wu, G.; Gao, J. Tanshinone IIA interacts with DNA by minor groove-binding. Biol. Pharm. Bull. 2008, 31, 2342–2345. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Song, J.S.; Lee, D.K.; Yang, C.H. Suppression of AP-1 activity by tanshinone and cancer cell growth inhibition. Bull. Korean Chem. Soc. 1999, 20, 925–928. [Google Scholar]
- Tsai, M.H.; Lin, Z.C.; Liang, C.J.; Yen, F.L.; Chiang, Y.C.; Lee, C.W. Eupafolin inhibits PGE2 production and COX2 expression in LPS-stimulated human dermal fibroblasts by blocking JNK/AP-1 and Nox2/p47phoxpathway. Toxicol. Appl. Pharmacol. 2014, 279, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, J.; Wang, Y.; Song, T.; Zhang, J.; Wu, G.; Zhang, T.; Du, G. Tanshinone IIA triggers p53 responses and apoptosis by RNA polymerase II upon DNA minor groove binding. Biochem. Pharmacol. 2009, 78, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.; Núñez, L.-E.; Magistri, M.; Brambilla, L.; Jovic, S.; Carbone, G.M.; Morís, F.; Catapano, C.V. Modulation of the Activity of Sp Transcription Factors by Mithramycin Analogues as a New Strategy for Treatment of Metastatic Prostate Cancer. PLoS ONE 2012, 7, e35130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Zhang, H.; Wang, H.B.; Li, Y.X.; Liu, G.H.; Xing, S.; Li, M.Z.; Zeng, M.S. Down-regulation of Sp1 suppresses cell proliferation, clonogenicity and the expressions of stem cell markers in nasopharyngeal carcinoma. J. Transl. Med. 2014, 12, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, M.; Patel, D.J. Solution Structure of the Mithramycin Dimer–DNA Complex. Biochemistry 1993, 32, 6588–6604. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Payet, D.; Travers, A.A.; Waring, M.J. PCR-based development of DNA substrates containing modified bases: An efficient system for investigating the role of the exocyclic groups in chemical and structural recognition by minor groove binding drugs and proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 13623–13628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, R.; Snyder, R.C.; Thomas, S.; Koller, C.A.; Miller, D.M. Mithramycin blocks protein binding and function of the SV40 early promoter. J. Clin. Investig. 