
A Combined Approach of NMR and Mass Spectrometry Techniques Applied to the α-Cyclodextrin/Moringin Complex for a Novel Bioactive Formulation †

 , , , ,
, , , , 
Abstract
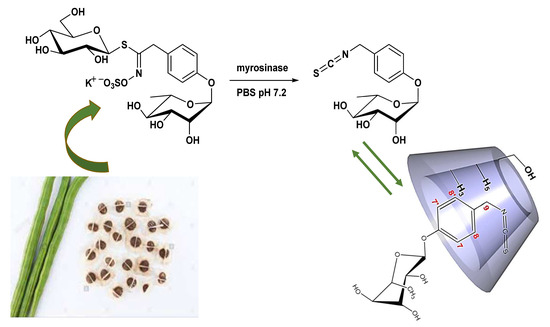
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
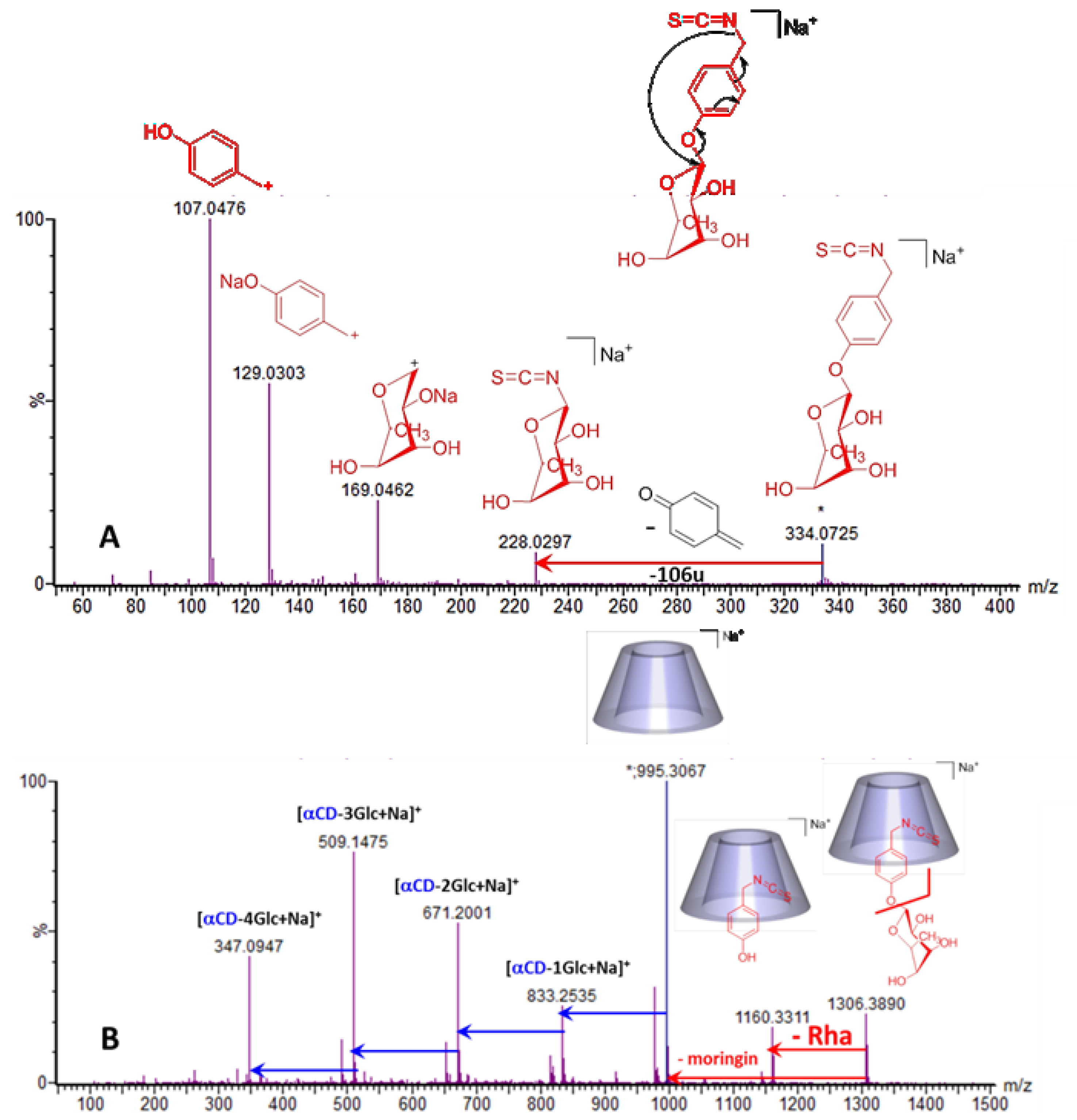{kind=link}
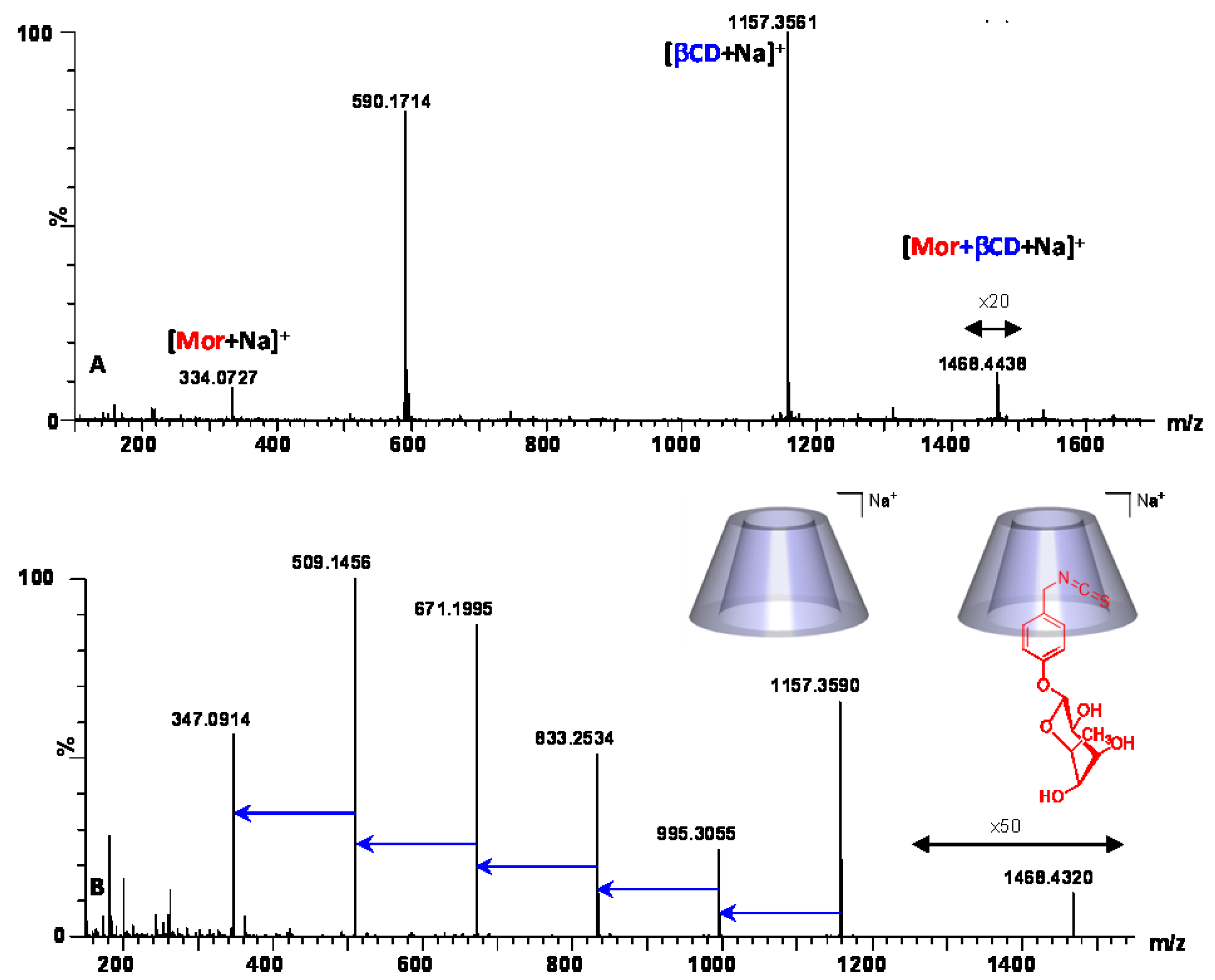{kind=link}
1. Introduction
2. Results and Discussion
2.1. Investigation of Anti-Inflammatory Properties
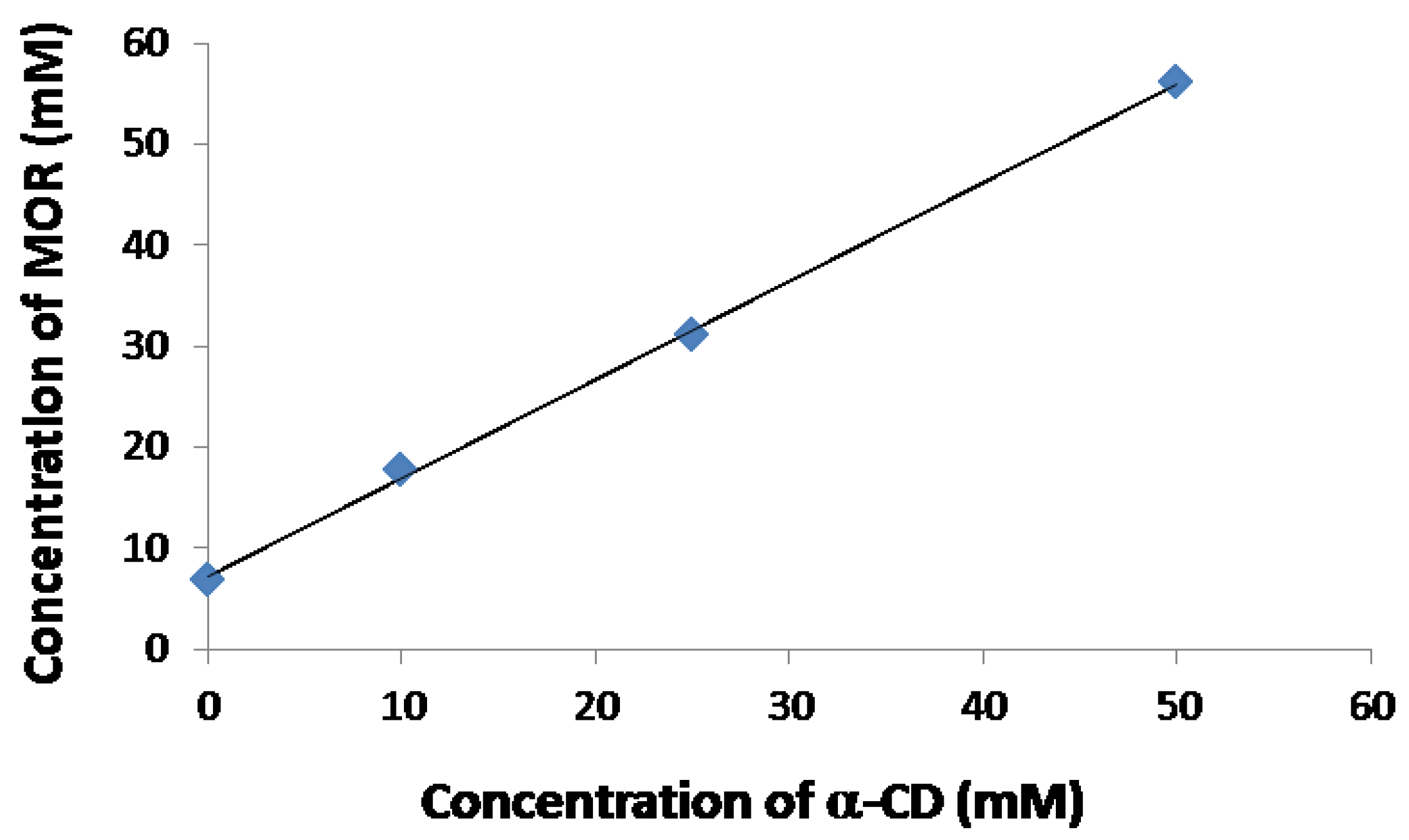
2.2. Physico-Chemical Studies of α-CD/MOR Mixture
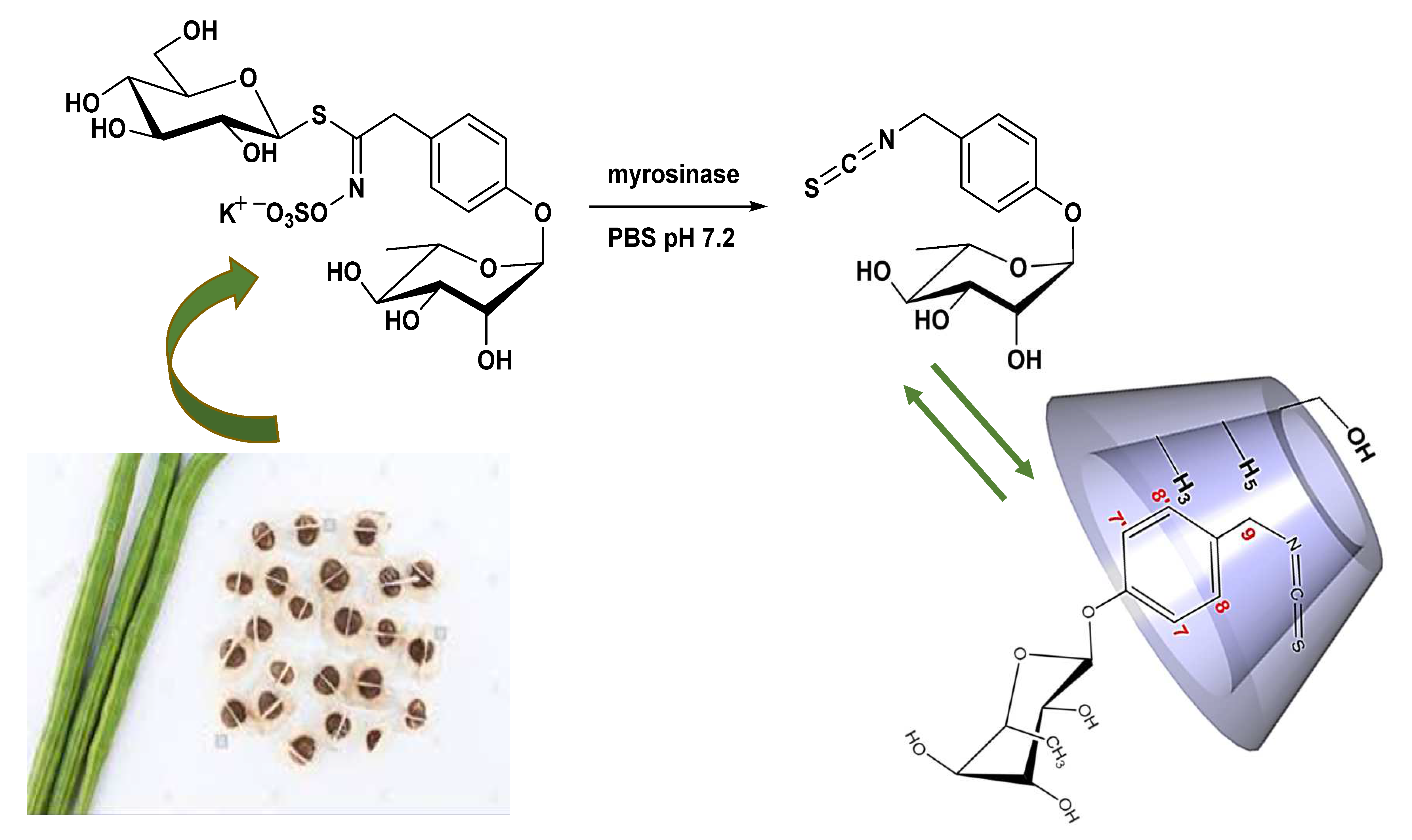
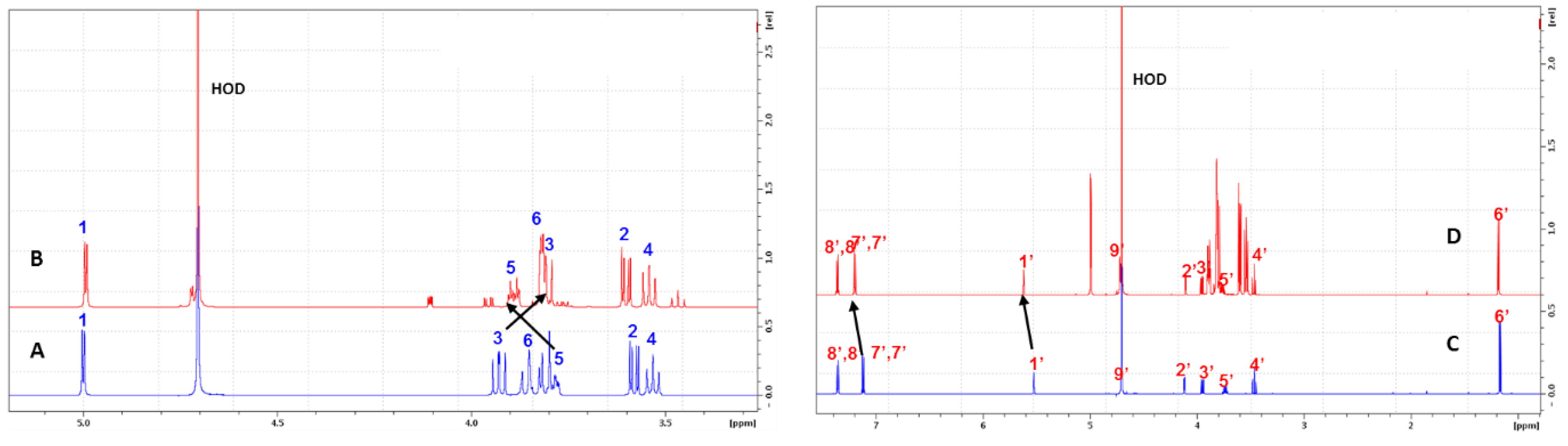
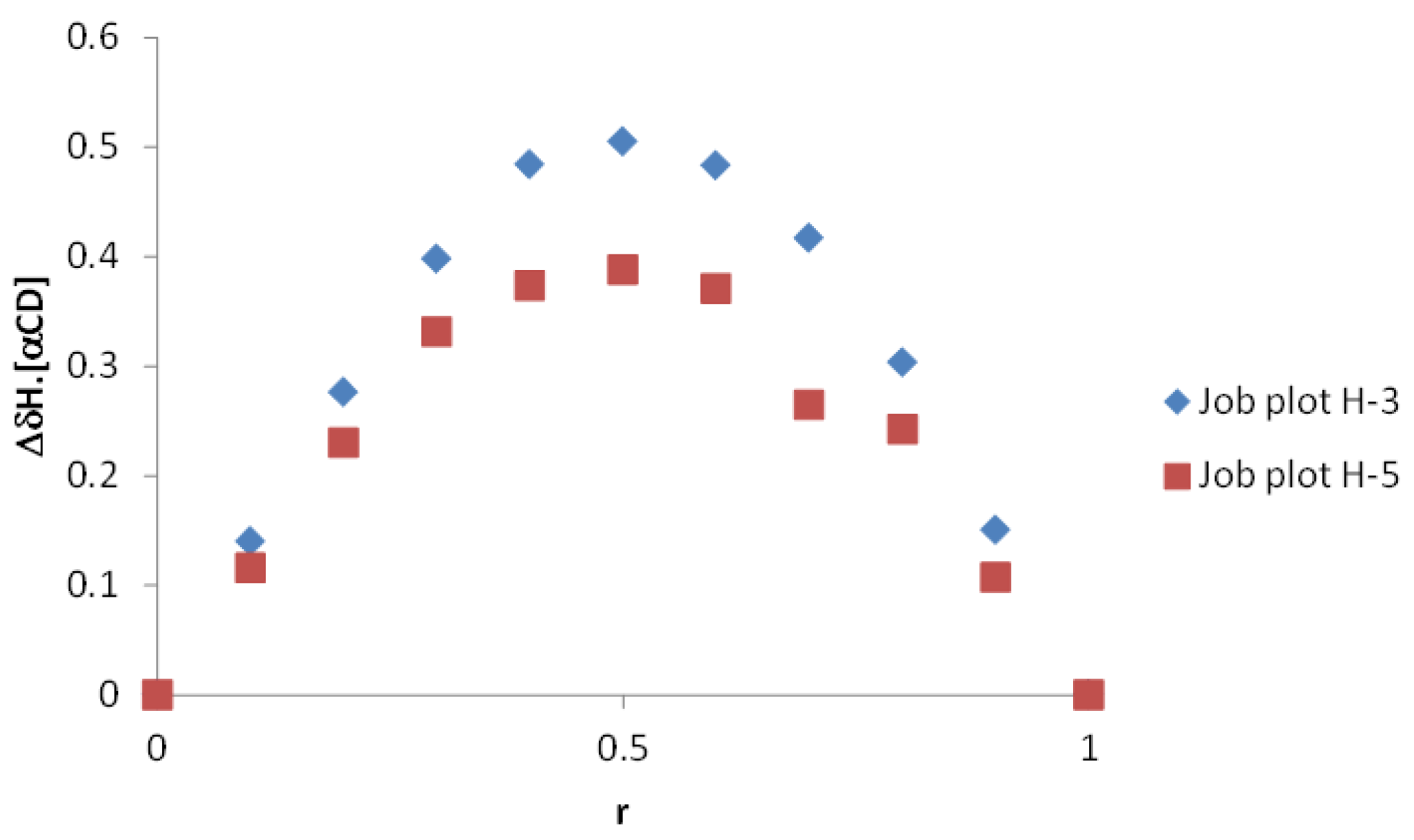
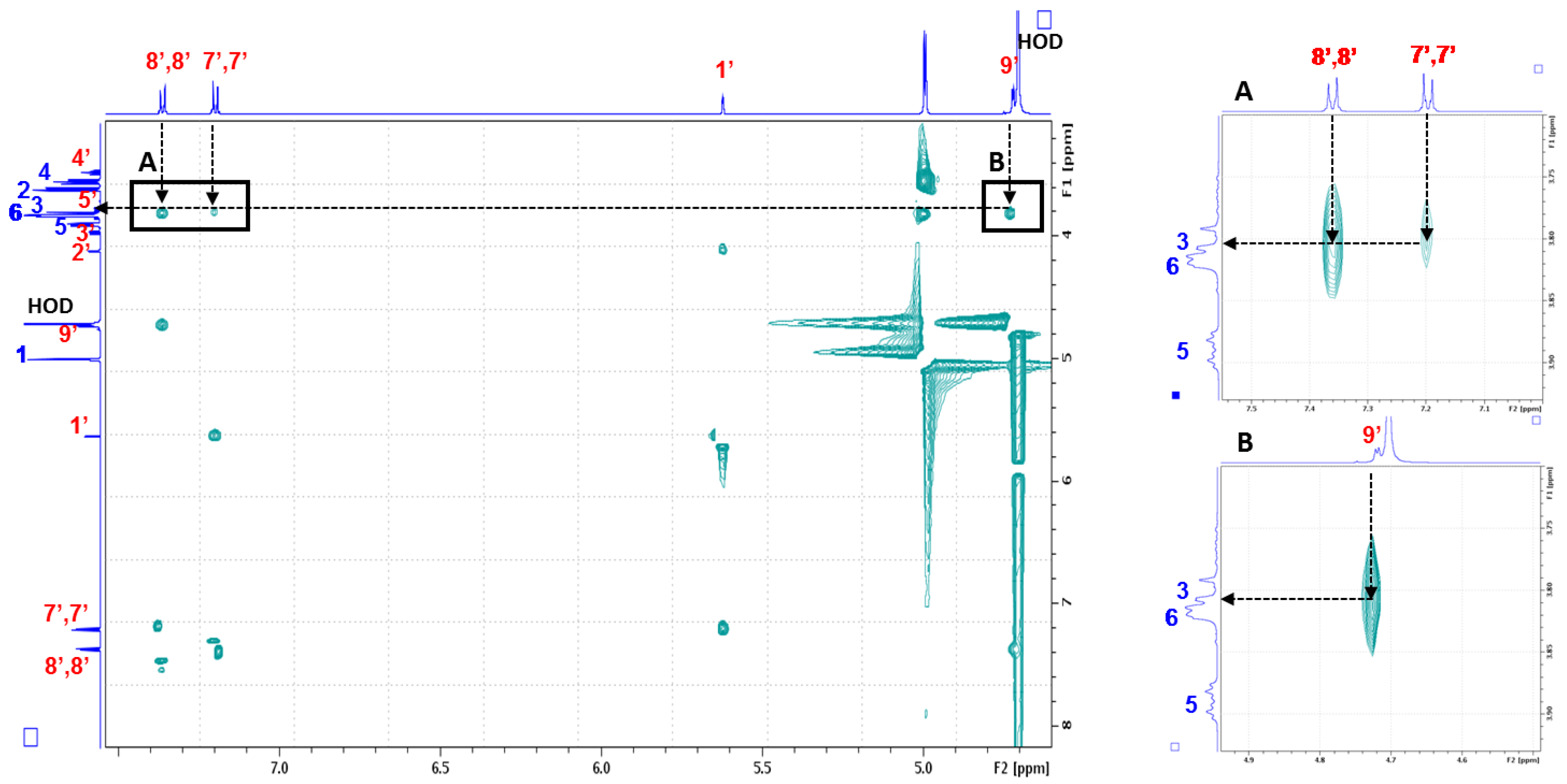
2.2.1. Evidence on the α-Cyclodextrin/Moringin (α-CD/MOR) Interactions
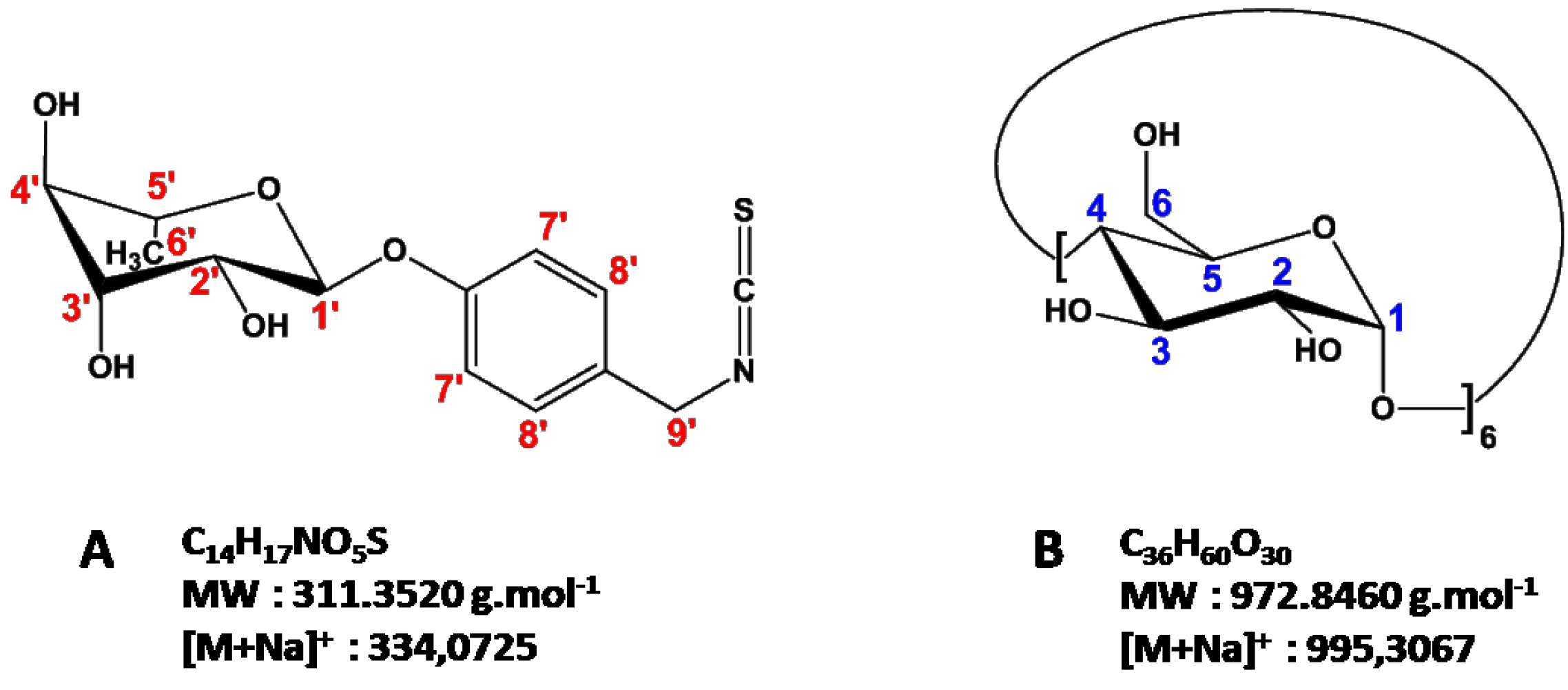
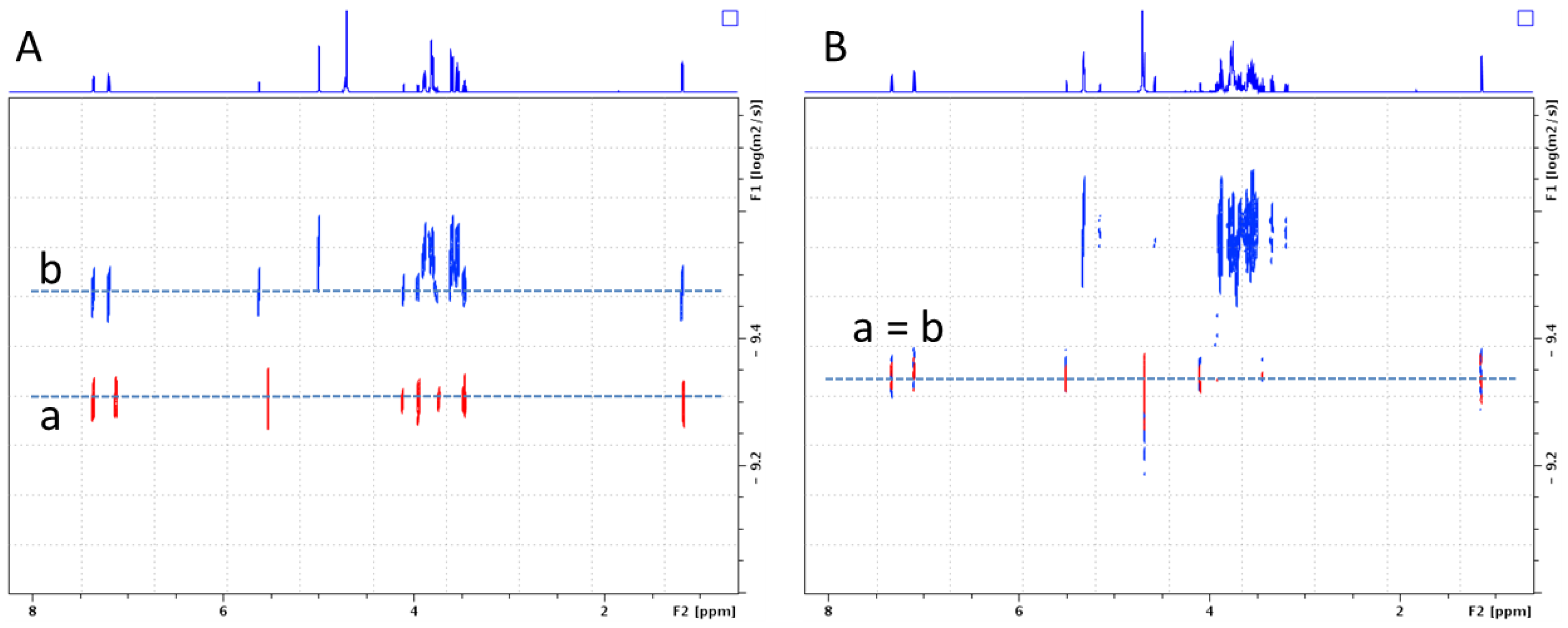
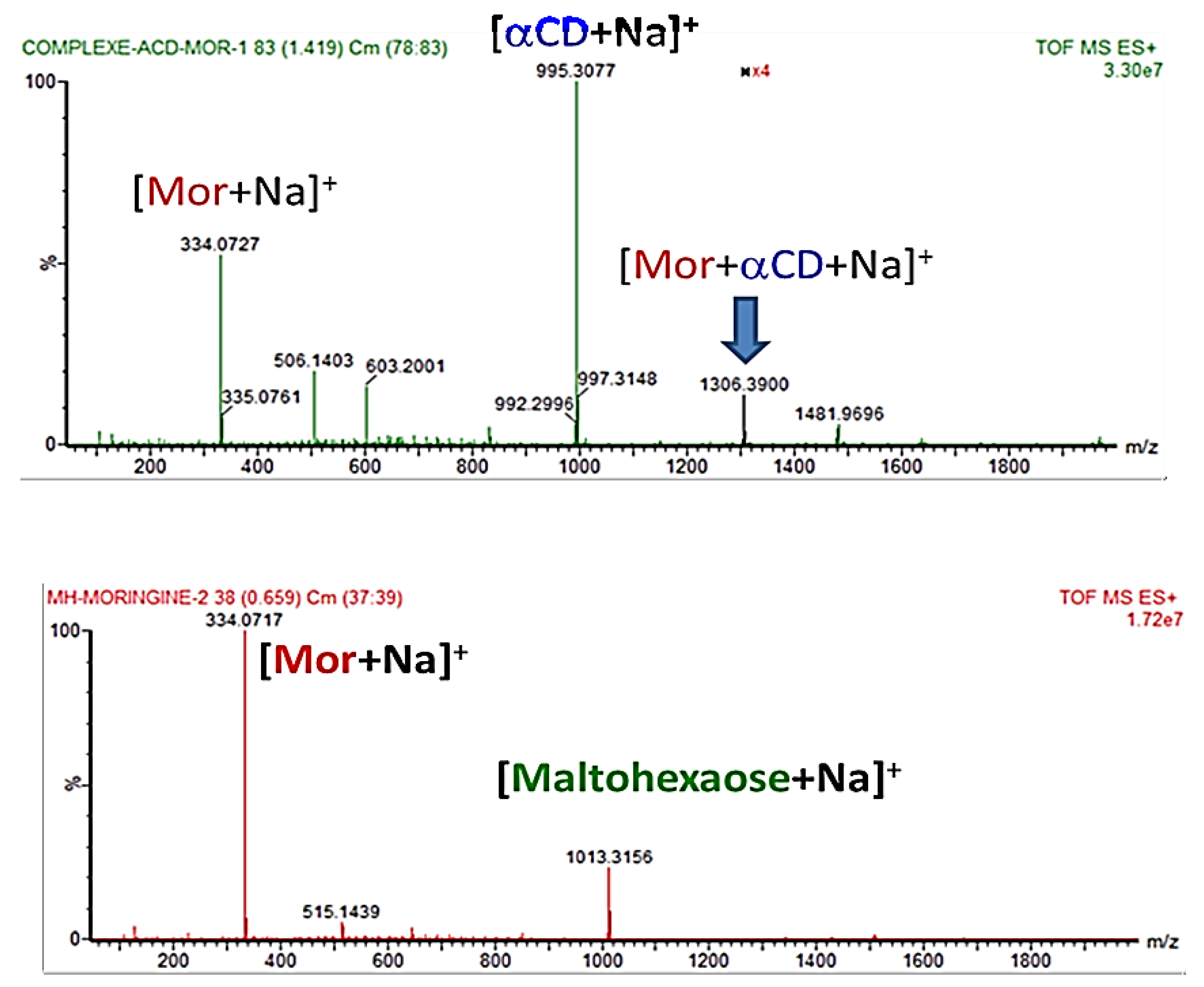
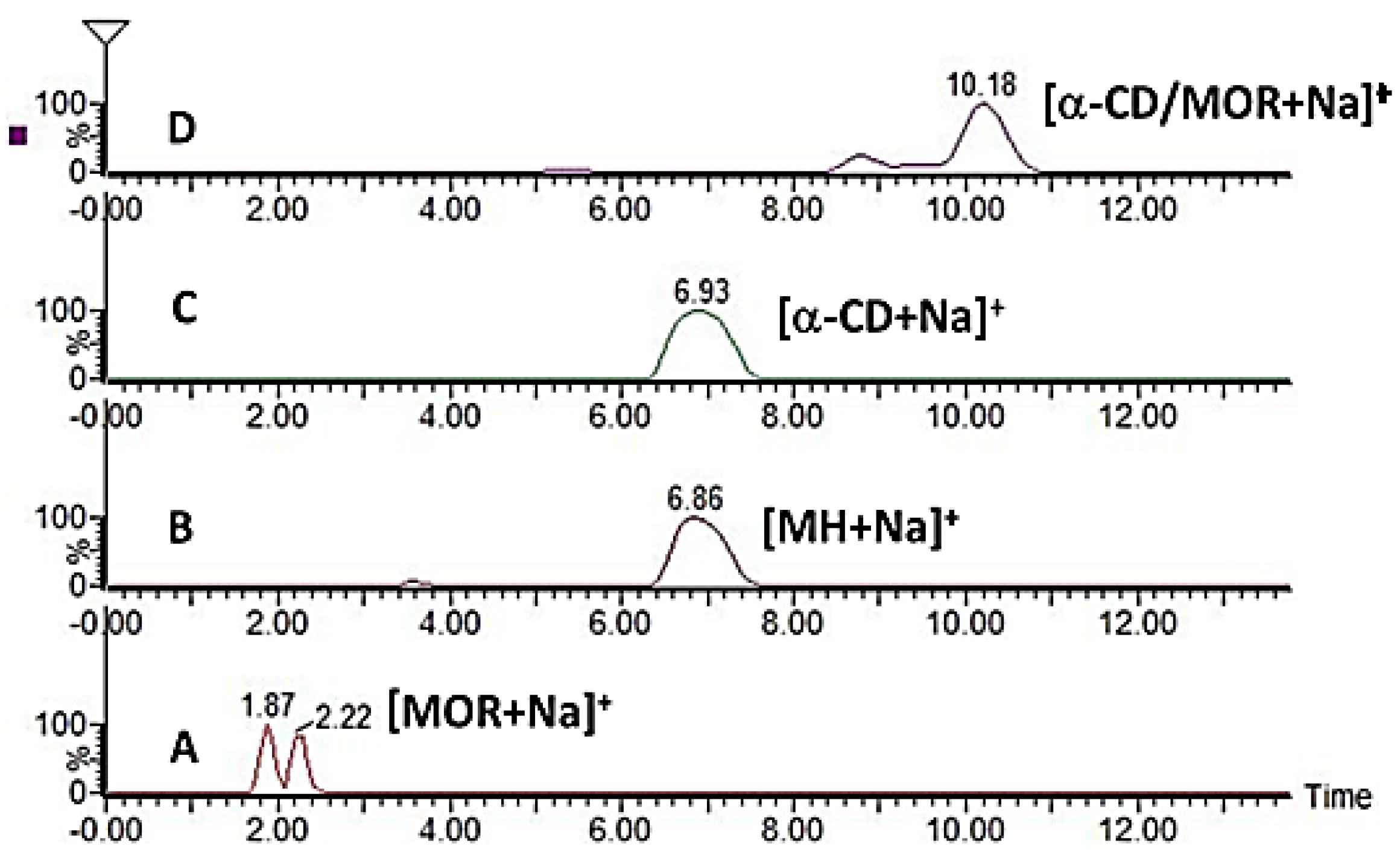
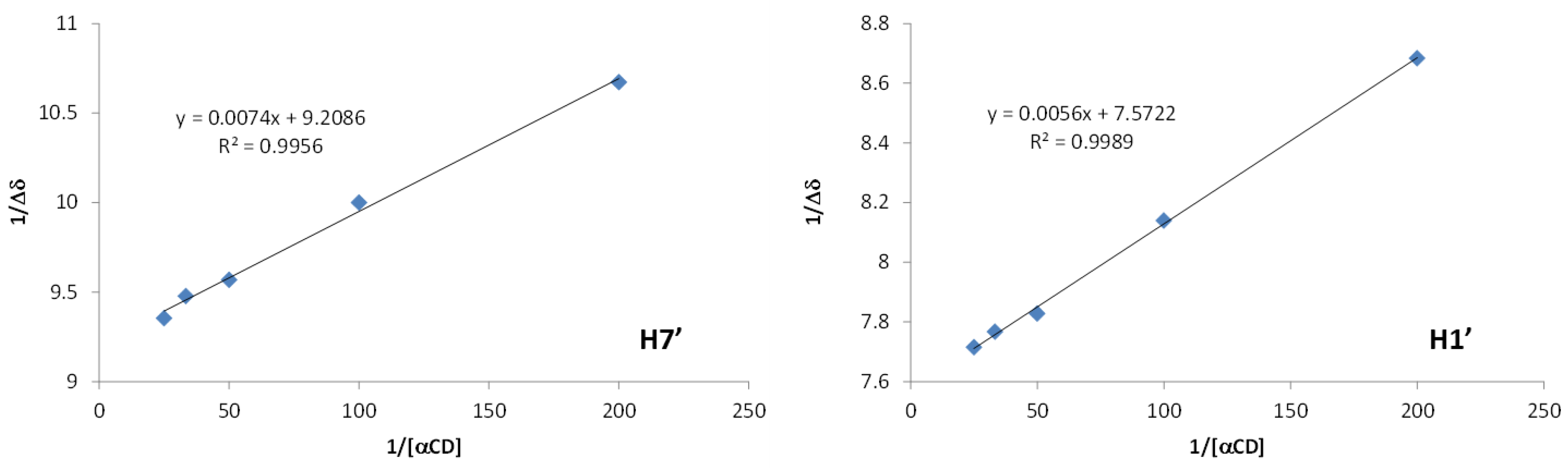
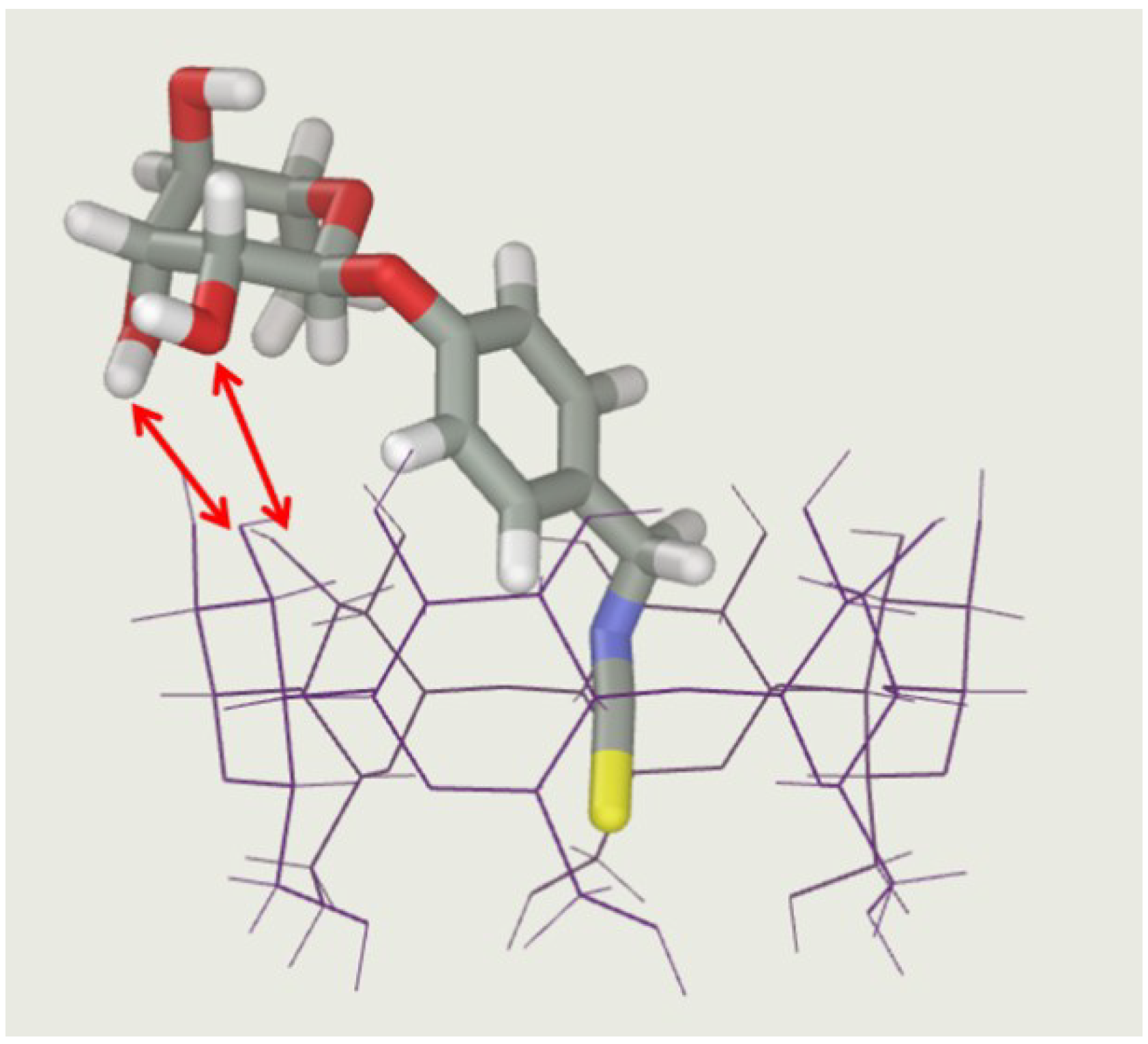
2.2.2. Characterization of the α-CD/MOR Inclusion Complex
3. Materials and Methods
3.1. Materials
3.2. In Vitro RAW Macrophages Culture Conditions and Drug Treatment
3.3. Eosin and Hematoxylin (E&H) Staining
3.4. Nuclear Magnetic Resonance (NMR) Studies
3.5. Solubility Studies
3.6. Determination of the Stoichiometry
3.7. Determination of the Associate Constant Value
3.8. Mass Spectrometry (MS) Studies
3.9. Modelization Studies
Author Contributions
Funding
Conflicts of Interest
References
