Facile Preparation of N-Glycosylated 10-Piperazinyl Artemisinin Derivatives and Evaluation of Their Antimalarial and Cytotoxic Activities

 , ,
, ,  , and
, and 
Abstract
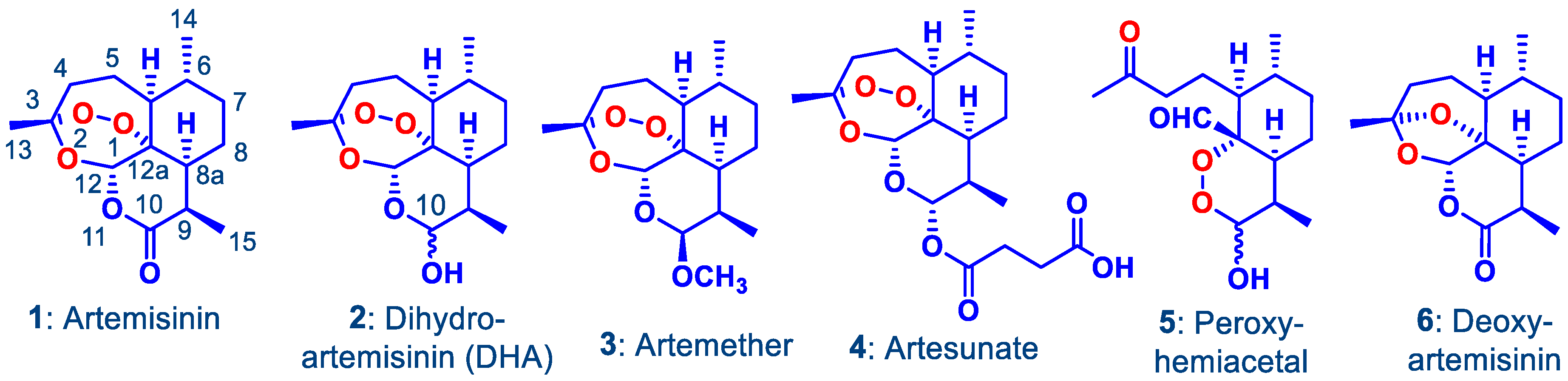
:1. Introduction
2. Results and Discussion
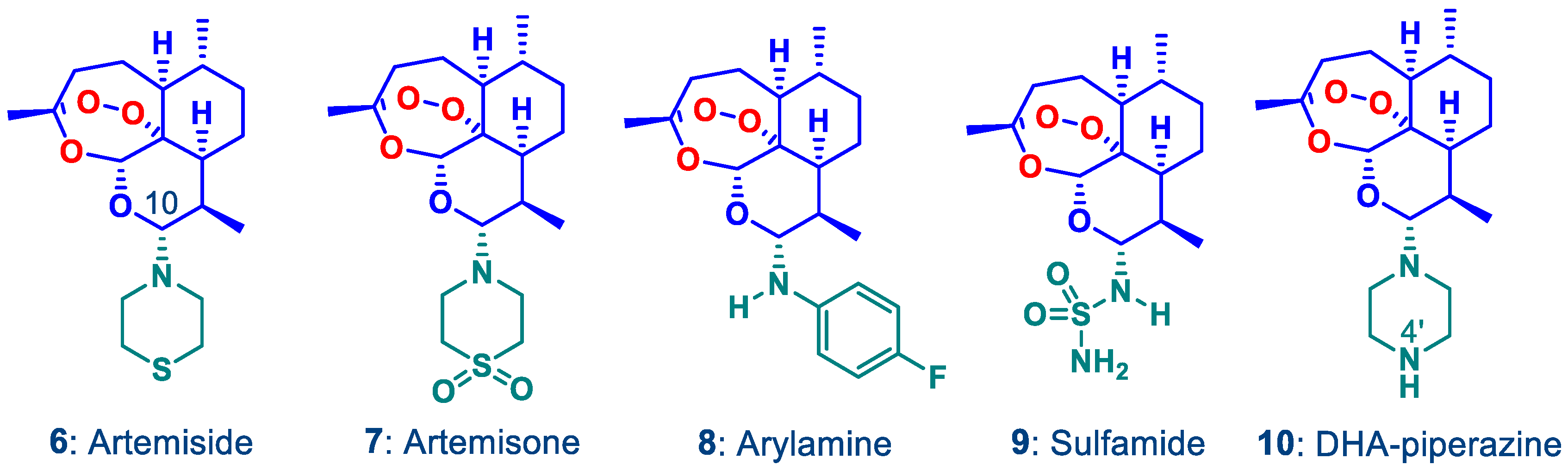
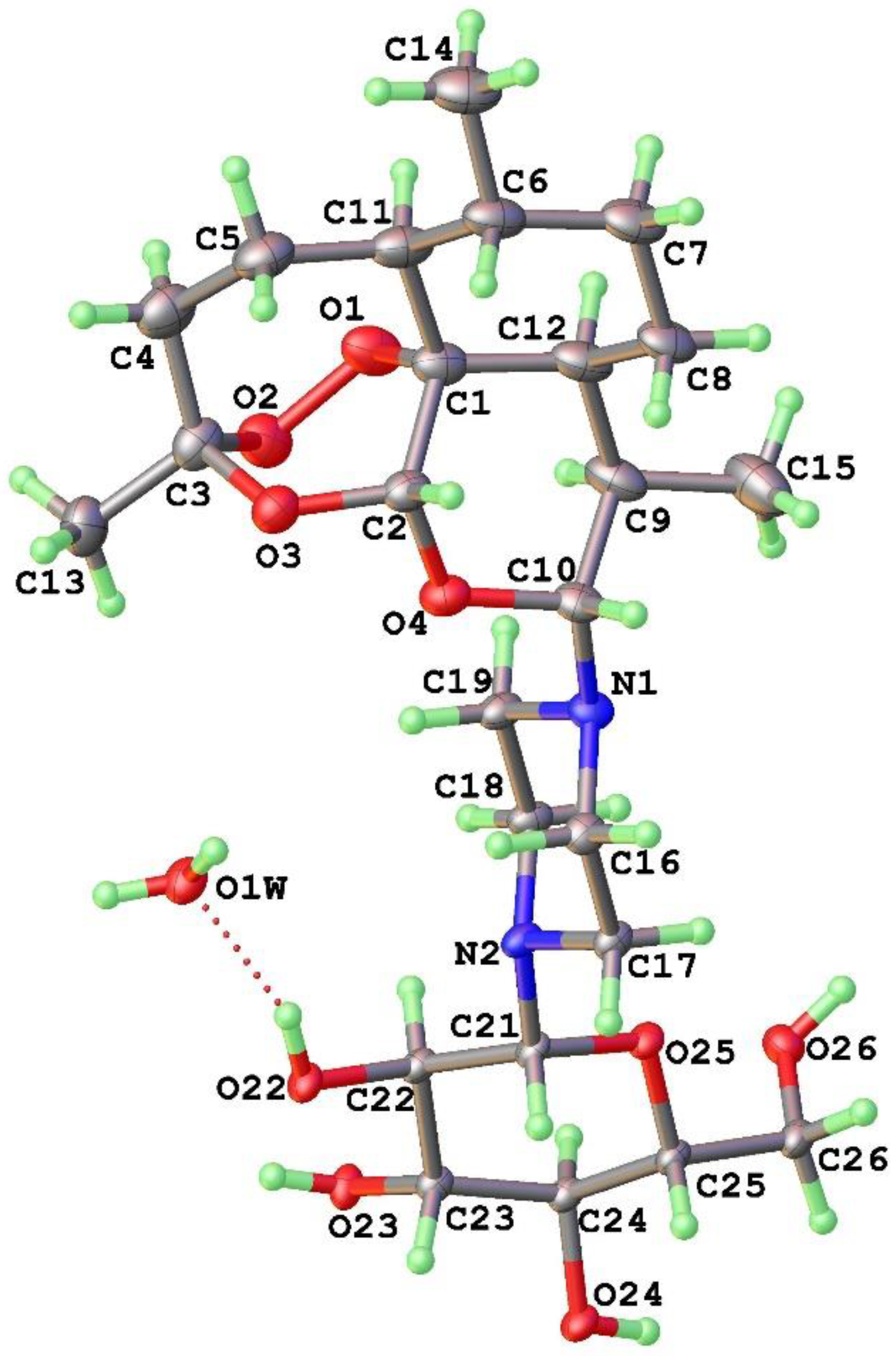
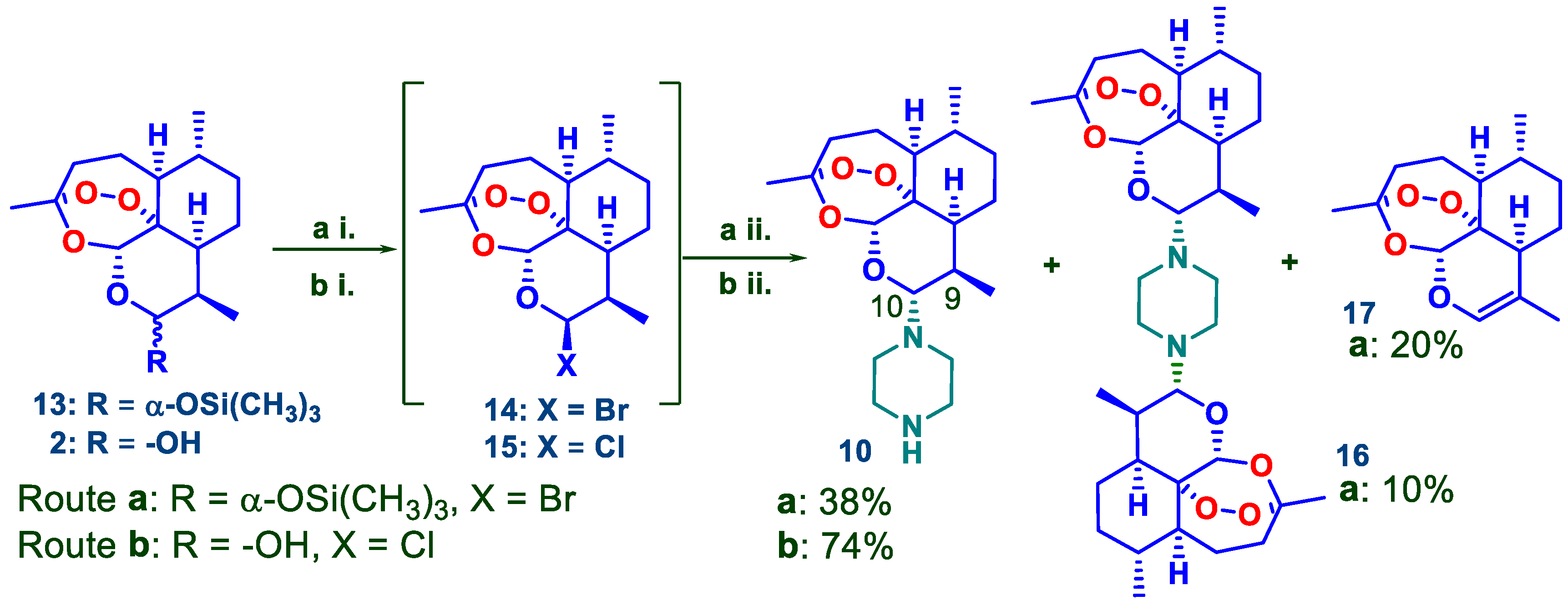
2.1. Synthetic Chemistry: Preparation of DHA-Piperazine Derivative 10