1989, 83, 2003–2007. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.C.; Ray, R.; Blume, S.; Miller, D.M. Mithramycin Blocks Transcriptional Initiation of the c-myc P1 and P2 Promoters. Biochemistry 1991, 30, 4290–4297. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, V.; Costa, R.H.; Lau, L.F.; Raychaudhuri, P.; Tyner, A.L. Negative regulation of the oncogenic transcription factor FoxM1 by thiazolidinediones and mithramycin. Cancer Biol. Ther. 2010, 9, 1008–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihara, N.; Chiba, T.; Yamaguchi, K.; Sudo, H.; Yagishita, H.; Imai, K. Minimal essential region for krüppel-like factor 5 expression and the regulation by specificity protein 3-GC box binding. Gene 2017, 601, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhi, X.; Zhou, Z.; Zhang, H.; Yang, R.; Zou, T.; Chen, C. Mithramycin A suppresses basal triple-negative breast cancer cell survival partially via down-regulating Krüppel-like factor 5 transcription by Sp1. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, H.; Meng, L.; Yin, L. Mithramycin inhibits epithelial-tomesenchymal transition and invasion by downregulating SP1 and SNAI1 in salivary adenoid cystic carcinoma. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Koutsodontis, G.; Kardassis, D. Inhibition of p53-mediated transcriptional responses by mithramycin A. Oncogene 2004, 23, 9190–9200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estève, P.O.; Hang, G.C.; Pradhan, S. Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J. Biol. Chem. 2007, 282, 2615–2625. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Qian, G.W.; Li, W.; Chen, F.F.; Di, J.H.; Zhang, B.F.; Pei, D.S.; Ma, P.; Zheng, J.N. A critical role of Sp1 transcription factor in regulating the human Ki-67 gene expression. Tumor Biol. 2011, 32, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, C.; Núñez, L.-E.; Morís, F.; Portugal, J. Genome-wide modulation of gene transcription in ovarian carcinoma cells by a new mithramycin analogue. PLoS ONE 2014, 9, e104687. [Google Scholar] [CrossRef] [PubMed]