- Montaut, S.; Rollin, P. Glucosinolates and Their Distribution; Juurlink, B.H.J., Ed.; Nova Science Publishers: New York, NY, USA, 2016; pp. 9–32. ISBN 978-1-63484-314-0. [Google Scholar]
- Blažević, I.; Montaut, S.; Burčul, F.; Rollin, P. Glucosinolates: Novel sources and biological potential. In Glucosinolates; Reference Series in Phytochemistry; Ramawat, K., Mérillon, J.-M., Eds.; Springer International Publishing: Basel, Switzerland, 2016; pp. 1–58. ISBN 978-3-319-25461-6. [Google Scholar]
- De Graaf, R.M.; Krosse, S.; Swolfs, A.E.M.; te Brinke, E.; Prill, N.; Leimu, R.; van Galen, P.M.; Wang, Y.; Aarts, M.G.M.; van Dam, N.M. Isolation and identification of 4-α-rhamnosyloxy benzyl glucosinolate in Noccaea caerulescens showing intraspecific variation. Phytochemistry 2015, 110, 166–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.; van Loon, J.; Ruschioni, S.; De Nicola, G.R.; Olsen, C.E.; Iori, R.; Agerbirk, N. Taste detection of the non-volatile isothiocyanate moringin results in deterrence to glucosinolate-adapted insect larvae. Phytochemistry 2015, 118, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Galuppo, M.; Montaut, S.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. An overview on neuroprotective effects of isothiocyanates for the treatment of neurodegenerative diseases. Fitoterapia 2015, 106, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Galuppo, M.; De Nicola, G.R.; Iori, R.; Bramanti, P.; Mazzon, E. 4(α-l-Rhamnosyloxy)-benzyl isothiocyanate, a bioactive phytochemical that attenuates secondary damage in an experimental model of spinal cord injury. Bioorg. Med. Chem. 2015, 23, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; Iori, R.; De Nicola, G.R.; Milardi, D.; Bramanti, P.; Mazzon, E. 4(α-l-Rhamnosyloxy)-benzyl isothiocyanate, a bioactive phytochemical that defends cerebral tissue and prevents severe damage induced by focal ischemia/reperfusion. J. Biol. Regul. Homeostatic Agents 2015, 29, 343–356. [Google Scholar]
- Michl, C.; Vivarelli, F.; Weigl, J.; De Nicola, G.R.; Canistro, D.; Paolini, M.; Iori, R.; Rascle, A. The Chemopreventive Phytochemical Moringin Isolated from Moringa oleifera Seeds Inhibits JAK/STAT Signaling. PLoS ONE 2016, 11, e0157430. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, D.; Tavecchio, M.; Falcioni, C.; Frapolli, R.; Erba, E.; Iori, R.; Rollin, P.; Barillari, J.; Manzotti, C.; Morazzoni, P.; et al. The isothiocyanate produced from glucomoringin inhibits NF-kB and reduces myeloma growth in nude mice in vivo. Biochem. Pharmacol. 2010, 79, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Rajan, T.S.; De Nicola, G.R.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. Anticancer activity of glucomoringin isothiocyanate in human malignant astrocytoma cells. Fitoterapia 2016, 110, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Eilert, U.; Wolters, B.; Nahrstedt, A. The Antibiotic Principle of Seeds of Moringa oleifera and Moringa stenopetala. Planta Med. 1981, 42, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; De Nicola, G.R.; Iori, R.; Dell’Utri, P.; Bramanti, P.; Mazzon, E. Antibacterial activity of glucomoringin bioactivated with myrosinase against two important pathogens affecting the health of long-term patients in hospitals. Molecules 2013, 18, 14340–14348. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; De Nicola, G.R.; Iori, R.; Navarra, M.; Lombardo, G.E.; Bramanti, P.; Mazzon, E. Antiinflammatory activity of glucomoringin isothiocyanate in a mouse model of experimental autoimmune encephalomyelitis. Fitoterapia 2014, 95, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Sicard-Roselli, C.; Perly, B.; Le Bas, G. The respective benefits of X-ray crystallography and NMR for the structural determination of the inclusion complex between butyl-isothiocyanate and alpha-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2001, 39, 333–337. [Google Scholar] [CrossRef]
- Ohta, Y.; Takatani, K.; Kawakishi, S. Kinetic and thermodynamic analyses of the cyclodextrin-allyl isothiocyanate inclusion complex in an aqueous solution. Biosci. Biotechnol. Biochem. 1999, 63, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, X.; Yang, Q.; Zhang, N.; Du, Y.; Zhu, H. Preparation and characterization of inclusion complex of benzyl isothiocyanate extracted from papaya seed with b-cyclodextrin. Food Chem. 2015, 184, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Rajan, T.S.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. The α-cyclodextrin complex of the Moringa isothiocyanate suppresses lipopolysaccharide-induced inflammation in RAW 264.7 macrophage cells through Akt and p38 inhibition. Inflamm. Res. 2017, 66, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Maldini, M.; Maksoud, S.A.; Natella, F.; Montoro, P.; Petretto, G.L.; Foddai, M.; De Nicola, G.R.; Chessa, M.; Pintorea, G. Moringa oleifera: Study of phenolics and glucosinolates by mass spectrometry. J. Mass Spectrom. 