Phosphocholine-Modified Lipooligosaccharides of Haemophilus influenzae Inhibit ATP-Induced IL-1β Release by Pulmonary Epithelial Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Nicotine and PC Inhibit IL-1β Release from A549 Cells via nAChRs
2.2. Nicotine and PC Inhibit IL-1β Release from Calu-3 Cells via nAChRs
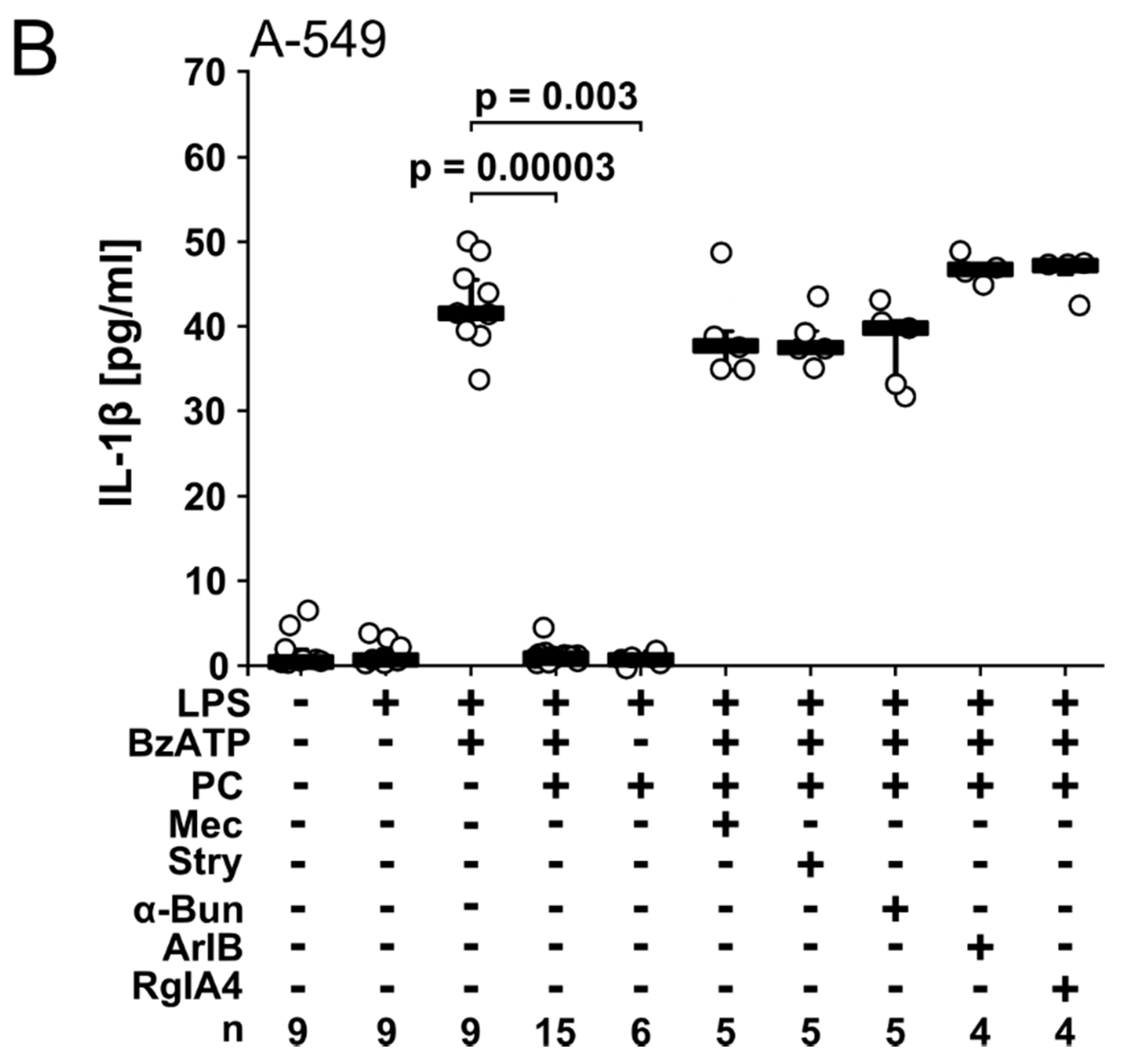
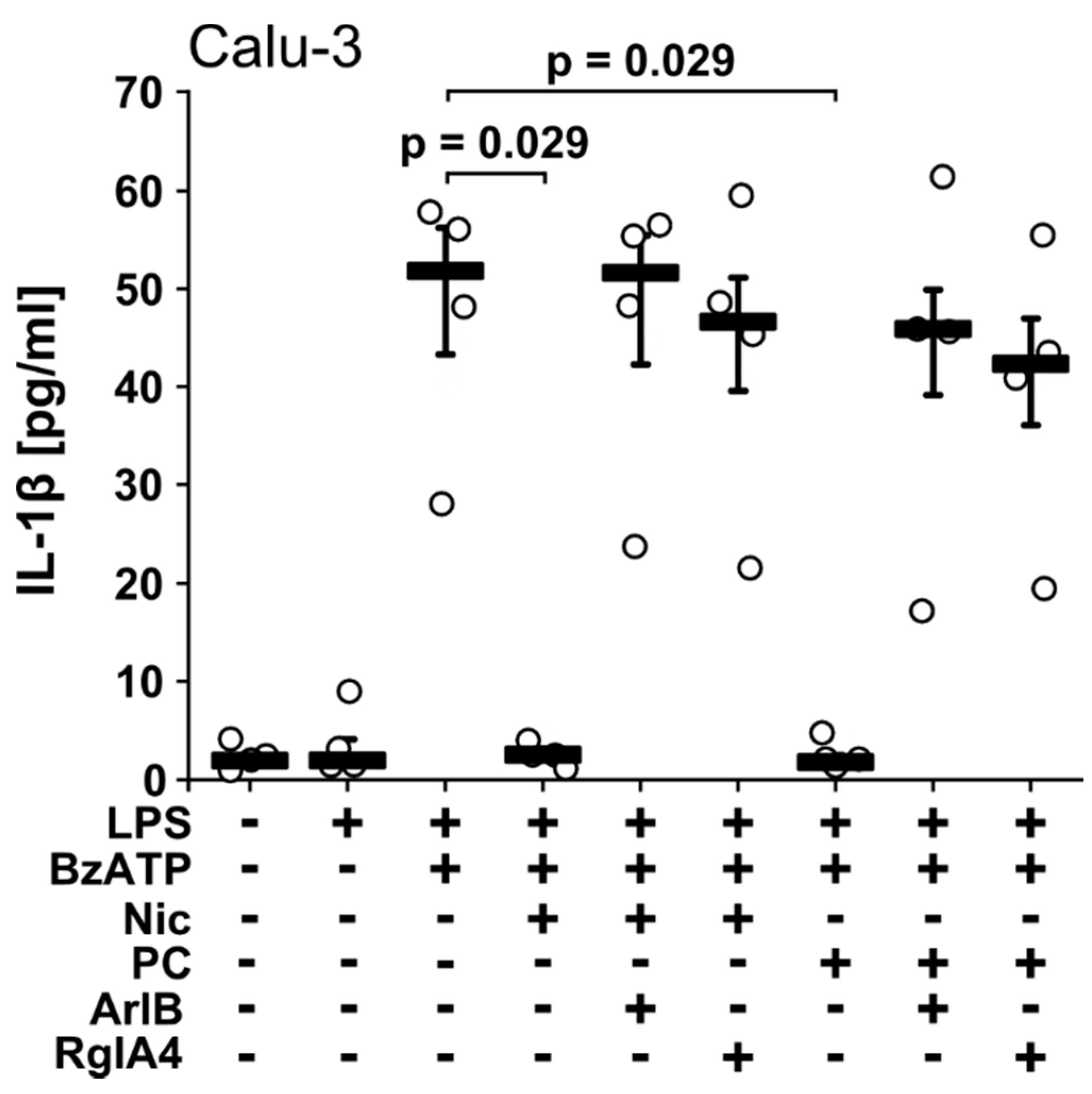
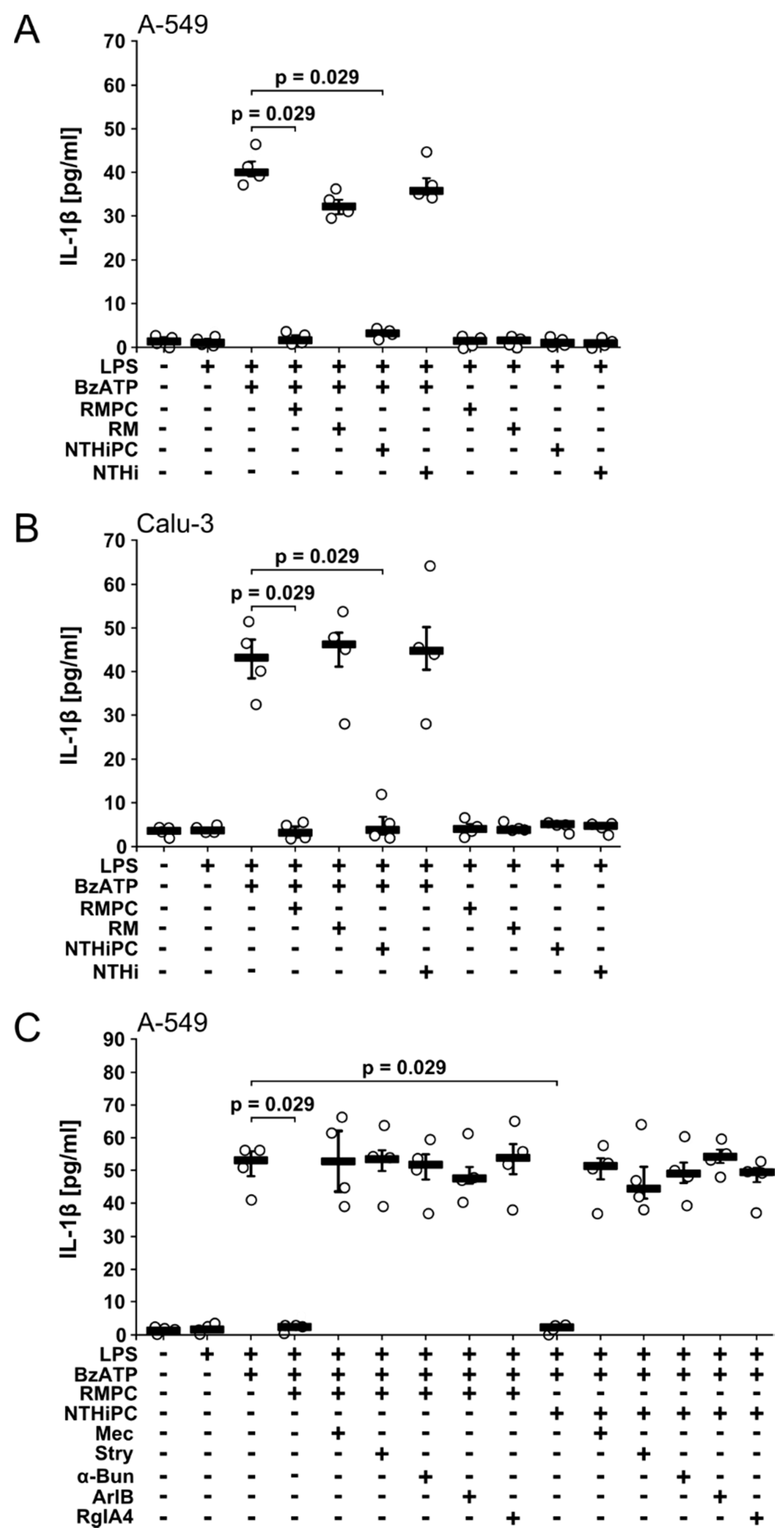
2.3. PC-LOS Inhibit BzATP-Mediated IL-1β Release from A549 and Calu-3 Cells
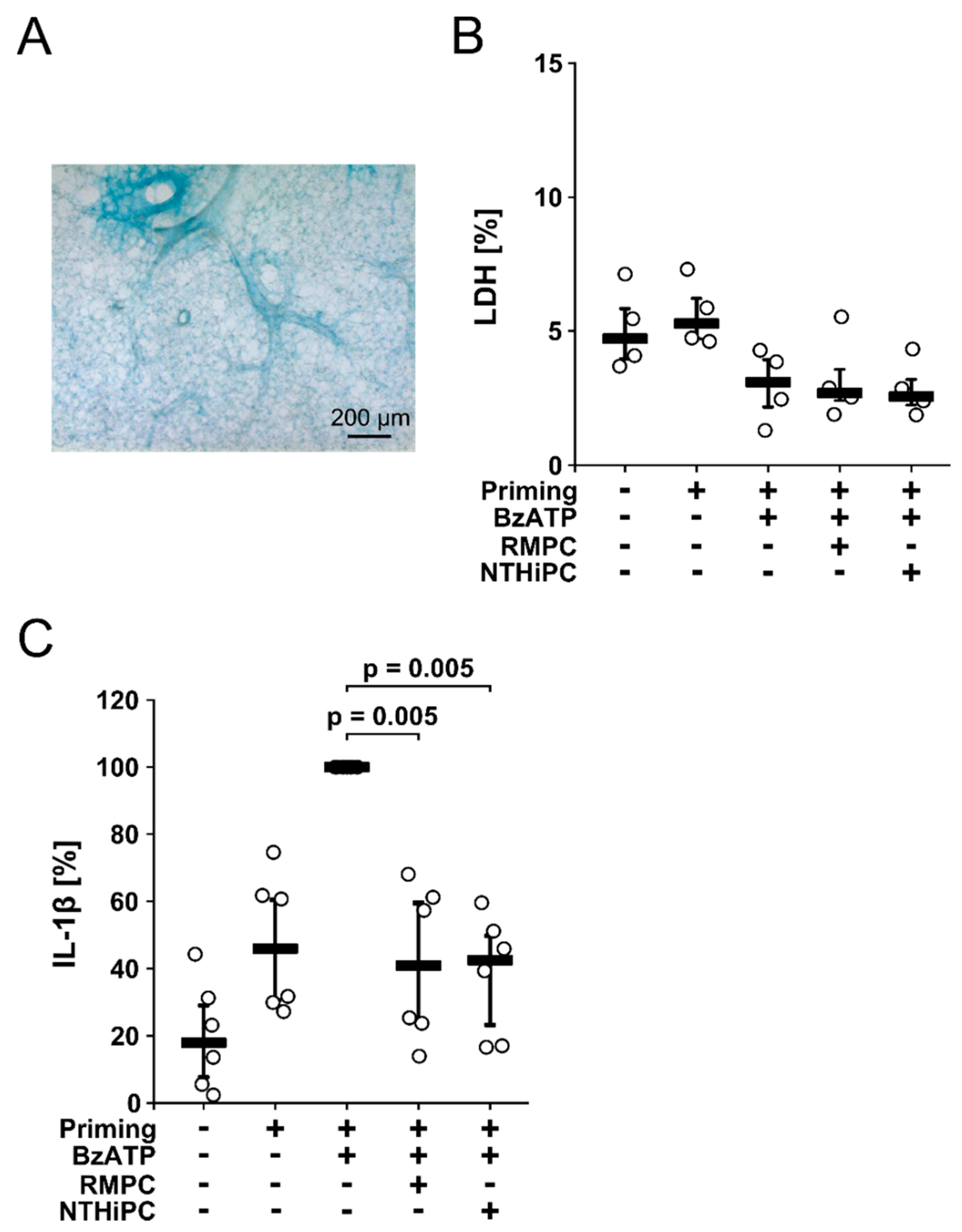
2.4. PC-LOS Inhibit the BzATP-Mediated IL-1β Release from Mouse Precision Cut Lung Slices (PCLS)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Purification and Characterization of PC-LOS
4.3. Pulmonary Epithelial Cell Lines
4.4. Mouse PCLS
4.5. Cytokine Measurement
4.6. LDH Measurement
4.7. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- King, P.T.; Sharma, R. The lung immune response to nontypeable Haemophilus influenzae (lung immunity to NTHi). J. Immunol. Res. 2015, 2015, 706376. [Google Scholar] [CrossRef] [PubMed]
- Duell, B.L.; Su, Y.-C.; Riesbeck, K. Host-pathogen interactions of nontypeable Haemophilus influenzae: From commensal to pathogen. FEBS Lett. 2016, 590, 3840–3853. [Google Scholar] [CrossRef] [PubMed]
- Risberg, A.; Masoud, H.; Martin, A.; Richards, J.C.; Moxon, E.R.; Schweda, E.K.H. Structural analysis of the lipopolysaccharide oligosaccharide epitopes expressed by a capsule-deficient strain of Haemophilus influenzae Rd. Eur. J. Biochem. 1999, 261, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Schweda, E.K.H.; Brisson, J.-R.; Alvelius, G.; Martin, A.; Weiser, J.N.; Hood, D.W.; Moxon, E.R.; Richards, J.C. Characterization of the phosphocholine-substituted oligosaccharide in lipopolysaccharides of type b Haemophilus influenzae. Eur. J. Biochem. 2000, 267, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Månsson, M.; Hood, D.W.; Moxon, E.R.; Schweda, E.K.H. Structural diversity in lipopolysaccharide expression in nontypeable Haemophilus influenzae. Eur. J. Biochem. 2003, 270, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Winn, D.; Johnson, R.; Hong, W.; West-Barnette, S.; Kock, N.; Swords, W.E. Lipooligosaccharides containing phosphorylcholine delay pulmonary clearance of nontypeable Haemophilus influenzae. Infect. Immun. 2008, 76, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Grabitzki, J.; Lochnit, G. Immunomodulation by phosphocholine—Biosynthesis, structures and immunological implications of parasitic PC-epitopes. Mol. Immunol. 2009, 47, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Weiser, J.N. Microbial modulation of host immunity with the small molecule phosphorylcholine. Infect. Immun. 2013, 81, 392–401. [Google Scholar] [CrossRef] [PubMed]