- Logotheti, S.; Michalopoulos, I.; Sideridou, M.; Daskalos, A.; Kossida, S.; Spandidos, D.A.; Field, J.K.; Vojtesek, B.; Liloglou, T.; Gorgoulis, V.; et al. Sp1 binds to the external promoter of the p73 gene and induces the expression of TAp73γ in lung cancer. FEBS J. 2010, 277, 3014–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Xu, P.; Cai, W.-J.; Wang, H.-G.; Li, B.-B.; Huang, G.-L.; He, Z.-W.; Chen, G.; Ye, C.-G. ZBP-89 and Sp1 contribute to Bak expression in hepatocellular carcinoma cells. BMC Cancer 2018, 18, 419. [Google Scholar] [CrossRef] [PubMed]
- Krikun, G.; Schatz, F.; Mackman, N.; Guller, S.; Demopoulos, R.; Lockwood, C.J. Regulation of tissue factor gene expression in human endometrium by transcription factors Sp1 and Sp3. Mol. Endocrinol. 2000, 14, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Aslam, F.; Palumbo, L.; Augenlicht, L.H.; Velcich, A. The Sp family of transcription factors in the regulation of the human and mouse MUC2 gene promoters. Cancer Res. 2001, 61, 570–576. [Google Scholar] [PubMed]
- Lee, J.J.; Park, K.; Shin, M.H.; Yang, W.J.; Song, M.J.; Park, J.H.; Yong, T.S.; Kim, E.K.; Kim, H.P. Accessible chromatin structure permits factors Sp1 and Sp3 to regulate human TGFBI gene expression. Biochem. Biophys. Res. Commun. 2011, 409, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Zeng, X.; Coimbra, R.; Eliceiri, B.P.; Baird, A. Counter regulation of ECRG4 gene expression by hypermethylation-dependent inhibition and the Sp1 transcription factor-dependent stimulation of the c2orf40 promoter. Gene 2017, 636, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Ferrari, A.C. Mithramycin targets sp1 and the androgen receptor transcription level-potential therapeutic role in advanced prostate cancer. Transl. Oncogenom. 2006, 1, 19–31. [Google Scholar]