2014, 49, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Roselli, C.; Perly, B.; Cassel, S.; Rollin, P.; Iori, R.; Manici, L.; Palmieri, S. Cyclodextrin assistance in the enzymatic degradation of the moringa glucosinolate. In Proceedings of the Ninth International Cyclodextrin Symposium, Santiago de Compostela, Spain, 31 May–3 June 1998; pp. 533–536. [Google Scholar]
- Giacoppo, S.; Rajan, T.S.; De Nicola, G.R.; Iori, R.; Bramanti, P.; Mazzon, E. Moringin activates Wnt canonical pathway by inhibiting GSK3β in a mouse model of experimental autoimmune encephalomyelitis. Drug Des. Devel. Ther. 2016, 10, 3291–3304. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Rajan, T.S.; De Nicola, G.R.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. The isothiocyanate isolated from Moringa oleifera shows potent anti-inflammatory activity in the treatment of murine subacute Parkinson’s disease. Rejuvenation Res. 2017, 20, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Roselli, C.; Perly, B.; Rollin, P. Complexes for Immobilising Isothiocyanate Natural Precursors in Cyclodextrins. Preparation and Use. U.S. Patent 6,716,827, 6 April 2014. [Google Scholar]
- Oliva, E.; Mathiron, D.; Bertaut, E.; Landy, D.; Cailleu, D.; Pilard, S.; Clément, C.; Courot, E.; Bonnet, V.; Djedaïni-Pilard, F. Physico-chemical studies of resveratrol, methyl-jasmonate and cyclodextrin interactions: an approach to resveratrol bioproduction optimization. RSC Adv. 2018, 8, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Gulbakan, B.; Weidmann, S.; Fagerer, S.R.; Ibanez, A.J.; Zenobi, R. Applying mass spectrometry to study non-covalent biomolecule complexes. Mass Spectrom. Rev. 2016, 35, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Galic, N.; De Pauw, E. On the specificity of cyclodextrin complexes detected by electrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 2002, 13, 946–953. [Google Scholar] [CrossRef] [Green Version]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Zimnicka, M.; Troc, A.; Ceborska, M.; Jakubczak, M.; Kolinski, M.; Danikiewicz, W. Structural elucidation of specific noncovalent association of folic acid with native cyclodextrins using an ion mobility mass spectrometry and theoretical approach. Anal. Chem. 2014, 86, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Mathiron, D.; Marçon, F.; Dubaele, J.-M.; Cailleu, D.; Pilard, S.; Djedaïni-Pilard, F. Benefits of methylated cyclodextrins in the development of midazolam pharmaceutical formulations. J. Pharm. Sci. 2013, 102, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.F.; Jiang, Z.T.; Li, R. Complexation of allyl isothiocyanate with β-cyclodextrin and its derivatives and molecular microcapsule of allyl isothiocyanate in β-cyclodextrin. Eur. Food Res. Technol. 2007, 225, 407–413. [Google Scholar] [CrossRef]
- Péan, C.; Creminon, C.; Wijkhuisen, A.; Perly, B.; Djedaïni-Pilard, F. Reliable NMR experiments for the study of β-cyclodextrin/prostaglandin E2 inclusion complex. J. Chim. Phys. 1999, 96, 1486–1493. [Google Scholar] [CrossRef]