- McSorley, H.J.; Hewitson, J.P.; Maizels, R.M. Immunomodulation by helminth parasites: Defining mechanisms and mediators. Int. J. Parasitol. 2013, 43, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.N.; Pan, N.; McGowan, K.L.; Musher, D.; Martin, A.; Richards, J. Phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. J. Exp. Med. 1998, 187, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cox, A.D.; Li, J.; McCready, W.; Ulanova, M. Activation of innate immune responses by Haemophilus influenzae lipooligosaccharide. Clin. Vaccine Immunol. 2014, 21, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Vladimer, G.I.; Marty-Roix, R.; Ghosh, S.; Weng, D.; Lien, E. Inflammasomes and host defenses against bacterial infections. Curr. Opin. Microbiol. 2013, 16, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkerton, J.W.; Kim, R.Y.; Robertson, A.A.B.; Hirota, J.A.; Wood, L.G.; Knight, D.A.; Cooper, M.A.; O’Neill, L.A.J.; Horvat, J.C.; Hansbro, P.M. Inflammasomes in the lung. Mol. Immunol. 2017, 86, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. A new lead to NLRP3 inhibition. J. Exp. Med. 2017, 214, 3147–3149. [Google Scholar] [CrossRef] [PubMed]
- Hecker, A.; Küllmar, M.; Wilker, S.; Richter, K.; Zakrzewicz, A.; Atanasova, S.; Mathes, V.; Timm, T.; Lerner, S.; Klein, J.; et al. Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1β release. J. Immunol. 2015, 195, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Mathes, V.; Fronius, M.; Althaus, M.; Hecker, A.; Krasteva-Christ, G.; Padberg, W.; Hone, A.J.; McIntosh, J.M.; Zakrzewicz, A.; et al. Phosphocholine—An agonist of metabotropic but not of ionotropic functions of α9-containing nicotinic acetylcholine receptors. Sci. Rep. 2016, 6, 28660. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, A.; Richter, K.; Agné, A.; Wilker, S.; Siebers, K.; Fink, B.; Krasteva-Christ, G.; Althaus, M.; Padberg, W.; Hone, A.J.; et al. Canonical and novel non-canonical cholinergic agonists inhibit ATP-induced release of monocytic interleukin-1β via different combinations of nicotinic acetylcholine receptor subunits α7, α9 and α10. Front. Cell. Neurosci. 