- Yang, W.J.; Song, M.J.; Park, E.Y.; Lee, J.J.; Park, J.H.; Park, K.; Park, J.H.; Kim, H.P. Transcription factors Sp1 and Sp3 regulate expression of human ABCG2 gene and chemoresistance phenotype. Mol. Cells 2013, 36, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Yerges-Armstrong, L.M.; Chu, Y.; Zmuda, J.M.; Zhang, Y. Transcriptional regulation of frizzled-1 in human osteoblasts by Sp1. PLoS ONE 2016, 11, e163277. [Google Scholar] [CrossRef] [PubMed]
- Blume, S.W.; Snyder, R.C.; Ray, R.; Thomas, S.; Koller, C.A.; Miller, D.M. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J. Clin. Investig. 1991, 88, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Kaluz, S.; Kaluzová, M.; Stanbridge, E.J. Expression of the hypoxia marker carbonic anhydrase IX is critically dependent on SP1 activity. Identification of a novel type of hypoxia-responsive enhancer. Cancer Res. 2003, 63, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.R.; Xi, S.; Shukla, V.; Hong, J.A.; Chen, H.; Xiong, Y.; Ripley, R.T.; Hoang, C.D.; Schrump, D.S. The Pluripotency Factor Musashi-2 Is a Novel Target for Lung Cancer Therapy. Ann. Am. Thorac. Soc. 2018, 15, S124. [Google Scholar] [CrossRef] [PubMed]
- Jun, D.Y.; Lee, J.Y.; Park, H.S.; Lee, Y.H.; Kim, Y.H. Tumor suppressor protein p53-mediated repression of human mitotic centromere-associated kinesin gene expression is exerted via down-regulation of Sp1 level. PLoS ONE 2017, 12, e189698. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Weidenbach, S.; Cano, K.E.; Wang, Z.; Mitra, P.; Ivanov, D.N.; Rohr, J.; Tsodikov, O.V. Structures of mithramycin analogues bound to DNA and implications for targeting transcription factor FLI1. Nucleic Acids Res. 2016, 44, 8990–9004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Glod, J.; Peer, C.J.; Sissung, T.M.; Arnaldez, F.I.; Long, L.; Figg, W.D.; Whitcomb, P.; Helman, L.J.; Widemann, B.C. A phase I/II trial and pharmacokinetic study of mithramycin in children and adults with refractory Ewing sarcoma and EWS–FLI1 fusion transcript. Cancer Chemother. Pharmacol. 