- Sforza, S.; Galaverna, G.; Corradini, R.; Dossena, A.; Marchelli, R. ESI-mass spectrometry analysis of unsubstituted and disubstituted β-cyclodextrins: Fragmentation mode and identification of the AB, AC, AD regioisomers. J. Am. Soc. Mass Spectrom. 2003, 14, 124–135. [Google Scholar] [CrossRef]
- Mele, A.; Panzeri, W.; Selva, A. Fast-atom bombardment mass spectrometric and tandem mass spectrometric study of (−)-menthol-β-(d)-glucopyranoside, neohesperidin dihydrochalcone and their non-covalent association with β-cyclodextrin. Two examples of interaction of a carbohydrate host with glycoconjugate guests. Eur. Mass Sprectom. 1997, 3, 347–354. [Google Scholar]
- Qi, Y.; Volmer, D.A. β-Cyclodextrin affects the formation of isomerization products during peptide deamidation. Eur. Mass Sprectom. 2015, 21, 701–705. [Google Scholar]
- Jurkowska, H.; Wrobel, M.; Szlezak, D.; Jasek-Gajda, E. New aspects of antiproliferative activity of 4-hydroxybenzyl isothiocyanate, a natural H2S-donor. Amino Acids 2018, 50, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Ekanayake, A.; Zoutendam, P.H.; Strife, R.J.; Fu, X.; Jayatilake, G.S. Development of white mustard (Sinapis alba L.) essential oil, a food preservative. Food Chem. 2012, 133, 767–774. [Google Scholar] [CrossRef]
- Borek, V.; Morra, M.J. Ionic Thiocyanate (SCN−) Production from 4-hydroxybenzyl glucosinolate contained in Sinapis alba seed meal. J. Agric. Food Chem. 2005, 53, 8650–8654. [Google Scholar] [CrossRef] [PubMed]