2017, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Message, S.D.; Johnston, S.L. Host defense function of the airway epithelium in health and disease: Clinical background. J. Leukoc. Biol. 2004, 75, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G.; Provoost, S.; Bracke, K.R.; Kuchmiy, A.; Lamkanfi, M. Inflammasomes in respiratory disease: From bench to bedside. Chest 2014, 145, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Peeters, P.M.; Wouters, E.F.; Reynaert, N.L. Immune homeostasis in epithelial cells: Evidence and role of inflammasome signaling reviewed. J. Immunol. Res. 2015, 2015, 828264. [Google Scholar] [CrossRef] [PubMed]
- Kostadinova, E.; Chaput, C.; Gutbier, B.; Lippmann, J.; Sander, L.E.; Mitchell, T.J.; Suttorp, N.; Witzenrath, M.; Opitz, B. NLRP3 protects alveolar barrier integrity by an inflammasome-independent increase of epithelial cell adherence. Sci. Rep. 2016, 6, 30943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana, P.T.; Martel, J.; Lai, H.-C.; Perfettini, J.-L.; Kanellopoulos, J.M.; Young, J.D.; Coutinho-Silva, R.; Ojcius, D.M. Is the inflammasome relevant for epithelial cell function? Microbes Infect. 2016, 18, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Sun, H.; Casbon, A.-J.; Lim, E.; Francis, K.P.; Hellman, J.; Prakash, A. NLRP3 inflammasome mediates dormant neutrophil recruitment following sterile lung injury and protects against subsequent bacterial pneumonia in mice. Front. Immunol. 2017, 8, 1337. [Google Scholar] [CrossRef] [PubMed]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors2. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.T. Cell line A549: A model system for the study of alveolar type II cell function. Am. Rev. Respir. Dis. 1977, 115, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.Q.; Finkbeiner, W.E.; Wine, J.J.; Mrsny, R.J.; Widdicombe, J.H. Calu-3: A human airway epithelial cell line that shows cAMP-dependent Cl-secretion. Am. J. Physiol. 1994, 266, L493–L501. [Google Scholar] [CrossRef] [PubMed]
- Orr-Urtreger, A.; Göldner, F.M.; Saeki, M.; Lorenzo, I.; Goldberg, L.; de Biasi, M.; Dani, J.A.; Patrick, J.W.; Beaudet, A.L. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack α-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci. 1997, 17, 9165–9171. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.R.; Zwart, R.; Sher, E.; Millar, N.S. Pharmacological properties of α9α10 nicotinic acetylcholine receptors revealed by heterologous expression of subunit chimeras. Mol. Pharmacol. 