2017, 80, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Morales, C.P.; Souza, R.F.; Spechler, S.J. Hallmarks of cancer progression in Barrett’s oesophagus. Lancet 2002, 360, 1587–1589. [Google Scholar] [CrossRef]
- De Palma, M.; Hanahan, D. The biology of personalized cancer medicine: Facing individual complexities underlying hallmark capabilities. Mol. Oncol. 2012, 6, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.M.; Bernstein, C.R.; West, C.M.L.; Homer, J.J. Molecular and cellular processes underlying the hallmarks of head and neck cancer. Eur. Arch. Otorhinolaryngol. 2013, 270, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Kohrman, N.; Gore, S.D.; Kim, T.K.; Zeidan, A.M.; Prebet, T. Epigenetics in Cancer: A Hematological Perspective. PLoS Genet. 2016, 12, e1006193. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Aftabuddin, M.; Gupta, D.K.; Raha, S.; Sen, P. Human Prostate Cancer Hallmarks Map. Sci. Rep. 2016, 6, 30691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A.; Karin, M.; Hunter, T.; Karin, M.; Ben-neriah, Y.; Hsu, H.; Al, E.; et al. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Huang, C.C.; Liu, H.; Delisi, C.; Nevalainen, M.T.; Szalma, S.; Bhanot, G. Robust gene network analysis reveals alteration of the STAT5a network as a hallmark of prostate cancer. Genome Inf. 2010, 24, 139–153. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Kasai, K. GLI1, a master regulator of the hallmark of pancreatic cancer. Pathol. Int. 2016, 66, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ouf, H.; Zhao, L.; Bismar, T.A. ERG expression in prostate cancer: Biological relevance and clinical implication. J. Cancer Res. Clin. Oncol. 2016, 142, 1781–1793. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Nimmo, R. RUNX transcription factors at the interface of stem cells and cancer. Biochem. J. 2017, 474, 1755–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.K.; Chauhan, A.S.; Zhuang, L.; Gan, B. FoxO transcription factors in cancer metabolism. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Monterisi, S.; Riso, P.L.; Russo, K.; Bertalot, G.; Vecchi, M.; Testa, G.; Di Fiore, P.P.; Bianchi, F. HOXB7 overexpression in lung cancer is a hallmark of acquired stem-like phenotype. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Jin, G.; Zhou, X. Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. 2015, 43, 3873–3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
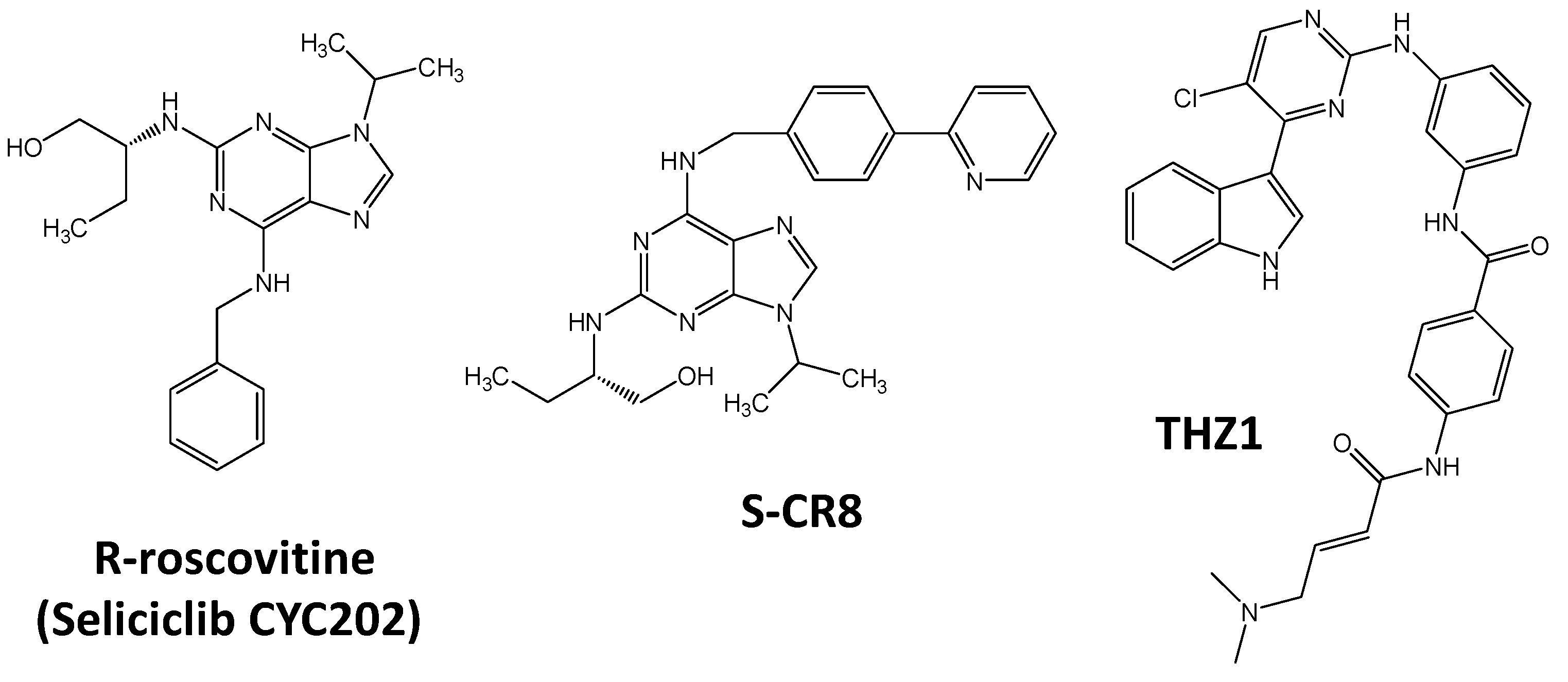{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
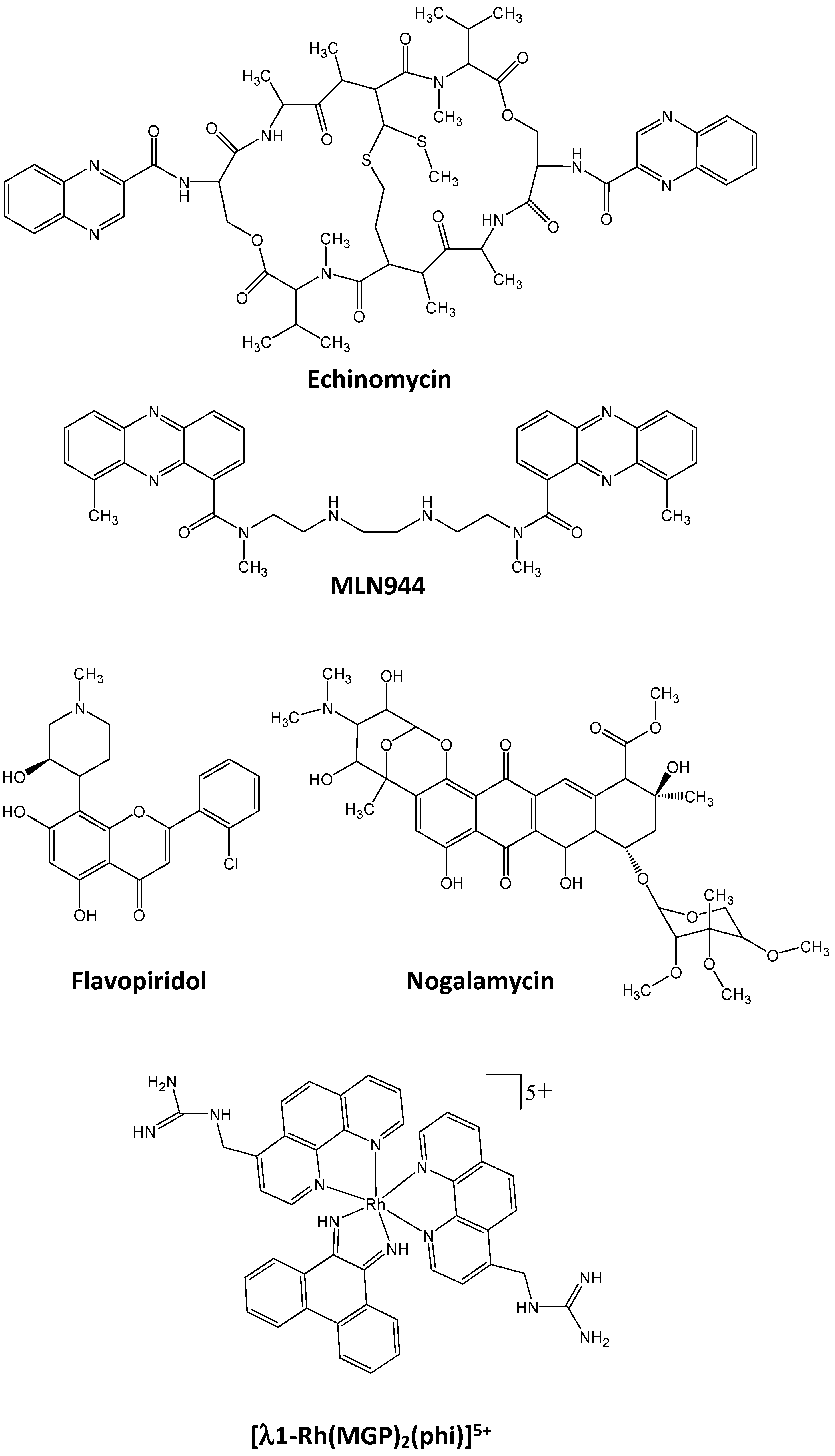{kind=link}
{kind=link}
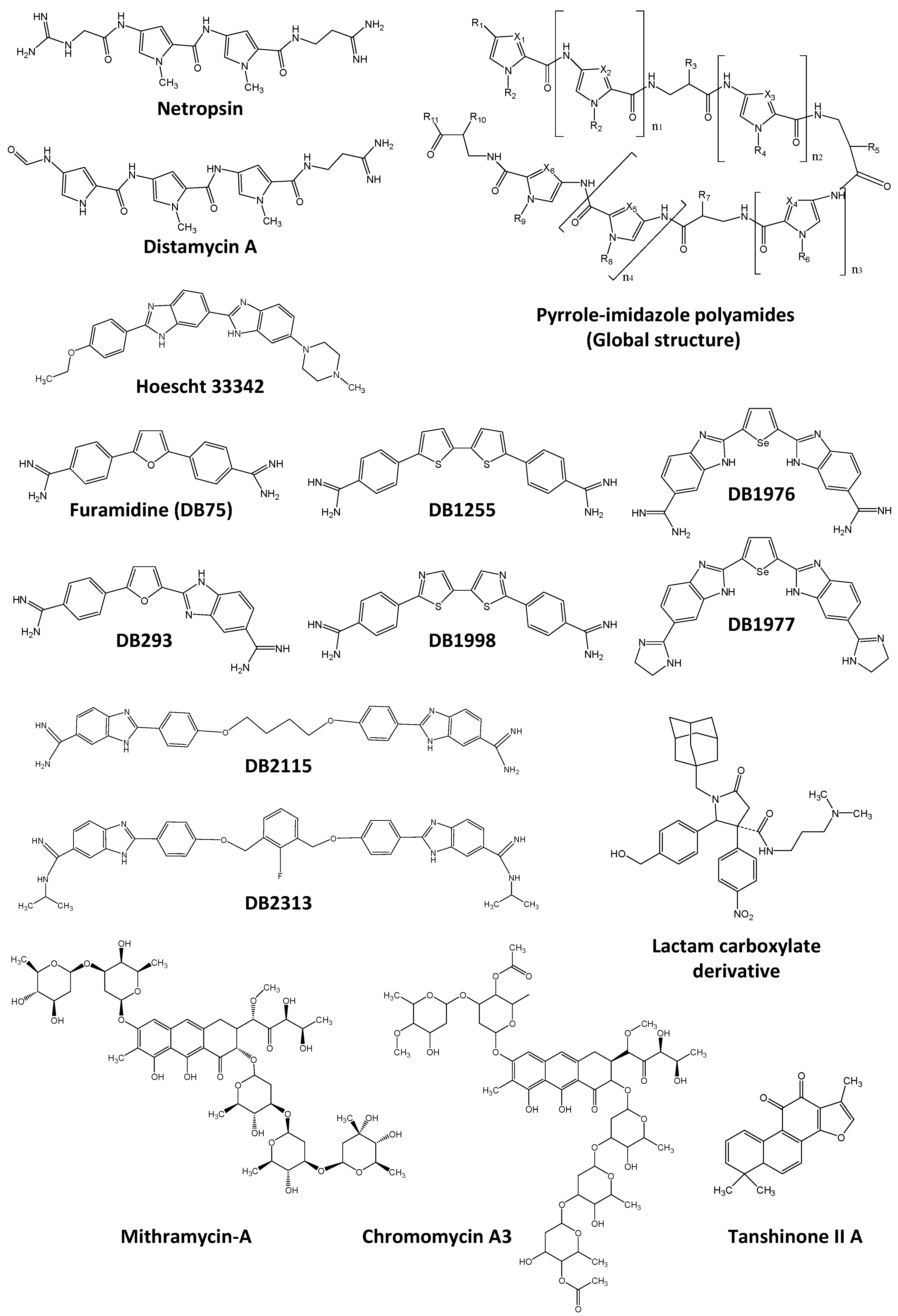{kind=link}
| ABL1 | CEBPA | ERCC3 | HIST1H2BE | MDM4 | PAX7 | SMARCA4 | TFPT |
| AFF1 | CHD1 | ERCC6 | HIST1H2BG | MED12 | PAX8 | SMARCB1 | THRAP3 |
| AFF3 | CHD2 | ERF | HLF | MEF2B | PBX1 | SMARCD1 | TLX1 |
| AFF4 | CHD4 | ERG | HMGA1 | MEF2C | PEG3 | SMARCE1 | TLX3 |
| APC | CHD5 | ESPL1 | HMGA2 | MEN1 | PER1 | SMURF2 | TNFAIP3 |
| AR | CHD7 | ESR1 | HOXA11 | MITF | PHF3 | SOX2 | TP53 |
| ARID1A | CIC | ETS1 | HOXA13 | MKL1 | PHF6 | SOX5 | TRIM24 |
| ARID1B | CIITA | ETV1 | HOXA7 | MLLT1 | PHOX2B | SOX9 | TRIM33 |
| ARID3B | CNOT3 | ETV4 | HOXA9 | MLLT10 | PLAG1 | SRCAP | TRIP11 |
| ARID5B | CREB1 | ETV5 | HOXC11 | MLLT3 | PML | SS18L1 | TRPS1 |
| ARNT | CREB3L1 | ETV6 | HOXC13 | MLLT6 | PMS1 | SSB | TRRAP |
| ARNT2 | CREBBP | EWSR1 | HOXD11 | MYB | PNN | SSX1 | TSC22D1 |
| ASB15 | CRTC1 | EYA4 | HOXD13 | MYBL1 | POU2AF1 | SSX2 | TSHZ3 |
| ASXL1 | CSDE1 | EZH2 | ID3 | MYC | POU2F2 | SSX4 | VHL |
| ATF1 | CTCF | FEV | IRF2 | MYCN | POU5F1 | STAT3 | WHSC1 |
| ATF7IP | CTNNB1 | FLI1 | IRF4 | MYOD1 | PPARG | STAT4 | WHSC1L1 |
| ATM | DACH1 | FOXA1 | IRF6 | NCOA1 | PRDM1 | STAT5B | WT1 |
| ATRX | DACH2 | FOXE1 | IRF8 | NCOA2 | PRDM16 | STAT6 | WWP1 |
