
Immunomodulatory Action of Substituted 1,3,4-Thiadiazines on the Course of Myocardial Infarction

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Possibility of Influencing the Functions of the Immune System via the Central Mechanisms of the Stress Response
2.2. The Compounds from the Group of Substituted 1,3,4-Thiadiazines
2.3. The Action of the Compounds from the Group of Substituted 1,3,4-Thiadiazines on Myocardial Ischemia and Myocardial Infarction
3. Experiment
3.1. Pharmacological Evaluation
3.2. In Silico Studies
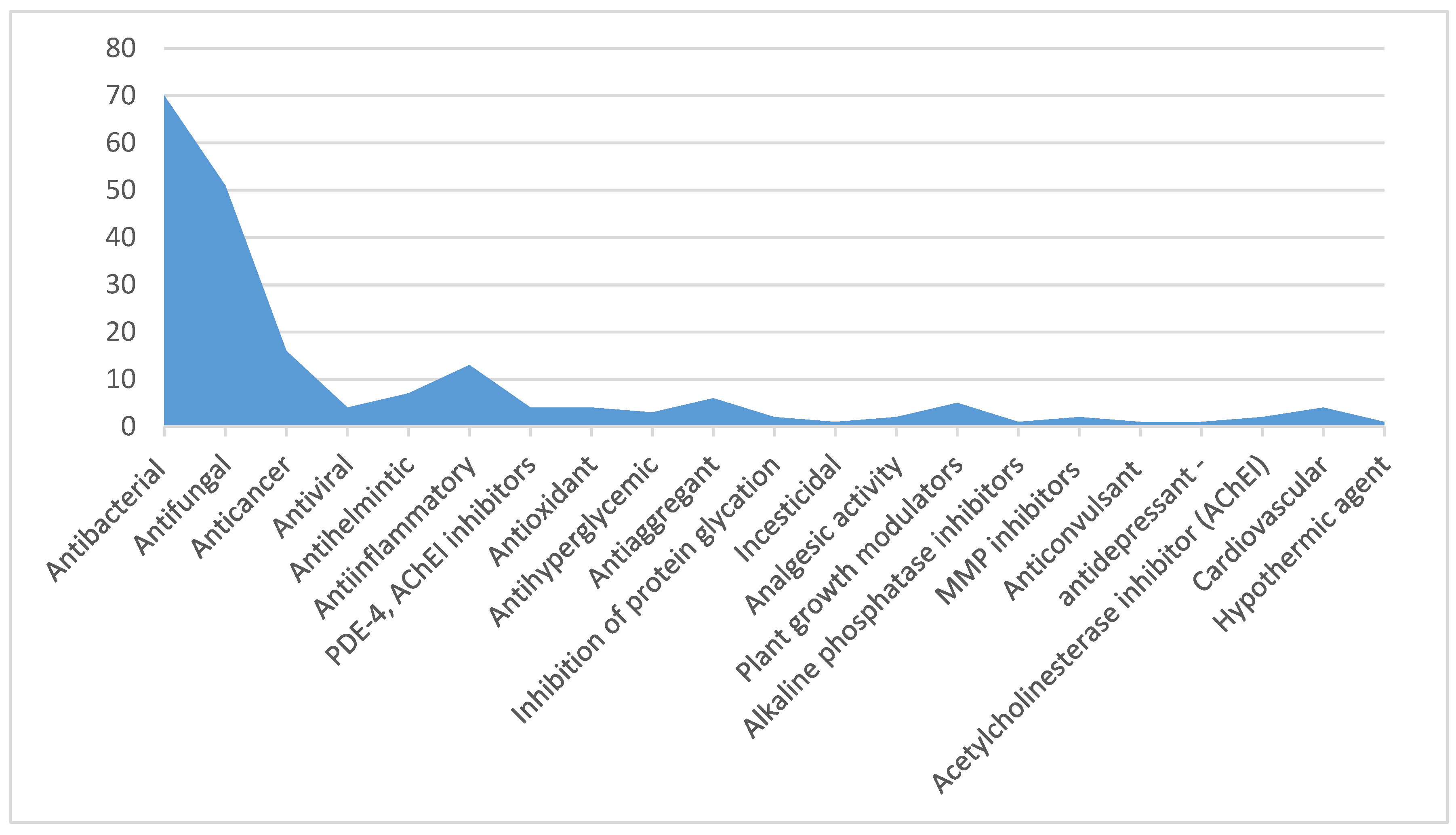
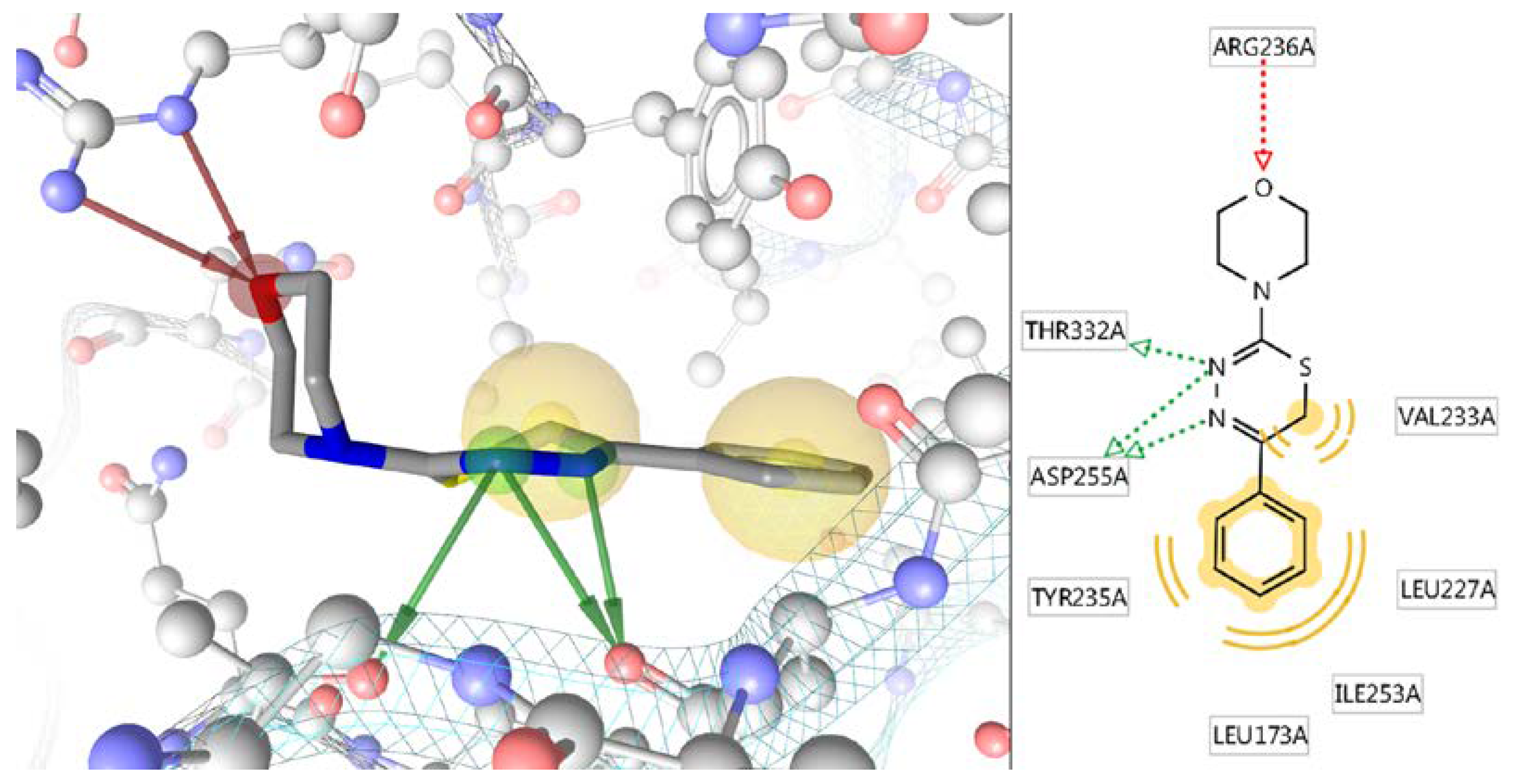
In Silico Study of the Targeted Mechanism of Anti-Inflammatory Action of Compound L-17