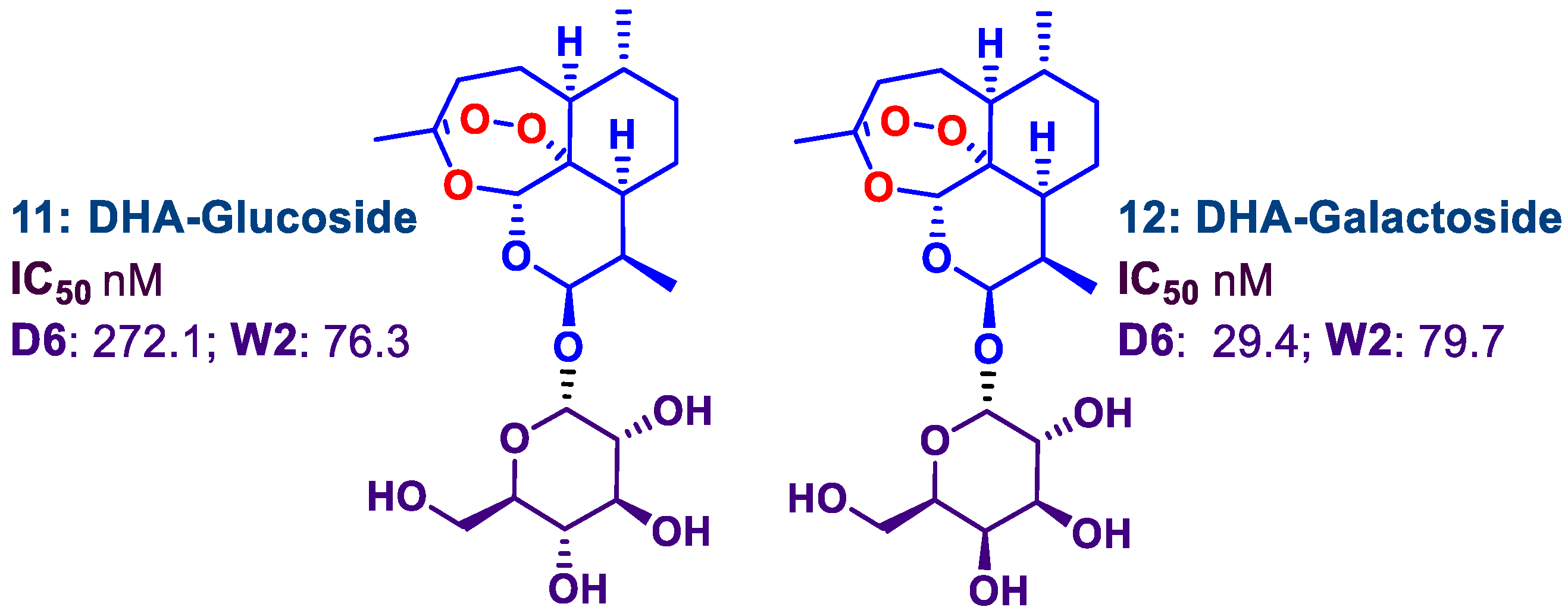
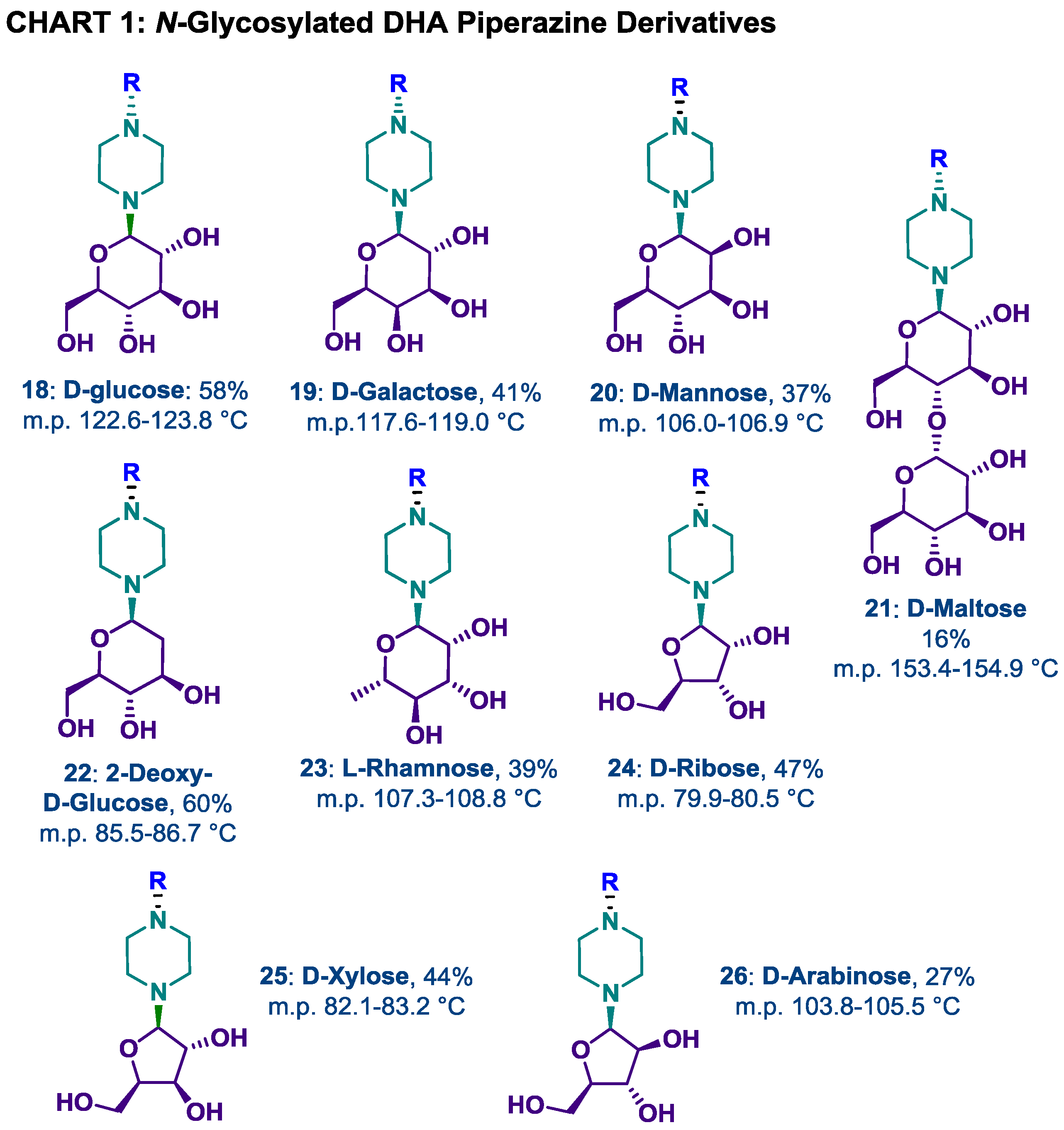
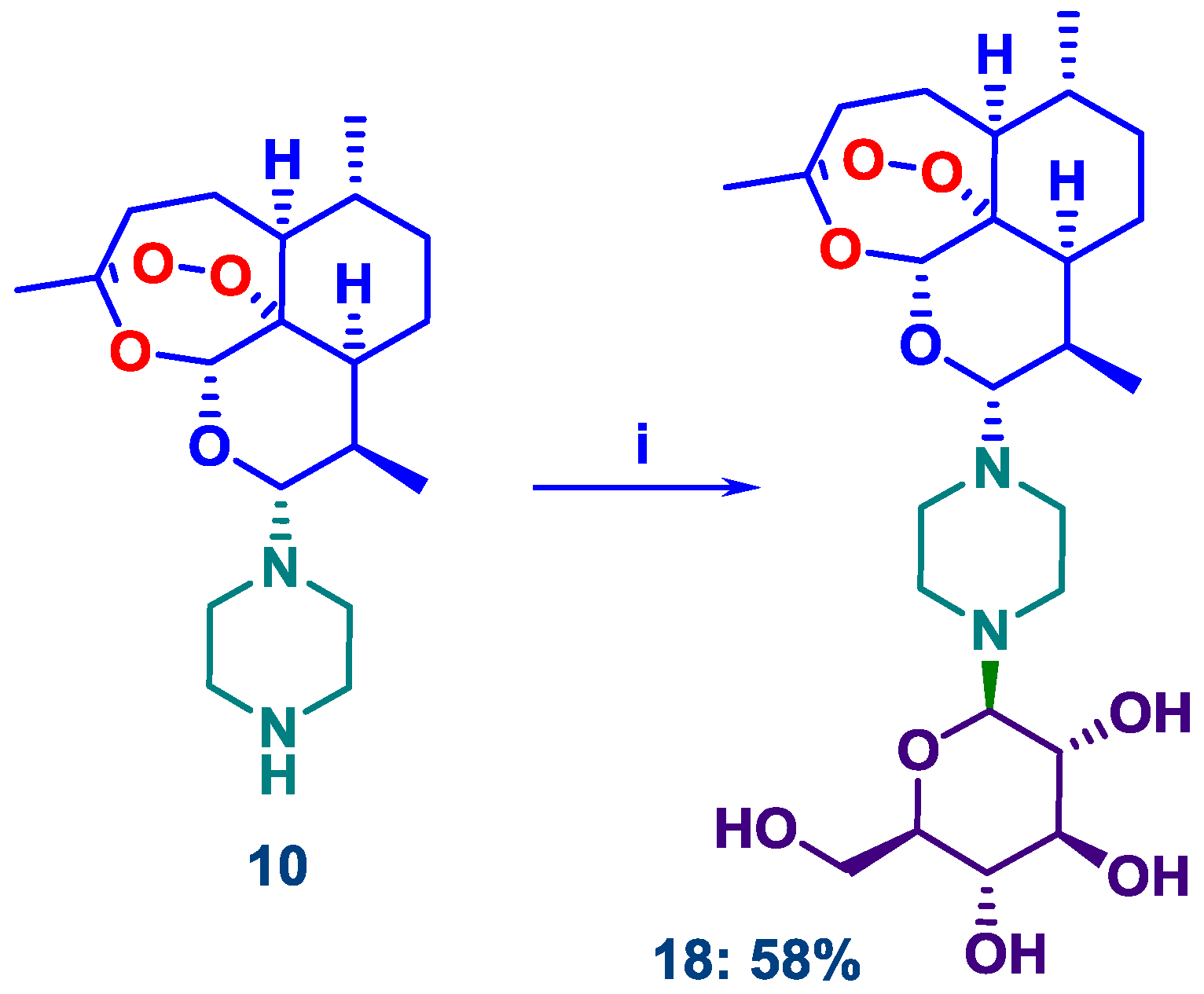
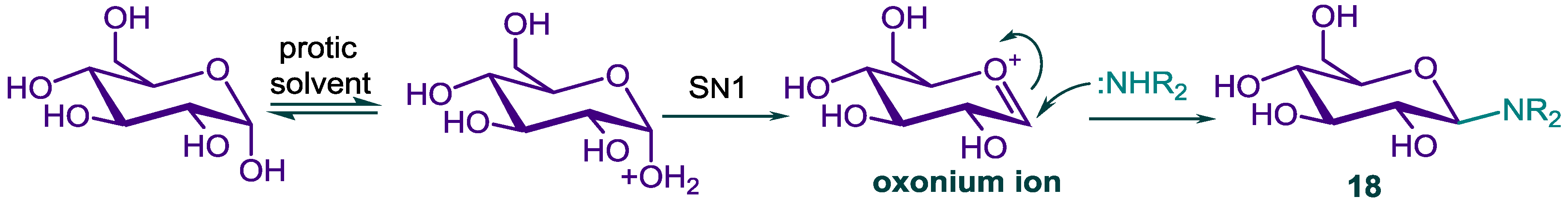
2.2. N-Glycosylation of DHA-Piperazine 10
2.3. Antimalarial Activities against Different P. falciparum Strains and Cytotoxicities
3. Conclusions
4. Materials and Methods
4.1. General
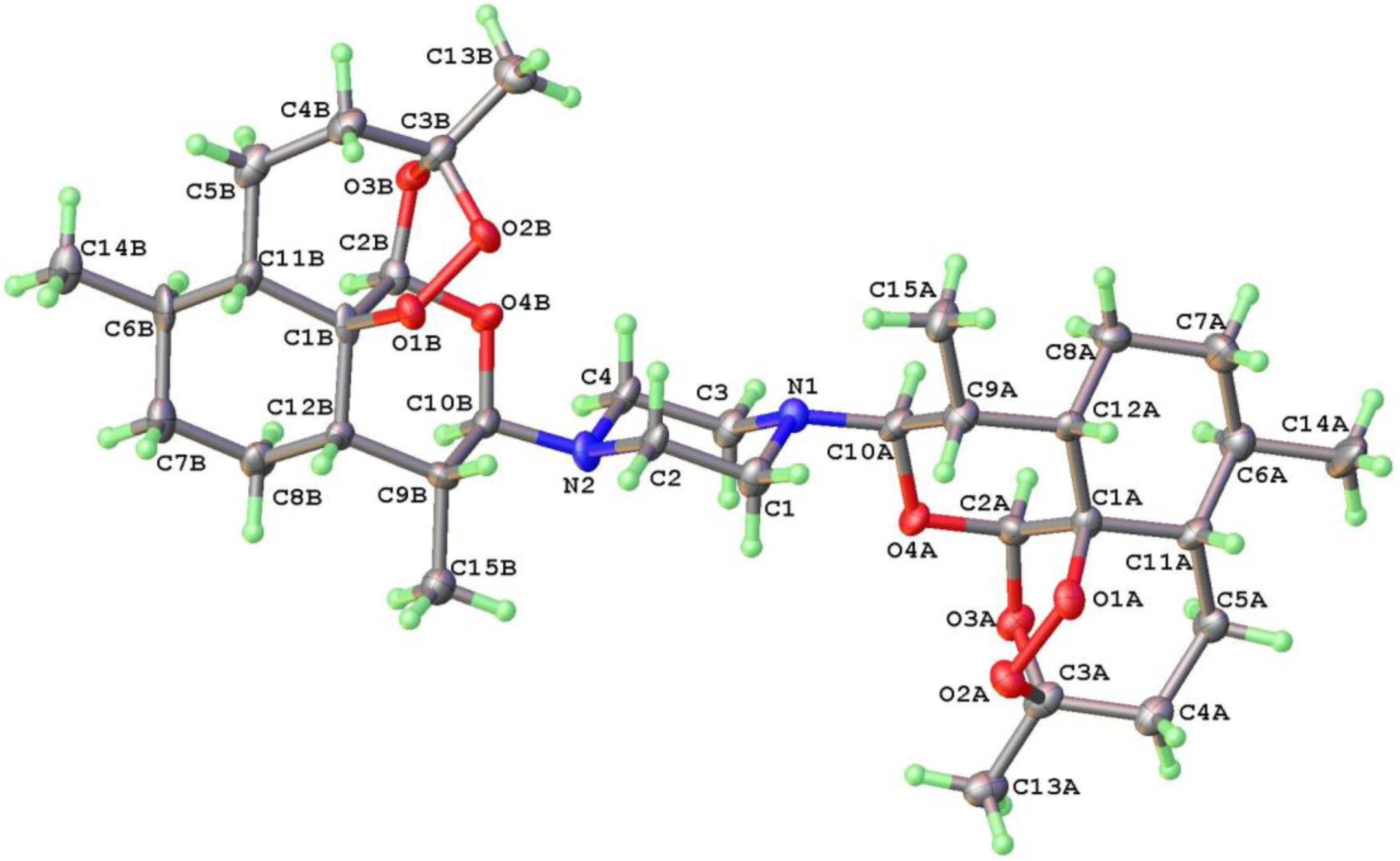