2004, 65, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, P.; Christensen, S.; Yoshikami, D.; Dowell, C.; Watkins, M.; Gulyas, J.; Rivier, J.; Olivera, B.M.; McIntosh, J.M. Discovery, synthesis, and structure activity of a highly selective α7 nicotinic acetylcholine receptor antagonist. Biochemistry 2007, 46, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Innocent, N.; Livingstone, P.D.; Hone, A.; Kimura, A.; Young, T.; Whiteaker, P.; McIntosh, J.M.; Wonnacott, S. Alpha-conotoxin Arenatus IBV11L, V16D corrected is a potent and selective antagonist at rat and human native α7 nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 2008, 327, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switalla, S.; Lauenstein, L.; Prenzler, F.; Knothe, S.; Förster, C.; Fieguth, H.-G.; Pfennig, O.; Schaumann, F.; Martin, C.; Guzman, C.A.; et al. Natural innate cytokine response to immunomodulators and adjuvants in human precision-cut lung slices. Toxicol. Appl. Pharmacol. 2010, 246, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Henjakovic, M.; Sewald, K.; Switalla, S.; Kaiser, D.; Müller, M.; Veres, T.Z.; Martin, C.; Uhlig, S.; Krug, N.; Braun, A. Ex vivo testing of immune responses in precision-cut lung slices. Toxicol. Appl. Pharmacol. 2008, 231, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Siebers, K.; Fink, B.; Zakrzewicz, A.; Agné, A.; Richter, K.; Konzok, S.; Hecker, A.; Zukunft, S.; Küllmar, M.; Klein, J.; et al. Alpha-1 antitrypsin inhibits ATP-mediated release of interleukin-1β via CD36 and nicotinic acetylcholine receptors. Front. Immunol. 2018, 9, 633. [Google Scholar] [CrossRef] [PubMed]
- Swords, W.E.; Ketterer, M.R.; Shao, J.; Campbell, C.A.; Weiser, J.N.; Apicella, M.A. Binding of the non-typeable Haemophilus influenzae lipooligosaccharide to the PAF receptor initiates host cell signalling. Cell. Microbiol. 2001, 3, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Branger, J.; Wieland, C.W.; Florquin, S.; Maris, N.A.; Pater, J.M.; Speelman, P.; Shimizu, T.; Ishii, S.; van der Poll, T. Plattelet-activating factor receptor-deficient mice show an unaltered clearance of nontypeable Haemophilus influenzae from their respiratory tract. Shock 2004, 22, 543–547. [Google Scholar] [CrossRef] [PubMed]
- West-Barnette, S.; Rockel, A.; Swords, W.E. Biofilm growth increases phosphorylcholine content and decreases potency of nontypeable Haemophilus influenzae endotoxins. Infect. Immun. 2006, 74, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Swords, W.E. Nontypeable Haemophilus influenzae biofilms: Role in chronic airway infections. Front. Cell. Infect. Microbiol. 2012, 2, 97. [Google Scholar] [CrossRef] [PubMed]