3.3. In Vivo Studies
3.3.1. Action of Substituted 1,3,4-Thiadiazines on the Course of Stress Reaction
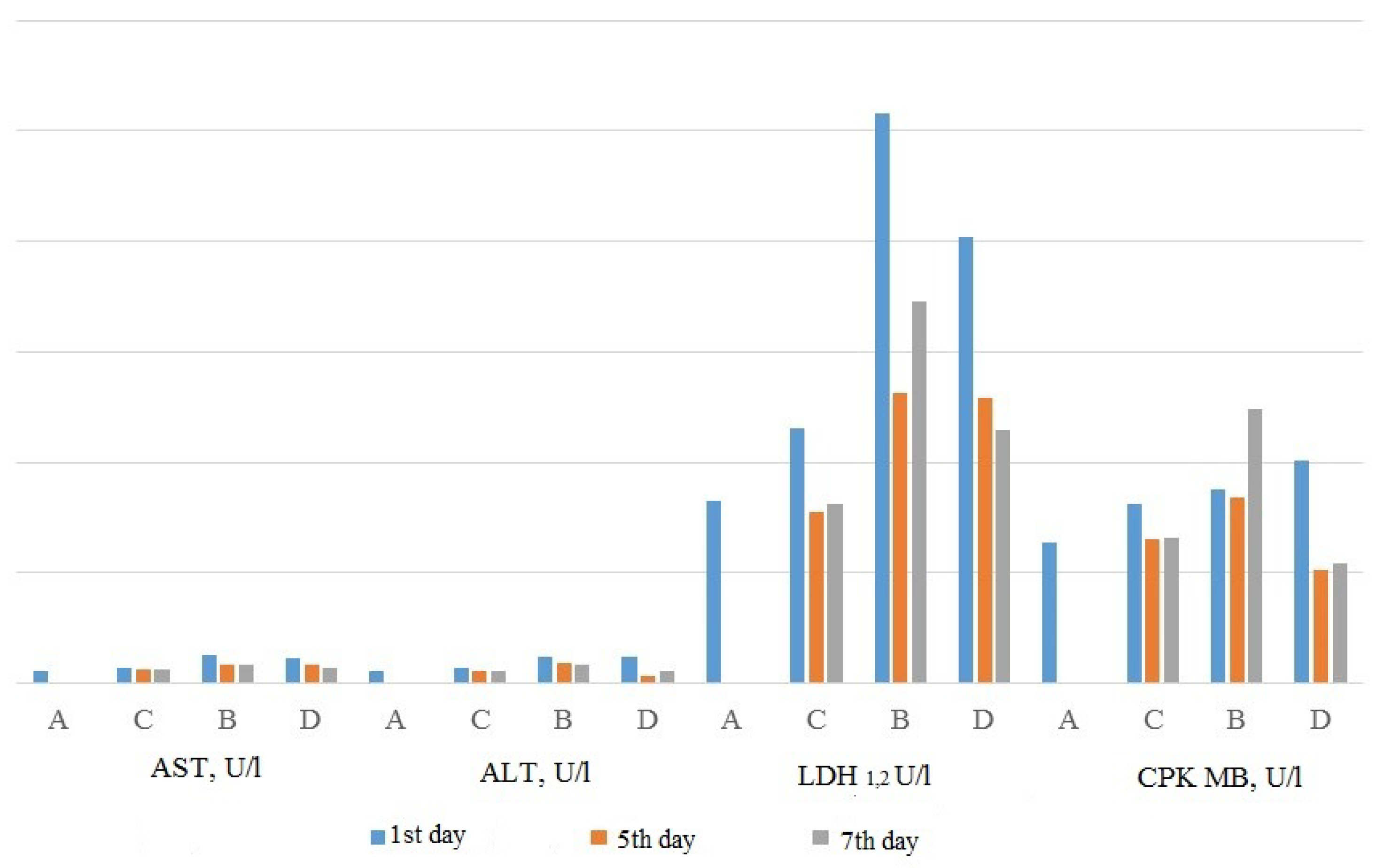
3.3.2. Action of Substituted 1,3,4-Thiadiazines on the Course of Myocardial Infarction
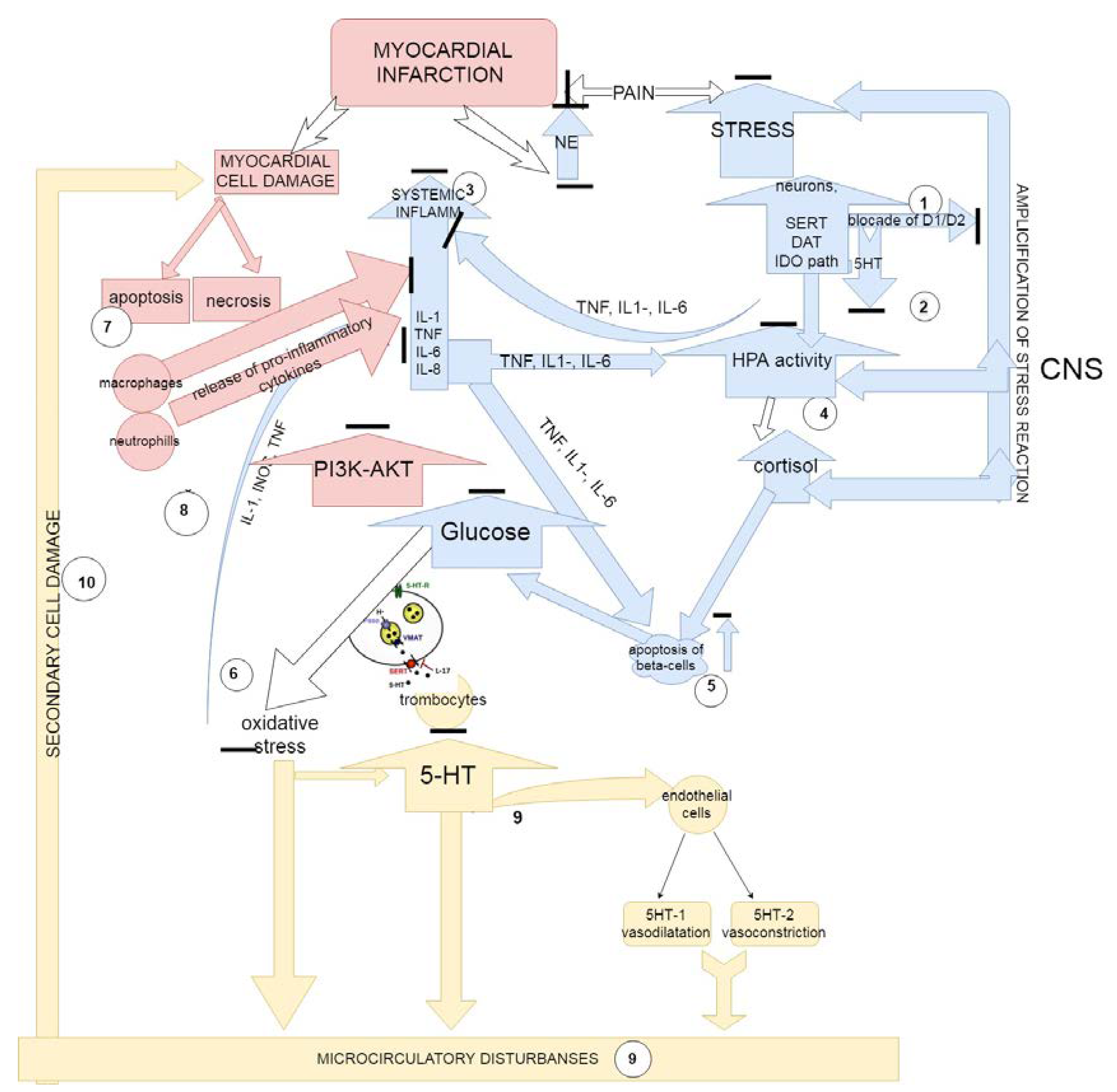
4. Discussion
The Proposed Mechanisms of Action of Substituted 1,3,4-Thiadiazines
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- The Immunomodulatory/Immunosuppressive Classification System|Moderate-to-Severe Psoriasis, Third Edition|Taylor and Francis Group. Available online: https://www.taylorfrancis.com/books/e/9781420088687/chapters/10.3109%2F9781420088687-18 (accessed on 2 April 2018).
- Lebish, I.J.; Moraski, R.M. Mechanisms of immunomodulation by drugs. Toxicol. Pathol. 1987, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Tzianabos, A.O. Polysaccharide immunomodulators as therapeutic agents: Structural aspects and biologic function. Clin. Microbiol. Rev. 2000, 13, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Khaitov, R.M.; Pinegin, В.V. Immunomodulators: Action mechanisms and clinical application. Immunologiya 2003, 24, 196–203. [Google Scholar]
- Frequently Asked Questions about Therapeutic Biological Products. Available online: https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/ucm113522.htm (accessed on 1 May 2018).