| BAZ2B | DAXX | FOXL2 | IRX6 | NCOA4 | PRDM9 | SUFU | WWTR1 |
| BCL11A | DDB2 | FOXP1 | JUN | NCOR1 | PRRX1 | SUZ12 | XBP1 |
| BCL11B | DDIT3 | FOXQ1 | KHDRBS2 | NCOR2 | PSIP1 | TAF1 | XPC |
| BCL3 | DDX5 | FUBP1 | KHSRP | NEUROG2 | RARA | TAF15 | ZBTB16 |
| BCL6 | DEK | FUS | KLF2 | NFE2L2 | RB1 | TAL1 | ZBTB20 |
| BCLAF1 | DIP2C | FXR1 | KLF4 | NFE2L3 | RBM15 | TAL2 | ZFP36L1 |
| BCOR | DNMT1 | GATA1 | KLF5 | NFIB | RBMX | TBX18 | ZFX |
| BRCA1 | DNMT3A | GATA2 | KLF6 | NFKB2 | REL | TBX22 | ZHX2 |
| BRCA2 | DOT1L | GATA3 | LDB1 | NFKBIA | RUNX1 | TBX3 | ZIC3 |
| BRD7 | EED | GLI3 | LMO1 | NONO | RUNX1T1 | TCEA1 | ZIM2 |
| BRD8 | EGR2 | GTF2I | LMO2 | NOTCH2 | RXRA | TCEB1 | ZNF208 |
| BRIP1 | ELAVL2 | HDAC9 | LMX1A | NOTCH3 | SALL3 | TCERG1 | ZNF226 |
| BRPF3 | ELF3 | HEY1 | LYL1 | NPM1 | SATB2 | TCF12 | ZNF331 |
| BTG1 | ELF4 | HIST1H1B | LZTR1 | NR3C2 | SETBP1 | TCF3 | ZNF384 |
| BTG2 | ELK4 | HIST1H1C | MAF | NR4A3 | SFPQ | TCF7L2 | ZNF469 |
| CBFA2T3 | ELL | HIST1H1D | MAFA | NSD1 | SIN3A | TFAP2D | ZNF595 |
| CBFB | EP300 | HIST1H1E | MAFB | OLIG2 | SMAD2 | TFDP1 | ZNF638 |
| CDX2 | EPC1 | HIST1H2BC | MAML1 | PAX3 | SMAD4 | TFE3 | |
| CDX4 | ERCC2 | HIST1H2BD | MAX | PAX5 | SMARCA1 | TFEB |
| Transcription Factor Family | Transcription Factor Target | Tumor Model | References |
|---|---|---|---|
| AT-rich binder | TBP | - | [323] |
| LEF-1 | Colon | [324] | |
| Hormone/Steroid receptor | estrogen receptor | Breast | [325] |
| androgen receptor | Prostate | [326,327] | |
| glucocorticoid receptor | - | [327,328] | |
| B-Zip | GCN-4 | - | [328,329] |
| AP-1 | - | [330,331,332] | |
| ETS-domain | ETS-1 | - | [333,334] |
| EVI1 | Leukemia | [335] | |
| ELK-1 | Breast | [336] | |
| PU.1/SPI1 | - | [337] | |
| Zn-Finger | TFIIIA | - | [338,339] |
| Zif268 | - | [340] | |
| Others | NF-Y | Lung | [341,342,343,344,345] |
| NFκB | Lung, Osteosarcoma | [346,347] | |
| HIF1 | Kidney, Glioblastoma, Multiple myeloma | [348,349,350,351] | |