- Morey, P.; Viadas, C.; Euba, B.; Hood, D.W.; Barberán, M.; Gil, C.; Grilló, M.J.; Bengoechea, J.A.; Garmendia, J. Relative contributions of lipooligosaccharide inner and outer core modifications to nontypeable Haemophilus influenzae pathogenesis. Infect. Immun. 2013, 81, 4100–4111. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Snow, J.; Li, J.; Zola, T.A.; Weiser, J.N. Phosphorylcholine allows for evasion of bactericidal antibody by Haemophilus influenzae. PLoS Pathog. 2012, 8, e1002521. [Google Scholar] [CrossRef] [PubMed]
- Humphries, H.E.; High, N.J. The role of licA phase variation in the pathogenesis of invasive disease by Haemophilus influenzae type b. FEMS Immunol. Med. Microbiol. 2002, 34, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Garlanda, C.; Doni, A.; Bottazzi, B. Pentraxins in innate immunity: From C-reactive protein to the long pentraxin PTX3. J. Clin. Immunol. 2008, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Faire, U.; Frostegård, J. Natural antibodies against phosphorylcholine in cardiovascular disease. Ann. N. Y. Acad. Sci. 2009, 1173, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J. Low level natural antibodies against phosphorylcholine: A novel risk marker and potential mechanism in atherosclerosis and cardiovascular disease. Clin. Immunol. 2010, 134, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Briles, D.E.; Shackelford, P.G.; Smith, D.S.; Nahm, M.H. Human antibodies to phosphocholine. IgG anti-PC antibodies express restricted numbers of V and C regions. J. Immunol. 1987, 138, 3325–3331. [Google Scholar] [PubMed]
- Nishinarita, S.; Sawada, S.; Horie, T. Phosphorylcholine antibodies in pulmonary infection. Med. Microbiol. Immunol. 1990, 179, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Hörkkö, S.; Chang, M.K.; Curtiss, L.K.; Palinski, W.; Silverman, G.J.; Witztum, J.L. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Investig. 2000, 105, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Fiskesund, R.; Steen, J.; Amara, K.; Murray, F.; Szwajda, A.; Liu, A.; Douagi, I.; Malmström, V.; Frostegård, J. Naturally occurring human phosphorylcholine antibodies are predominantly products of affinity-matured B cells in the adult. J. Immunol. 2014, 192, 4551–4559. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.N.; Shchepetov, M.; Chong, S.T. Decoration of lipopolysaccharide with phosphorylcholine: A phase-variable characteristic of Haemophilus influenzae. Infect. Immun. 1997, 65, 943–950. [Google Scholar] [PubMed]