- Selye, H. A syndrome produced by diverse nocuous agents. J. Neuropsychiatry Clin. Neurosci. 1998, 10, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Reiche, E.M.V.; Nunes, S.O.V.; Morimoto, H.K. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004, 5, 617–625. [Google Scholar] [CrossRef]
- Dantzer, R.; Kelley, K.W. Stress and immunity: An integrated view of relationships between the brain and the immune system. Life Sci. 1989, 44, 1995–2008. [Google Scholar] [CrossRef]
- Marketon, W.J.I.; Glaser, R. Stress hormones and immune function. Cell. Immunol. 2008, 252, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Adamo, S.A. The stress response and immune system share, borrow, and reconfigure their physiological network elements: Evidence from the insects. Horm. Behav. 2017, 88, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S. Effects of stress on immune function: The good, the bad, and the beautiful. Immunol. Res. 2014, 58, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Adamo, S.A. Comparative psychoneuroimmunology/ecoimmunology: Lessons from simpler model systems. Oxf. Handb. Psychoneuroimmunol. 2012. [Google Scholar] [CrossRef]
- Boonstra, R. Reality as the leading cause of stress: Rethinking the impact of chronic stress in nature. Funct. Ecol. 2013, 27, 11–23. [Google Scholar] [CrossRef]
- Blalock, J.E. The syntax of immune-neuroendocrine communication. Immunol. Today 1994, 15, 504–511. [Google Scholar] [CrossRef]
- Chrousos, G.P. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 1995, 332, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Kim, Y.-K. The role of IL-12 and TGF-beta1 in the pathophysiology of major depressive disorder. Int. Immunopharmacol. 2006, 6, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Janssen, D.G.A.; Caniato, R.N.; Verster, J.C.; Baune, B.T. A psychoneuroimmunological review on cytokines involved in antidepressant treatment response. Hum. Psychopharmacol. 2010, 25, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Kenis, G.; Maes, M. Effects of antidepressants on the production of cytokines. Int. J. Neuropsychopharmacol. 2002, 5, 401–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubera, M.; Lin, A.H.; Kenis, G.; Bosmans, E.; van Bockstaele, D.; Maes, M. Anti-Inflammatory effects of antidepressants through suppression of the interferon-gamma/interleukin-10 production ratio. J. Clin. Psychopharmacol. 2001, 21, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Kubera, M.; Maes, M.; Holan, V.; Basta-Kaim, A.; Roman, A.; Shani, J. Prolonged desipramine treatment increases the production of interleukin-10, an anti-inflammatory cytokine, in C57BL/6 mice subjected to the chronic mild stress model of depression. J. Affect. Disord. 2001, 63, 171–178. [Google Scholar] [CrossRef]
- Kubera, M.; Maes, M.; Kenis, G.; Kim, Y.-K.; Lasoń, W. Effects of serotonin and serotonergic agonists and antagonists on the production of tumor necrosis factor alpha and interleukin-6. Psychiatry Res. 2005, 134, 251–258. [Google Scholar] [CrossRef] [PubMed]
- De Berardis, D.; Conti, C.M.V.; Serroni, N.; Moschetta, F.S.; Olivieri, L.; Carano, A.; Salerno, R.M.; Cavuto, M.; Farina, B.; Alessandrini, M.; et al. The effect of newer serotonin-noradrenalin antidepressants on cytokine production: A review of the current literature. Int. J. Immunopathol. Pharmacol. 2010, 23, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Rocchi, G.; Escelsior, A.; Fornaro, M. Immunomodulation mechanism of antidepressants: Interactions between serotonin/norepinephrine balance and Th1/Th2 balance. Curr. Neuropharmacol. 2012, 10, 97–123. [Google Scholar] [CrossRef] [PubMed]
- Maes, M. Depression is an inflammatory disease, but cell-mediated immune activation is the key component of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Nazimek, K.; Strobel, S.; Bryniarski, P.; Kozlowski, M.; Filipczak-Bryniarska, I.; Bryniarski, K. The role of macrophages in anti-inflammatory activity of antidepressant drugs. Immunobiology 2017, 222, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Nazimek, K.; Kozlowski, M.; Bryniarski, P.; Strobel, S.; Bryk, A.; Myszka, M.; Tyszka, A.; Kuszmiersz, P.; Nowakowski, J.; Filipczak-Bryniarska, I. Repeatedly administered antidepressant drugs modulate humoral and cellular immune response in mice through action on macrophages. Exp. Biol. Med. 2016, 241, 1540–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, R.; Kiecolt-Glaser, J.K. Stress-induced immune dysfunction: Implications for health. Nat. Rev. Immunol. 2005, 5, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Malinin, A.; Oshrine, B.; Serebruany, V. Treatment with selective serotonin reuptake inhibitors for enhancing wound healing. Med. Hypotheses 2004, 63, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, E.P.; Ilkaya, F.; Yildiz, L.; Aydin, F.; Senturk, N.; Denizli, H.; Canturk, T.; Turanli, A.Y. Effects of paroxetine on cutaneous wound healing in healthy and diabetic rats. Adv. Skin Wound Care 2014, 27, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Bradaschia-Correa, V.; Josephson, A.M.; Mehta, D.; Mizrahi, M.; Neibart, S.S.; Liu, C.; Kennedy, O.D.; Castillo, A.B.; Egol, K.A.; Leucht, P. The selective serotonin reuptake inhibitor fluoxetine directly inhibits osteoblast differentiation and mineralization during fracture healing in mice. J. Bone Miner. Res. 2017, 32, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.F.; Hernandez, L.; Yadav, E.; Schwemberger, S.; Dugan, A. The burn wound inflammatory response is influenced by midazolam. Inflammation 2012, 35, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Dugan, A.L.; Gregerson, K.A.; Neely, A.; Gardner, J.; Noel, G.J.; Babcock, G.F.; Horseman, N.D. Mice treated with a benzodiazepine had an improved survival rate following Pseudomonas aeruginosa infection. J. Burn Care Res. 2010, 31, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kiecolt-Glaser, J.K.; Page, G.G.; Marucha, P.T.; MacCallum, R.C.; Glaser, R. Psychological influences on surgical recovery. Perspectives from psychoneuroimmunology. Am. Psychol. 1998, 53, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R.; Pike, J.L.; Cole, J.C.; Oxman, M.N. Effects of a behavioral intervention, Tai Chi Chih, on varicella-zoster virus specific immunity and health functioning in older adults. Psychosom. Med. 2003, 65, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.L.; Farrar, W.B.; Golden-Kreutz, D.M.; Glaser, R.; Emery, C.F.; Crespin, T.R.; Shapiro, C.L.; Carson, W.E. Psychological, behavioral, and immune changes after a psychological intervention: A clinical trial. J. Clin. Oncol. 2004, 22, 3570–3580. [Google Scholar] [CrossRef] [PubMed]