4.2. Synthesis of Glycoside Derivatives: 10α-(1-Piperazino)-10-deoxo-10-dihydroartemisinin 10 and (bis-11′,4-piperazino)-10α-deoxo-10-dihydroartemisinin 16
4.3. Synthesis of Glycoside Derivatives
4.3.1. D-Glucose
4.3.2. D-Galactose
4.3.3. D-Mannose
4.3.4. D-Maltose
4.3.5. 2-Deoxy-D-glucose
4.3.6. L-Rhamnose
4.3.7. D-Ribose
4.3.8. D-Xylose
4.3.9. D-Arabinose
4.4. Biological Assays
4.4.1. Cultures of Pf and In Vitro Antimalarial Assays
4.4.2. Cell Cytotoxicity Assays
4.4.3. IC50 Calculation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Geneva 2016 World Malaria Report; WHO: Geneva, Switzerland, 2016; ISBN 978-92-4-151171-1. [Google Scholar]
- Navaratnam, V.; Mansor, S.M.; Sit, N.W.; Grace, J.; Li, Q.; Olliaro, P. Pharmacokinetics of artemisinin-type compounds. Clin. Pharmacokinet. 2000, 39, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.W.; Lung, C.M.; Ng, N.C.; Wong, H.N.; Shek, L.Y.; Williams, I.D.; Gomes, M.F.; Cartwright, A. Artesunate and dihydroartemisinin (DHA): Unusual decomposition products formed under mild conditions and comments on the fitness of DHA as an antimalarial drug. ChemMedChem 2007, 2, 1448–1463. [Google Scholar] [CrossRef] [PubMed]
- Olliaro, P.L.; Nair, N.K.; Haynes, R.K.; Tang, M.M.K.; Cheu, C.K.W.; Zanolari, B.; Decosterd, L.A.; Navaratnam, V. Unpublished work.