- Liang, X.; Zhang, D.; Liu, W.; Yan, Y.; Zhou, F.; Wu, W.; Yan, Z. Reactive oxygen species trigger NF-κB-mediated NLRP3 inflammasome activation induced by zinc oxide nanoparticles in A549 cells. Toxicol. Ind. Health 2017, 33, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liang, Z.; Li, H.; Li, C.; Yang, Z.; Li, Y.; She, D.; Cao, L.; Wang, W.; Liu, C.; et al. Perfluorocarbon reduces cell damage from blast injury by inhibiting signal paths of NF-κB, MAPK and Bcl-2/Bax signaling pathway in A549 cells. PLoS ONE 2017, 12, e0173884. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Uhlig, S.; Ullrich, V. Videomicroscopy of methacholine-induced contraction of individual airways in precision-cut lung slices. Eur. Respir. J. 1996, 9, 2479–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paddenberg, R.; König, P.; Faulhammer, P.; Goldenberg, A.; Pfeil, U.; Kummer, W. Hypoxic vasoconstriction of partial muscular intra-acinar pulmonary arteries in murine precision cut lung slices. Respir. Res. 2006, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Sorci, G.; Cornet, S.; Faivre, B. Immune evasion, immunopathology and the regulation of the immune system. Pathogens 2013, 2, 71–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Avondt, K.; van Sorge, N.M.; van Sorge, N.M.; Meyaard, L. Bacterial immune evasion through manipulation of host inhibitory immune signaling. PLoS Pathog. 2015, 11, e1004644. [Google Scholar] [CrossRef] [PubMed]
- Backhaus, S.; Zakrzewicz, A.; Richter, K.; Damm, J.; Wilker, S.; Fuchs-Moll, G.; Küllmar, M.; Hecker, A.; Manzini, I.; Ruppert, C.; et al. Surfactant inhibits ATP-induced release of interleukin-1β via nicotinic acetylcholine receptors. J. Lipid Res. 2017, 58, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Dushianthan, A.; Cusack, R.; Goss, V.; Postle, A.D.; Grocott, M.P.W. Clinical review: Exogenous surfactant therapy for acute lung injury/acute respiratory distress syndrome—Where do we go from here? Crit. Care 2012, 16, 238. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The discovery of α1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Franchini, L.F.; Elgoyhen, A.B. Adaptive evolution in mammalian proteins involved in cochlear outer hair cell electromotility. Mol. Phylogenet. Evol. 2006, 41, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pereira, E.F.; Maus, A.D.; Ostlie, N.S.; Navaneetham, D.; Lei, S.; Albuquerque, E.X.; Conti-Fine, B.M. Human bronchial epithelial and endothelial cells express α7 nicotinic acetylcholine receptors. Mol. Pharmacol. 2001, 60, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Murphy, T.F. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med. 2008, 359, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Essilfie, A.-T.; Simpson, J.L.; Dunkley, M.L.; Morgan, L.C.; Oliver, B.G.; Gibson, P.G.; Foster, P.S.; Hansbro, P.M. Combined Haemophilus influenzae respiratory infection and allergic airways disease drives chronic infection and features of neutrophilic asthma. Thorax 2012, 67, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Jalalvand, F.; Riesbeck, K. Haemophilus influenzae: Recent advances in the understanding of molecular pathogenesis and polymicrobial infections. Curr. Opin. Infect. Dis. 2014, 27, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Waters, V.; Klingel, M.; McDonald, N.; Dell, S.; Leahy, T.R.; Yau, Y.; Grasemann, H. Changes in airway inflammation during pulmonary exacerbations in patients with cystic fibrosis and primary ciliary dyskinesia. Eur. Respir. J. 2016, 47, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Daly, J.; Baines, K.J.; Yang, I.A.; Upham, J.W.; Reynolds, P.N.; Hodge, S.; James, A.L.; Hugenholtz, P.; Willner, D.; et al. Airway dysbiosis: Haemophilus influenzae and Tropheryma in poorly controlled asthma. Eur. Respir. J. 2016, 47, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Baines, K.J.; Horvat, J.C.; Essilfie, A.-T.; Brown, A.C.; Tooze, M.; McDonald, V.M.; Gibson, P.G.; Hansbro, P.M. COPD is characterized by increased detection of Haemophilus influenzae, Streptococcus pneumoniae and a deficiency of Bacillus species. Respirology 2016, 21, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Bogner, P.N.; Thatcher, T.H.; Sime, P.J.; Phipps, R.P.; Thanavala, Y. Cigarette smoke exposure exacerbates lung inflammation and compromises immunity to bacterial infection. J. Immunol. 2014, 192, 5226–5235. [Google Scholar] [CrossRef] [PubMed]
- Earl, C.S.; Keong, T.W.; An, S.-Q.; Murdoch, S.; McCarthy, Y.; Garmendia, J.; Ward, J.; Dow, J.M.; Yang, L.; O’Toole, G.A.; et al. Haemophilus influenzae responds to glucocorticoids used in asthma therapy by modulation of biofilm formation and antibiotic resistance. EMBO Mol. Med. 2015, 7, 1018–1033. [Google Scholar] [CrossRef] [PubMed]