- Pessina, A.; Thomas, R.M.; Palmieri, S.; Luisi, P.L. An improved method for the purification of myrosinase and its physicochemical characterization. Arch. Biochem. Biophys. 1990, 280, 383–389. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds glucomoringin and moringin are available from the authors. |













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathiron, D.; Iori, R.; Pilard, S.; Soundara Rajan, T.; Landy, D.; Mazzon, E.; Rollin, P.; Djedaïni-Pilard, F. A Combined Approach of NMR and Mass Spectrometry Techniques Applied to the α-Cyclodextrin/Moringin Complex for a Novel Bioactive Formulation †. Molecules 2018, 23, 1714. https://doi.org/10.3390/molecules23071714
Mathiron D, Iori R, Pilard S, Soundara Rajan T, Landy D, Mazzon E, Rollin P, Djedaïni-Pilard F. A Combined Approach of NMR and Mass Spectrometry Techniques Applied to the α-Cyclodextrin/Moringin Complex for a Novel Bioactive Formulation †. Molecules. 2018; 23(7):1714. https://doi.org/10.3390/molecules23071714
Chicago/Turabian StyleMathiron, David, Renato Iori, Serge Pilard, Thangavelu Soundara Rajan, David Landy, Emanuela Mazzon, Patrick Rollin, and Florence Djedaïni-Pilard. 2018. "A Combined Approach of NMR and Mass Spectrometry Techniques Applied to the α-Cyclodextrin/Moringin Complex for a Novel Bioactive Formulation †" Molecules 23, no. 7: 1714. https://doi.org/10.3390/molecules23071714
APA StyleMathiron, D., Iori, R., Pilard, S., Soundara Rajan, T., Landy, D., Mazzon, E., Rollin, P., & Djedaïni-Pilard, F. (2018). A Combined Approach of NMR and Mass Spectrometry Techniques Applied to the α-Cyclodextrin/Moringin Complex for a Novel Bioactive Formulation †. Molecules, 23(7), 1714. https://doi.org/10.3390/molecules23071714