- Parapini, S.; Olliaro, P.; Navaratnam, V.; Taramelli, D.; Basilico, N. Stability of the antimalarial drug dihydroartemisinin under physiologically-relevant conditions: Implications for clinical treatment, pharmacokinetic and in vitro assays. Antimicrob. Agents Chemother. 2015, 59, 4046–4052. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.H. The pharmaceutical death-ride of dihydroartemisinin. Malar. J. 2010, 9, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, P.M.; Scheinmann, F.; Stachulski, A.V.; Maggs, J.L.; Park, B.K. Efficient preparations of the ß-glucuronides of dihydroartemisinin and structural confirmation of the human glucuronide metabolite. J. Med. Chem. 2001, 44, 1467–1470. [Google Scholar] [CrossRef] [PubMed]
- Ilett, K.F.; Ethell, B.T.; Maggs, J.L.; Davis, T.M.E.; Batty, K.T.; Burchell, B.; Binh, T.Q.; Thu, L.T.A.; Hung, N.C.; Pirmohamed, M.; et al. Glucuronidation of dihydro-artemisinin in vivo and by human liver microsomes and expressed UDP-glucuronosyltransferases. Drug Metab. Dispos. 2002, 30, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.-S.; Fleckenstein, L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batty, K.T.; Ilett, K.F.; Powell, S.M.; Martin, J.; Davis, T.M.E. Relative bioavailability of artesunate and dihydroartemisinin: Investigations in the isolated perfused rat liver and in healthy Caucasian volunteers. Am. J. Trop. Med. Hyg. 2002, 66, 130–136. [Google Scholar] [CrossRef]
- Van Agtmael, M.A.; Gupta, V.; van der Wösten, T.H.; Rutten, J.-P.B.; van Boxtel, C.J. Grapefruit juice increases the bioavailability of artemether. Eur. J. Clin. Pharmacol. 1999, 55, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Wesche, D.L.; DeCoster, M.A.; Tortella, F.C.; Brewer, T.G. Neurotoxicity of artemisinin analogs in vitro. Antimicrob. Agents Chemother. 1994, 38, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, G.; Roehrdanz, E.; Haynes, R.K.; Kahl, R. Neurotoxic mode of action of artemisinin. Antimicrob. Agents. Chemother. 2002, 46, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, G.; Temerowski, M.; Haynes, R.K.; Fugmann, B. Identification of non-neurotoxic artemisinin derivatives in vivo and in vitro. Res. Adv. Antimicrob. Agents Chemother. 2003, 3, 35–47. [Google Scholar]
- Paloque, L.; Ramadani, A.P.; Mercereau-Puijalon, O.; Augereau, J.M.; Benoit-Vical, F. Plasmodium falciparum: Multifaceted resistance to artemisinins. Malar. J. 2016, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Culleton, R.; Zhang, M.; Ramaprasad, A.; von Seidlein, L.; Zhou, H.; Zhu, G.; Tang, J.; Liu, Y.; Wang, W.; et al. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N. Engl. J. Med. 2017, 376, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K. From artemisinin to new artemisinin antimalarials: Biosynthesis, extraction, old and new derivatives, stereochemistry and medicinal chemistry requirements. Curr. Top. Med. Chem. 2006, 6, 509–537. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.-W.; Ho, W.-Y.; Ko, C.K.F.; Gerena, L.; Kyle, D.E.; Peters, W.; Robinson, B.L. Convenient access both to highly antimalaria active 10-arylaminoartemisinins, and to 10-alkyl ethers including artemether, arteether, and artelinate. ChemBioChem 2005, 6, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Ho, W.-Y.; Chan, H.-W.; Fugmann, B.; Stetter, J.; Croft, S.L.; Vivas, L.; Peters, W.; Robinson, B.L. Highly antimalaria-active artemisinin derivatives: Biological activity does not correlate with chemical reactivity. Angew. Chem. Int. Ed. 2004, 43, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Fugmann, B.; Stetter, J.; Rieckmann, K.; Heilmann, H.D.; Chan, H.W.; Cheung, M.K.; Lam, W.L.; Wong, H.N.; Croft, S.L.; et al. Artemisone-a highly active antimalarial drug of the artemisinin class. Angew. Chem. Int. Ed. 2006, 45, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K. Strategies in the development and chemical modification of the new artemisinin antimalarial artemisone. Synform 2008, 3, A34–A36. [Google Scholar]
- Haynes, R.K.; Wong, H.N.; Wu, Y.; Wu, W.K.; Cheu, K.W.; Williams, I.D.; Krishna, S.; Slavic, K.; Gravett, A.M.; Liu, W.M. Methylene homologues of artemisone: An unexpected structure–activity relationship and a possible implication for the design of C10-substituted artemisinins. ChemMedChem 2016, 11, 1469–1479. [Google Scholar]
- Ramharter, M.; Burkhardt, D.; Nemeth, J.; Adegnika, A.A.; Kremsner, P.G. In vitro activity of artemisone compared with artesunate against Plasmodium falciparum. Am. J. Trop. Med. Hyg. 2006, 75, 637–639. [Google Scholar] [PubMed]