- Almajan, G.L.; Barbuceanu, S.-F.; Saramet, I.; Draghici, C. New 6-amino-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines and [1,2,4]triazolo[3,4-b] [1,3,4]thiadiazin-6-ones: Synthesis, characterization and antibacterial activity evaluation. Eur. J. Med. Chem. 2010, 45, 3191–3195. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziem, A. Synthesis and antimicrobial activity of some novel thiazoles, 1,3,4-thiadiazines, 1,3,4-thiadiazoles incorporating coumarin moiety. J. Heterocycl. Chem. 2014, 52, 251–259. [Google Scholar] [CrossRef]
- Prakash, O.; Aneja, D.K.; Hussain, K.; Lohan, P.; Ranjan, P.; Arora, S.; Sharma, C.; Aneja, K.R. Synthesis and biological evaluation of dihydroindeno and indeno [1,2-e] [1,2,4]triazolo [3,4-b] [1,3,4]thiadiazines as antimicrobial agents. Eur. J. Med. Chem. 2011, 46, 5065–5073. [Google Scholar] [CrossRef] [PubMed]
- Purohit, D.H.; Dodiya, B.L.; Ghetiya, R.M.; Vekariya, P.B.; Joshi, H.S. Synthesis and antimicrobial activity of some new 1,3,4-thiadiazoles and 1,3,4-thiadiazines containing 1,2,4-Triazolo nucleus. Acta Chim. Slov. 2011, 58, 53–59. [Google Scholar] [PubMed]
- Puthiyapurayil, P.; Poojary, B.; Chikkanna, C.; Buridipad, S.K. Synthesis, spectral characterization and biological evaluation of a novel series of 6-arylsubstituted-3-[2-(4-substitutedphenyl)propan-2-yl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Eur. J. Med. Chem. 2012, 57, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Wagle, S.; Adhikari, A.V.; Kumari, N.S. Antimicrobial and antiinflammatory studies on some 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines and 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles containing quinoxaline. Asian J. Chem. 2008, 20, 629–641. [Google Scholar]
- Karegoudar, P.; Prasad, D.J.; Ashok, M.; Mahalinga, M.; Poojary, B.; Holla, B.S. Synthesis, antimicrobial and anti-inflammatory activities of some 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles and 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines bearing trichlorophenyl moiety. Eur. J. Med. Chem. 2008, 43, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.A.; Shaker, R.M.; Ameen, M.A.; Mohammed, M.F. Synthesis, anti-inflammatory, analgesic, and antibacterial activities of some triazole, triazolothiadiazole, and triazolothiadiazine derivatives. Arch. Pharm. Res. 2011, 34, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Iradyan, M.A.; Iradyan, N.S.; Paronikyan, R.V.; Stepanyan, G.M. Synthesis and biological activity of substituted 6-alkyl(6H)-3-phenyl-1,2, 4-triazolo[3,4-b]-1,3,4-thiadiazines. Pharm. Chem. J. 2010, 44, 413–417. [Google Scholar] [CrossRef]
- Shiradkar, M.; Bhaskara, B.P.; Kaur, R.; Dighe, R.; Burange, P.; Shah, R. Microwave assisted synthesis and bioactivity of s-Triazolo[3,4-b][1,3,4]thiadiazoles, s-Triazolo[3,4-b][1,3,4]thiadiazines and s-Triazolo[3′, 4′:2,3]thiadiazino [5,6-b]quinoxaline: Part-III. Asian J. Chem. 2007, 19, 2603–2608. [Google Scholar]
- Logvinova, Y.S.; Vasilyeva, T.M.; Makarov, V.A.; Chupakhin, O.N.; Sidorova, L.P.; Perova, N.M.; Rusinov, V.L. Experimental study of antiaggregation effect of 2-cycloalkylamino-5-phenyl-1,3,4-5H(C2H5)-thiadiazines in vitro. Gematol. Transfuziol. 2008, 53, 12–15. [Google Scholar]
- Vasil’eva, T.M.; Makarov, V.A.; Chupakhin, O.N.; Sidorova, L.P.; Perov, N.M.; Rusinov, V.L. Antiaggregant properties of new 1,3,4-thiadiazine derivatives. Eksp. Klin. Farmakol. 2009, 72, 27–30. [Google Scholar] [PubMed]
- Logvinova, O.S.; Vasil’eva, T.M.; Makarov, V.A.; Chupakhin, O.N.; Sidorova, L.P.; Perova, N.M.; Rusinov, V.L. Influence of new 1,3,4-thiadiazines on platelet aggregation in vitro and ex vivo. Eksp. Klin. Farmakol. 2010, 73, 21–25. (In Russian) [Google Scholar] [PubMed]
- Sidorova, L.P.; Tseitler, T.A.; Perova, N.M.; Emel’yanov, V.V.; Savateeva, E.A.; Maksimova, N.E.; Mochul’skaya, N.N.; Chereshnev, V.A.; Chupakhin, O.N. Synthesis of new 1,3,4-thiadiazines capable of inhibiting nonenzymatic glycosylation of proteins. Pharm. Chem. J. 2015, 49, 501–505. [Google Scholar] [CrossRef]
- Shevelev, O.B.; Illarionova, N.B.; Petrovski, D.V.; Sarapultsev, A.P.; Chupakhin, O.N.; Moshkin, M.P. Effects of a compound from the group of substituted thiadiazines with hypothermia inducing properties on brain metabolism in rats, a study in vivo and in vitro. PLoS ONE 2017, 12, e0180739. [Google Scholar] [CrossRef] [PubMed]
- Chupakhin, O.N.; Sidorova, L.P.; Tarakhty, E.A.; Novikova, A.P.; Perova, N.M.; Vinogradov, V.A.; Van Ginkel, M.F. Substituted 6H-1,3,4-thiadiazine-2-amines, the Use Thereof as Anaesthetising, Cardiovascular and Hypometabolic Agents, and Pharmaceutical Composition Containing Them. U.S. Patent 6,313,111, 6 November 2001. [Google Scholar]
- Sarapultsev, A.P.; Chupakhin, O.N.; Sarapultsev, P.A.; Sidorova, L.P.; Tseitler, T.A. Pharmacologic evaluation of antidepressant activity and synthesis of 2-morpholino-5-phenyl-6h-1,3,4-thiadiazine hydrobromide. Pharmaceuticals 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Online BBB Predictor. 2018. Available online: http://www.cbligand.org/BBB/ (accessed on 1 May 2018).
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Druzhilovsky, D.S.; Rudik, A.V.; Filimonov, D.A.; Gawande, D.; Suresh, K.; Poroikov, V.V. Capacities of computer evaluation of hidden potential of phytochemicals of medicinal plants of the traditional Indian Ayurvedic medicine. Biochem. Suppl. Ser. B Biomed. Chem. 2016, 10, 43–54. [Google Scholar] [CrossRef]
- PDBe: Protein Data Bank in Europe. 2018. Available online: http://www.ebi.ac.uk/pdbe/ (accessed on 1 May 2018).
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrencik, J.E.; Patny, A.; Leung, I.K.; Korniski, B.; Emmons, T.L.; Hall, T.; Hirsch, J.L. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6. J. Mol. Biol. 2010, 400, 413–433. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hanzlik, R.P.; Weaver, R.F.; Schönbrunn, E. Molecular mode of action of a covalently inhibiting peptidomimetic on the human calpain protease core. Biochemistry 2006, 45, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.B.; Kilpatrick, J.M.; Chu, N.; Babu, Y.S. Structure of 3,4-dichloroisocoumarin-inhibited factor D. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 711–717. [Google Scholar] [CrossRef]
- Maibaum, J.; Liao, S.M.; Vulpetti, A.; Ostermann, N.; Randl, S.; Rüdisser, S.; Lorthiois, E.; Erbel, P.; Kinzel, B.; Kolb, F.A.; et al. Small-molecule factor D inhibitors targeting the alternative complement pathway. Nat. Chem. Biol. 2016, 12, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- ModBase: Database of Comparative Protein Structure Models. 2018. Available online: https://modbase.compbio.ucsf.edu/modbase-cgi/index.cgi (accessed on 1 May 2018).