- Ulanova, M.; Tsang, R.S.W. Haemophilus influenzae serotype a as a cause of serious invasive infections. Lancet Infect. Dis. 2014, 14, 70–82. [Google Scholar] [CrossRef]
- MacVane, S.H. Antimicrobial resistance in the intensive care unit: A focus on Gram-negative bacterial infections. J. Intensive Care Med. 2017, 32, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Schweda, E.K.H.; Richards, J.C. Structural profiling of short-chain lipopolysaccharides from Haemophilus influenzae. Methods Mol. Med. 2003, 71, 161–183. [Google Scholar] [PubMed]
- Paddenberg, R.; Mermer, P.; Goldenberg, A.; Kummer, W. Videomorphometric analysis of hypoxic pulmonary vasoconstriction of intra-pulmonary arteries using murine precision cut lung slices. J. Vis. Exp. 2014, 83, e50970. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.C.; Jarett, L.; Finke, E.H. Embedding in epoxy resins for ultrathin sectioning in electron microscopy. Stain Technol. 1960, 35, 313–323. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, K.; Koch, C.; Perniss, A.; Wolf, P.M.; Schweda, E.K.H.; Wichmann, S.; Wilker, S.; Magel, I.; Sander, M.; McIntosh, J.M.; et al. Phosphocholine-Modified Lipooligosaccharides of Haemophilus influenzae Inhibit ATP-Induced IL-1β Release by Pulmonary Epithelial Cells. Molecules 2018, 23, 1979. https://doi.org/10.3390/molecules23081979
Richter K, Koch C, Perniss A, Wolf PM, Schweda EKH, Wichmann S, Wilker S, Magel I, Sander M, McIntosh JM, et al. Phosphocholine-Modified Lipooligosaccharides of Haemophilus influenzae Inhibit ATP-Induced IL-1β Release by Pulmonary Epithelial Cells. Molecules. 2018; 23(8):1979. https://doi.org/10.3390/molecules23081979
Chicago/Turabian StyleRichter, Katrin, Christian Koch, Alexander Perniss, Philipp M. Wolf, Elke K. H. Schweda, Sven Wichmann, Sigrid Wilker, Ilona Magel, Michael Sander, J. Michael McIntosh, and et al. 2018. "Phosphocholine-Modified Lipooligosaccharides of Haemophilus influenzae Inhibit ATP-Induced IL-1β Release by Pulmonary Epithelial Cells" Molecules 23, no. 8: 1979. https://doi.org/10.3390/molecules23081979
APA StyleRichter, K., Koch, C., Perniss, A., Wolf, P. M., Schweda, E. K. H., Wichmann, S., Wilker, S., Magel, I., Sander, M., McIntosh, J. M., Padberg, W., & Grau, V. (2018). Phosphocholine-Modified Lipooligosaccharides of Haemophilus influenzae Inhibit ATP-Induced IL-1β Release by Pulmonary Epithelial Cells. Molecules, 23(8), 1979. https://doi.org/10.3390/molecules23081979