- Vivas, L.; Rattray, L.; Stewart, L.B.; Robinson, B.; Fugmann, B.; Haynes, R.K.; Peters, W.; Croft, S.L. Antimalarial efficacy and drug interactions of the novel semi-synthetic endoperoxide artemisone in vitro and in vivo. J. Antimicrob. Chemother 2007, 59, 658–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeer, K.; Breyer, M.; Hucke, K.; Gaefke, D.; Kern, A.; Schmuck, G. Artemifone, a New Anti-Malarial Artemisinin Derivative: Metabolism In Vitro in Liver Microsomes and Hepatocyte Sandwich Cultures of Different Species Including Man. Presentation PO63. In Proceedings of the XVI International Congress for Tropical Medicine and Malaria: Medicine and Health in the Tropics, Marseille, France, 11–15 September 2005; Abstract Book, O-063. p. 144. [Google Scholar]
- Krudsood, S.; Wilairatana, P.; Chalermrut, K.; Leowattana, W.; Voith, B.; Hampel, B.; Looareesuwan, S. Artemifone, a New Anti-Malarial Artemisinin Derivative: Open Pilot Trial to Investigate the Antiparasitic Activity of Bay 44-9585 in Patients with Uncomplicated P. falciparum Malaria. In Proceedings of the XVI International Congress for Tropical Medicine and Malaria: Medicine and Health in the Tropics, Marseille, France, 11–15 September 2005; Abstract Book, P054. p. 142. [Google Scholar]
- Nagelschmitz, J.; Voith, B.; Römer, A. Artemifone, A New Anti- Malaria Artemisinin Derivative- Safety And Pharmacokinetics In Healthy Male Subjects. In Proceedings of the XVI International Congress for Tropical Medicine and Malaria: Medicine and Health in the Tropics, Marseille, France, 11–15 September 2005; Abstract Book, P047. p. 142. [Google Scholar]
- Nagelschmitz, J.; Voith, B.; Wensing, G.; Roemer, A.; Fugmann, B.; Haynes, R.K.; Kotecka, B.M.; Rieckmann, K.H.; Edstein, M.D. First assessment in humans of the safety, tolerability, pharmacokinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone Antimicrob. Agents Chemother. 2008, 52, 3085–3091. [Google Scholar] [CrossRef] [PubMed]
- Na-Bangchang, K.; Krudsood, S.; Silachamroon, U.; Molunto, P.; Tasanor, O.; Chalermrut, K.; Tangpukdee, N.; Matangkasombut, O.; Kano, S.; Looareesuwan, S. The pharmacokinetics of oral dihydroartemisinin and artesunate in healthy Thai volunteers. Southeast Asian J. Trop. Med. Public Health 2004, 35, 575–582. [Google Scholar] [PubMed]
- http://www.artemis-therapeutics.com/news/ (accessed on 30 June 2018).
- Efferth, T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr. Drug Targets 2006, 7, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Lai, H.; Singh, N.P.; Sasaki, T. Anticancer properties of artemisinin derivatives and their targeted delivery by transferrin conjugation. Int. J. Pharm. 2008, 354, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor activity of artemisinin and its derivatives: From a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012, 247597. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K. Anticancer effect of antimalarial artemisinin compounds. Ann. Med. Health Sci. Res. 2015, 5, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Ganapathi, S.; Ster, I.C.; Saeed, M.E.M.; Cowan, M.; Finlayson, C.; Kovacsevics, H.; Jansen, H.; Kremsner, P.G.; Efferth, T.; et al. A randomised, double blind, placebo-controlled pilot study of oral artesunate therapy for colorectal cancer. EBioMedicine 2015, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gravett, A.M.; Liu, W.M.; Krishna, S.; Chan, W.-C.; Haynes, R.K.; Wilson, N.L.; Dalgleish, A.G. In vitro study of the anti-cancer effects of artemisone alone or in combination with other chemotherapeutic agents. Cancer Chemother. Pharmacol. 2011, 67, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Van Huijsduijnen, R.H.; Guy, R.K.; Chibale, K.; Haynes, R.K.; Peitz, I.; Kelter, G.; Phillips, M.A.; Vennerstrom, J.L.; Yuthavong, Y.; Wells, T.N.C. Anticancer properties of distinct antimalarial drug classes. PLoS ONE 2013, 8, e82962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, A.; Mazumder, A.; du Plessis, L.; du Preez, J.L.; Haynes, R.K.; du Plessis, J. In vitro anti-cancer effects of artemisone nano-vesicular formulations on melanoma cells. Nanomedicine 2015, 11, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, A.; Mazumder, A.; Fox, L.T.; Brümmer, A.; Gerber, M.; du Preez, J.L.; Haynes, R.K.; du Plessis, J. In vitro Skin Permeation of Artemisone and its nano-vesicular formulations. Int. J. Pharm. 2016, 503, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; Chan, D.H.W.; Lee, K.W.; Tin, W.S.; Wong, H.N.; Haynes, R.K. Evaluation and optimization of synthetic routes from dihydroartemisinin to the alkylamino-artemisinins artemiside and artemisone: A test of N-glycosylation methodologies on a lipophilic peroxide. Tetrahedron 2018. [Google Scholar] [CrossRef]