- Price, K.L.; Lillestol, R.K.; Ulens, C.; Lummis, S.C.R. Palonosetron–5-HT3 receptor interactions as shown by a binding protein cocrystal structure. ACS Chem. Neurosci. 2016, 7, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Malo, M.; Brive, L.; Luthman, K.; Svensson, P. Investigation of D1 receptor-agonist interactions and D1/D2 agonist selectivity using a combination of pharmacophore and receptor homology modeling. Chem. Med. Chem. 2012, 7, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Salmas, R.E.; Yurtsever, M.; Stein, M.; Durdagi, S. Modeling and protein engineering studies of active and inactive states of human dopamine D2 receptor (D2R) and investigation of drug/receptor interactions. Mol. Divers. 2015, 19, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Indarte, M.; Madura, J.D.; Surratt, C.K. Dopamine transporter comparative molecular modeling and binding site prediction using the LeuT(Aa) leucine transporter as a template. Proteins 2008, 70, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Elena Silva, M.; Villa, L.; Vistoli, G. Binding site analysis of full-length alpha1a adrenergic receptor using homology modeling and molecular docking. Biochem. Biophys. Res. Commun. 2004, 319, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Koldso, H.; Grouleff, J.; Schiott, B. Insights to ligand binding to the monoamine transporters-from homology modeling to LeuBAT and dDAT. Front. Pharmacol. 2015, 6, 208. [Google Scholar] [CrossRef] [PubMed]
- Ci, S.; Ren, T.; Su, Z. Investigating the putative binding-mode of GABA and diazepam within GABA A receptor using molecular modeling. Protein J. 2008, 27, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Skovstrup, S.; Taboureau, O.; Bräuner-Osborne, H.; Jorgensen, F.S. Homology modelling of the GABA transporter and analysis of tiagabine binding. Chem. Med. Chem. 2010, 5, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- MarvinSketch. 2016. Available online: http://www.chemaxon.com/products/marvin/marvinsketch/ (accessed on 1 May 2018).
- MOPAC. 2018. Available online: http://openmopac.net (accessed on 1 May 2018).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.B.; Ahn, M.; Lee, S.M.; Koh, H.W.; Lee, S.H.; Kim, S.H.; Park, B.H. Inhibition of Janus activated kinase-3 protects against myocardial ischemia and reperfusion injury in mice. Exp. Mol. Med. 2013, 45, e23. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Amzel, L.M. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007, 318, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, J.M.; Eathiraj, S.; Vensel, D.; Liu, Y.; Bull, C.O.; Cornell-Kennon, S.; Makhija, S. Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): An orally bioavailable, selective, and potent allosteric AKT inhibitor. J. Med. Chem. 2016, 59, 6455–6469. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Adsit, F.G.; Boyington, J.C. The 1.5 Å crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J. Biol. Chem. 2010, 285, 40762–40770. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.W.; Rey, F.A.; Sodeoka, M.; Verdine, G.L.; Harrison, S.C. Structure of the NF-kappa B p50 homodimer bound to DNA. Nature 1995, 373, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Inte:ligand. 2018. Available online: http://www.inteligand.com/ (accessed on 1 May 2018).
- Kvetnansky, R. Stress: Neuroendocrine and Molecular Approaches, 1st ed.; CRC Press: Philadelphia, PA, USA, 1992; ISBN 978-2-88124-506-0. [Google Scholar]
- Sarapultsev, P.A.; Chupakhin, O.N.; Medvedeva, S.U.; Mukhlynina, E.A.; Brilliant, S.A.; Sidorova, L.P.; Danilova, I.G.; Sarapultsev, A.P. The impact of immunomodulator compound from the group of substituted thiadiazines on the course of stress reaction. Int. Immunopharmacol. 2015, 25, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, V.V. Stress and Pathology; Belorusskaya Nauka: Minsk, Belarus, 2007; ISBN 978-985-08-0829-5. (In Russian) [Google Scholar]
- Benelli, A.; De Pol, A.; Poggioli, R.; Cavazzuti, E.; Arletti, R.; Bertolini, A.; Vergoni, A.V. L-sulpiride, at antidepressant dosage, prevents conditioned-fear stress-induced gastric lesions in rats. Pharmacol. Res. 2000, 42, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Tagliavini, S.; Forgione, A.; Bazzani, C.; Botticelli, A.; Balugani, A.; Bertolini, A. Effect of sulpiride on ischemia- and reperfusion-induced heart damage, in rats. Riv. Eur. Sci. Med. Farmacol. 1992, 14, 411–415. [Google Scholar] [PubMed]
- Shirokikh, Y.V.; Kuznetsov, S.I.; Shapovalova, N.V. Cardiological Syndrome X: Diagnosis and Intensive Care. Gen. Reanimatol. 2007, 3, 83–87. [Google Scholar] [CrossRef]
- Esch, T. Health in stress: Change in the stress concept and its significance for prevention, health and life style. Gesundheitswesen 2002, 64, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Steptoe, A.; Kivimäki, M. Stress and cardiovascular disease. Nat. Rev. Cardiol. 2012, 9, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Sudakov, K.V.; Umryukhin, P.E. The System Basis of Emotional Stress; GEOTAR-Media: Moscow, Russia, 2009; ISBN 978-5-9704-1400-2. (In Russian) [Google Scholar]
- Sarapultsev, A.P.; Chupakhin, O.N.; Sarapultsev, P.A.; Rantsev, M.A.; Medvedeva, S.U.; Sidorova, L.P.; Abidov, M.T.; Danilova, I.G. Modulation of inflammatory response improves myocardial infarct healing in rats. Curr. Pharm. Des. 2014, 20, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Sarapultsev, A.P.; Chupakhin, O.N.; Sarapultsev, P.A.; Rantsev, M.A.; Medvedeva, S.U.; Sidorova, L.P. Effect of a new class of compounds of the group of substituted 5R1, 6H2-1,3,4-thiadiazine-2-amines on the inflammatory and cytokine response in experimental myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.J.; Pfeffer, J.M.; Finn, P.; Pfeffer, M.A. Characteristics of a model of myocardial infarction produced by coronary artery ligation in the rat. Cardiovasc. Pathol. 1995, 4, 189–194. [Google Scholar] [CrossRef]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.J.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrkin, A.L. Myocardial Infarction, 3rd ed.; MIA: Moscow, Russia, 2003; ISBN 5-89481-081-7. (In Russian) [Google Scholar]