| c-MYC | Osteosarcoma, Burkitt’s lymphoma | [352,353] | |
| OCT1 | Prostate | [354] | |
| RUNX | Acute myeloid leukemia | [355] | |
| E2F1 | Chronic myeloid leukemia | [355] |
| Type of Protein | Gene Promoter | Tumor Model | References |
|---|---|---|---|
| Transcription factors | c-MYC | Cervix carcinoma | [386] |
| FoxM1 | Liver | [387] | |
| KLF5 | Breast | [388,389] | |
| SNAI1 | Salivary adenoid cystic carcinoma | [390] | |
| SP1 | |||
| Regulators of cell proliferation | CDKN1A | Liver | [391] |
| Survivin | Colon | [392] | |
| Ki-67 | - | [393] | |
| CRABP1 | Ovary | [394] | |
| Tumor suppressors | KCNMA1 | Ovary | [394] |
| p73 | Lung | [395] | |
| Apoptotic genes | XIAP | Ovary | [394] |
| BAK1 | Liver | [396] | |
| Secreted factors | Collagen-alpha 1 | - | [397] |
| Tissue factor | - | [397] | |
| MUC2 | Colon | [398] | |
| TGFBI | Lung, Breast | [399] | |
| ECRG4 | Leukemia | [400] | |
| Membrane associated proteins (receptors, transporters) | Androgen receptor | Prostate | [401] |
| ABCG2 | Lung | [402] | |
| FZD1 | - | [403] | |
| Metabolic enzymes | Dihydrofolate reductase | Breast | [404] |
| Carbonic anhydrase IX | - | [405] | |
| Cell differentiation | MSI2 | Lung | [406] |
| Cell movement | KIF2C kinesin | Colon | [407] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.-H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. https://doi.org/10.3390/molecules23061479
Lambert M, Jambon S, Depauw S, David-Cordonnier M-H. Targeting Transcription Factors for Cancer Treatment. Molecules. 2018; 23(6):1479. https://doi.org/10.3390/molecules23061479
Chicago/Turabian StyleLambert, Mélanie, Samy Jambon, Sabine Depauw, and Marie-Hélène David-Cordonnier. 2018. "Targeting Transcription Factors for Cancer Treatment" Molecules 23, no. 6: 1479. https://doi.org/10.3390/molecules23061479
APA StyleLambert, M., Jambon, S., Depauw, S., & David-Cordonnier, M. -H. (2018). Targeting Transcription Factors for Cancer Treatment. Molecules, 23(6), 1479. https://doi.org/10.3390/molecules23061479