- Chupakhin, O.; Sarapultsev, A.; Chereshneva, M.; Gette, I.; Sidorova, L.; Danilova, I.; Sarapultsev, P. Influence of a biologically active compound from substituted thiadiazines on transaminase activity in myocardial homogenate in experimental myocardial infarction. Int. J. Pharm. Pharm. Sci. 2015, 7, 147–151. [Google Scholar]
- Lapshina, L.A.; Kopitsa, N.P.; Kravchun, P.G.; Petyunina, O.V. Cytokines and neurohormones level in patients with postinfarction cardiosclerosis depending on localization of myocardial infarction. Ukr. Cardiol. J. 2005, 5, 27–32. [Google Scholar]
- Gusev, E.Y.; Yurchenko, L.N.; Chereshnev, V.A.; Zotova, N.V. Methodology of research in systemic inflammation. Cytokines Inflamm. 2008, 1, 15–23. (In Russian) [Google Scholar]
- Zotova, N.V.; Chereshnev, V.A.; Gusev, E.Y. Systemic Inflammation: Methodological Approaches to Identification of the Common Pathological Process. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Mughal, R.S.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Mechanism of TNFalpha-induced IL-1alpha, IL-1beta and IL-6 expression in human cardiac fibroblasts: Effects of statins and thiazolidinediones. Cardiovasc. Res. 2007, 76, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Eryukhin, I.A.; Shlyapnikov, S.A. Extreme Condition of the Body. Elements of the Theory and Practical Problems on the Clinical Model of Combined Severe Trauma; Aesculap: Saint Petersburg, Russia, 1997; ISBN 5-8923-004-X. (In Russian) [Google Scholar]
- Vitkovskiĭ, I.A.; Kuznik, B.I. Effect of interleukin-1 on lymphocyte capacity of releasing factors affecting platelet adhesion and aggregation, blood coagulation and fibrinolysis. Ross. Fiziol. Zhurnal Im. IM Sechenova. 2002, 88, 468–475. [Google Scholar]
- Baudry, N.; Rasetti, C.; Vicaut, E. Differences between cytokine effects in the microcirculation of the rat. Am. J. Physiol. 1996, 271, H1186–H1192. [Google Scholar] [CrossRef] [PubMed]
- Bar, J.; Zosmer, A.; Hod, M.; Elder, M.G.; Sullivan, M.H. The regulation of platelet aggregation in vitro by interleukin-1beta and tumor necrosis factor-alpha: Changes in pregnancy and in pre-eclampsia. Thromb. Haemost. 1997, 78, 1255–1261. [Google Scholar] [PubMed]
- Hot, A.; Lenief, V.; Miossec, P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann. Rheum. Dis. 2012, 71, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Sarapultsev, P.A.; Sarapultsev, A.P.; Sidorova, L.P. The influence of apoptosis activation on healing after myocardial infarction. Bull. Ural. Med. Acad Sci. 2014, 3, 38–39. [Google Scholar]
- Chen, S.; Owens, G.C.; Edelman, D.B. Dopamine inhibits mitochondrial motility in hippocampal neurons. PLoS ONE 2008, 3, e2804. [Google Scholar] [CrossRef]
- Zhu, C.-B.; Lindler, K.M.; Owens, A.W.; Daws, L.C.; Blakely, R.D.; Hewlett, W.A. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 2010, 35, 2510–2520. [Google Scholar] [CrossRef] [PubMed]
- Bernik, T.R.; Friedman, S.G.; Ochani, M.; DiRaimo, R.; Susarla, S.; Czura, C.J.; Tracey, K.J. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J. Vasc. Surg. 2002, 36, 1231–1236. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, M.E.; Rubin, R.T.; McKlveen, J.M.; Karwoski, T.E.; Fulton, B.A.; Czambel, R.K. Pituitary-adrenal responses to oxotremorine and acute stress in male and female M1 muscarinic receptor knockout mice: Comparisons to M2 muscarinic receptor knockout mice. J. Neuroendocrinol. 2008, 20, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Ray, A.; Chakravarti, A.K.; Sen, P. Role of dopaminergic mechanisms in the regulation of stress responses in experimental animals. Pharmacol. Biochem. Behav. 1994, 48, 53–56. [Google Scholar] [CrossRef]
- Sarkar, C.; Das, S.; Chakroborty, D.; Chowdhury, U.R.; Basu, B.; Dasgupta, P.S.; Basu, S. Cutting Edge: Stimulation of dopamine D4 receptors induce T cell quiescence by up-regulating Kruppel-like factor-2 expression through inhibition of ERK1/ERK2 phosphorylation. J. Immunol. 2006, 177, 7525–7529. [Google Scholar] [CrossRef] [PubMed]
- Nomura, J.; Hosoi, T.; Okuma, Y.; Nomura, Y. The presence and functions of muscarinic receptors in human T cells: The involvement in IL-2 and IL-2 receptor system. Life Sci. 2003, 72, 2121–2126. [Google Scholar] [CrossRef]
- Hoeger, S.; Gottmann, U.; Liu, Z.; Schnuelle, P.; Birck, R.; Braun, C.; van der Woude, F.J.; Yard, B.A. Dopamine treatment in brain-dead rats mediates anti-inflammatory effects: The role of hemodynamic stabilization and D-receptor stimulation. Transpl. Int. 2007, 20, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Alagha, K.; Palot, A.; Sofalvi, T.; Pahus, L.; Gouitaa, M.; Tummino, C.; Martinez, S.; Charpin, D.; Bourdin, A.; Chanez, P. Long-acting muscarinic receptor antagonists for the treatment of chronic airway diseases. Ther. Adv. Chronic. Dis. 2014, 5, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani-Boroujerdi, S.; Behl, M.; Hahn, F.F.; Pena-Philippides, J.C.; Hutt, J.; Sopori, M.L. Role of muscarinic receptors in the regulation of immune and inflammatory responses. J. Neuroimmunol. 2008, 194, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Spurej, E. Serotonin reuptake inhibitors and cardiovascular diseases: A platelet connection. Cell. Mol. Life Sci. 2005, 62, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Schlienger, R.G.; Meier, C.R. Effect of selective serotonin reuptake inhibitors on platelet activation: Can they prevent acute myocardial infarction? Am. J. Cardiovasc. Drugs. 2003, 3, 149–162. [Google Scholar] [CrossRef] [PubMed]
- De Chaffoy de Courcelles, D.; Roevens, P.; Van Belle, H.; De Clerck, F. The synergistic effect of serotonin and epinephrine on the human platelet at the level of signal transduction. FEBS Lett. 1987, 219, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Treasure, C.B.; Vita, J.A.; Cox, D.A.; Fish, R.D.; Gordon, J.B.; Mudge, G.H.; Colucci, W.S.; Sutton, M.G.; Selwyn, A.P.; Alexander, R.W. Endothelium-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation 1990, 81, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J.; Miller, A.H. Pro-inflammatory cytokine-induced SAPK/MAPK and JAK/STAT in rheumatoid arthritis and the new anti-depression drugs. Exp. Opin. Ther. Targets 2008, 12, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, Q.; Li, P.; Lu, Q.; Pei, X.; Sun, Y.; Wang, G.; Hao, K. Magnesium isoglycyrrhizinate attenuates lipopolysaccharide-induced depressive-like behavior in mice. Biomed. Pharmacother. 2017, 86, 177–184. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the substituted 1,3,4-thadiazines compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  |  |
| Reaxys RN = 20515191 (Ref. [38]) | Reaxys RN = 20515189 (Ref. [38]) | Reaxys RN = 21926455 (Ref. [40]) |
 |  |  |
| Reaxys RN = 6006308 (Ref. [41]) | Reaxys RN = 22412039 (Ref. [41]) | Reaxys RN = 5094636 (Ref. [45]) |
 |  |  |
| Reaxys RN = 6535400 (Ref. [45]) | L-17, a 5-phenyl substituted-6H-1,3,4-thiadiazine-2-amine. (Ref. [48,49,50,51,52,53,54]) | 1,3,4-thiadiazine cycle |
 |  |
| CHEMBL2104951 | CHEMBL2440857 |
| ChEMBL ID, Name, Code | Assay Description | Activity ID | Activity |
|---|---|---|---|
| CHEMBL2104951 Aladorian ARM036 S36 | Induction of cardiac function improvement in myocardial infarction-induced acute heart failure mouse model at plasma concentration 200 nM after 7 days post myocardial infarction. | 16878971 | Active |
| Induction of cardiac function improvement in myocardial infarction-induced acute heart failure mouse model subjected to permanent ligation of left anterior descending coronary artery assessed as increase in fractional shortening at plasma concentration of 100 to 200 nM after 2 weeks post-myocardial infarction by echocardiography. | 16878972 | Active | |
| Induction of cardiac function improvement in myocardial infarction-induced acute heart failure mouse model subjected to permanent ligation of left anterior descending coronary artery assessed as increase in cardiac contractility at plasma concentration 200 nM after 7 days post-myocardial infarction. | 16878973 | Active | |
| CHEMBL2440857 JTV-519 | Reduction in PKA-phosphorylation of RyR2 in myocardial infarction-induced heart failure mouse model at 0.5 mg/kg/h after 28 days post-myocardial infarction. | 16878976 | Active |
| Improvement in soleus muscle fatigability in myocardial infarction-induced heart failure mouse model at 0.5 mg/kg/day relative to control. | 16878983 | Active | |
| Improvement in soleus muscle fatigability in myocardial infarction-induced heart failure calstabin2-deficient knockout mouse at 0.5 mg/kg/day relative to control. | 16878984 | Active | |
| Normalization of RYR1 in myocardial infarction-induced heart failure mouse soleus muscle assessed as increase in average channel open dwell time. | 16878985 | t = 1.5 ms | |
| Normalization of RYR1 in myocardial infarction-induced heart failure mouse soleus muscle assessed as decrease in average channel close dwell time. | 16878986 | t = 1567 ms | |
| Induction of calstabin-1 binding to RYR1 in myocardial infarction-induced heart failure wild type mouse soleus muscle at 0.5 mg/kg/day dosed via implantable osmotic minipumps. | 16878996 | Active | |
| Inhibition of of PKA-induced RyR1 phosphorylation in myocardial infarction-induced heart failure mouse soleus muscle at 0.5 mg/kg/day dosed via implantable osmotic minipumps. | 16878997 | Active | |
| Induction of calstabin-1 binding to RYR1 in myocardial infarction-induced heart failure calsiabin2−/− mouse soleus muscle at 0.5 mg/kg/day dosed via implantable osmotic minipumps. | 16878998 | Active |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarapultsev, A.P.; Vassiliev, P.M.; Sarapultsev, P.A.; Chupakhin, O.N.; Ianalieva, L.R.; Sidorova, L.P. Immunomodulatory Action of Substituted 1,3,4-Thiadiazines on the Course of Myocardial Infarction. Molecules 2018, 23, 1611. https://doi.org/10.3390/molecules23071611
Sarapultsev AP, Vassiliev PM, Sarapultsev PA, Chupakhin ON, Ianalieva LR, Sidorova LP. Immunomodulatory Action of Substituted 1,3,4-Thiadiazines on the Course of Myocardial Infarction. Molecules. 2018; 23(7):1611. https://doi.org/10.3390/molecules23071611
Chicago/Turabian StyleSarapultsev, Alexey P., Pavel M. Vassiliev, Petr A. Sarapultsev, Oleg N. Chupakhin, Laura R. Ianalieva, and Larisa P. Sidorova. 2018. "Immunomodulatory Action of Substituted 1,3,4-Thiadiazines on the Course of Myocardial Infarction" Molecules 23, no. 7: 1611. https://doi.org/10.3390/molecules23071611
APA StyleSarapultsev, A. P., Vassiliev, P. M., Sarapultsev, P. A., Chupakhin, O. N., Ianalieva, L. R., & Sidorova, L. P. (2018). Immunomodulatory Action of Substituted 1,3,4-Thiadiazines on the Course of Myocardial Infarction. Molecules, 23(7), 1611. https://doi.org/10.3390/molecules23071611