- De Lange, C.; Coertzen, D.; Smit, F.J.; Wentzel, J.F.; Wong, H.N.; Birkholtz, L.-M.; Haynes, R.K.; N’Da, D.D. Synthesis, in vitro antimalarial activities and cytotoxicities of amino-artemisinin-ferrocene derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Morake, M.; Coertzen, D.; Ngwane, A.; Wentzel, J.F.; Wong, H.N.; Smit, F.J.; Birkholtz, L.-M.; Pietersen, R.-D.; Baker, B.; Wiid, I.; et al. Preliminary evaluation of artemisinin–cholesterol conjugates as potential drugs for the treatment of intractable forms of malaria and tuberculosis. ChemMedChem 2018, 13, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kupferschmid, M.; AquinoGil, M.O.; Shams-Eldin, H.; Schmidt, J.; Yamakawa, N.; Krzewinski, F.; Schwarz, R.T.; Lefebvre, T. Identification of O-GlcNAcylated proteins in Plasmodium falciparum. Malar. J. 2017, 16, 485. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, L.-Q.; Andersen, S.L.; Klayman, D.L. Antimalarial activity of new dihydroartemisinin derivatives. 5. Sugar analogues. J. Med. Chem. 1992, 35, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.K.; Karle, J.M. Stereoelectronic properties of antimalarial artemisinin analogues in relation to neurotoxicity. Chem. Res. Toxicol. 1999, 12, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Rabinovich, G.A. Galectins as modulators of tumor progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.S.; Ashraf, G.M.; Banu, N. Galectins-Potential targets for cancer therapy. Cancer Lett. 2007, 253, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Sörme, P.; Arnoux, P.; Kahl-Knutsson, B.; Leffler, H.; Rini, J.M.; Nilsson, U.J. Structural and thermodynamic studies on galectin-3 in complex with synthetic inhibitors: Carbohydrate-protein affinity enhancements through fine–tuning of an arginine-arene interaction. J. Am. Chem. Soc. 2005, 127, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.C.; Kornfeld, S. Hepatic uptake of proteins coupled to fetuin glycopeptide. Biochem. Biophys. Res. Commun. 1971, 45, 622–629. [Google Scholar] [CrossRef]
- Witczak, Z.J. Carbohydrate therapeutics: New developments and strategies. ACS Symp. Ser. 2006, 932, 25–46. [Google Scholar]
- Garg, N.K.; Singh, B.; Jain, A.; Nirbhavane, P.; Sharma, R.; Tyagi, R.K.; Kushwah, V.; Jain, S.; Katare, O.P. Fucose decorated solid-lipid nanocarriers mediate efficient delivery of methotrexate in breast cancer therapeutics. Colloids Surf. B Biointerfaces 2016, 146, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K.; Chan, H.W.; Cheung, M.K.; Lam, W.L.; Soo, M.K.; Tsang, H.W.; Voerste, A.; Williams, I.D. C-10 Ester and ether derivatives of dihydroartemisinin: 10-α artesunate, preparation of authentic 10-β artesunate, and of other esters and ether derivatives bearing potential aromatic intercalating groups at C-10. Eur. J. Org. Chem. 2002, 2002, 113–132. [Google Scholar] [CrossRef]
- Likhosherstov, L.M.; Novikova, O.S.; Dervitskaya, V.A.; Kochetkov, N.K. A new simple synthesis of amino sugar β-D-glycosylamines. Carbohydr. Res. 1986, 146, C1–C5. [Google Scholar] [CrossRef]
- Oscarson, S. Conjugation of Monosaccharides-Synthesis of Glycosidic Linkages in Glycosides, Oligosaccharides and Polysaccharides. In Carbohydrates; Finch, P., Ed.; Springer: Dordrecht, The Netherlands, 1999; Chapter 4; pp. 150–186. ISBN 978-90-481-4033-6. [Google Scholar] [CrossRef]
- Chisholm, J.D.; Vrankem, D.L.V. Glycosylation of 2,2-indolylindolines. J. Org. Chem. 1995, 60, 6672–6673. [Google Scholar] [CrossRef]
- Jung, M.E.; Yang, E.C.; Vu, B.T.; Kiankarimi, M.; Spyrou, E.; Kaunitz, J. Glycosylation of fluoro-quinolones through direct and oxygenated polymethylene linkages as a sugar-mediated active transport system for antimicrobials. J. Med. Chem. 1999, 42, 3899–3909. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, J.D.; Van Vranken, D.L. Regio-controlled synthesis of the antitumor antibiotic AT2433-A1. J. Org. Chem. 2000, 65, 7541–7553. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, M.; Nishimura, T.; Kawamoto, H.; Nakano, M.; Honma, T.; Yoshinari, T.; Arakawa, H.; Suda, H.; Morishima, H.; Nishimura, S. Synthesis and biological activities of NB-506 analogues modified at the glucose group. Bioorg. Med. Chem. Lett. 2000, 10, 419–422. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Makler, M.T.; Ries, J.M.; Williams, J.A.; Bancroft, J.E.; Piper, R.C.; Gibbins, B.L.; Hinrichs, D.J. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993, 48, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Sparatore, A.; Basilico, N.; Parapini, S.; Romeo, S.; Novelli, F.; Sparatore, F.; Taramelli, D. 4-Aminoquinoline quinolizidinyl- and quinolizidinylalkyl-derivatives with antimalarial activity. Bioorg. Med. Chem. 2005, 13, 5338–5345. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, S.; Gelati, M.; Basilico, N.; Parati, E.A.; Haynes, R.K.; Taramelli, D. Differential effects on angiogenesis of two antimalarial compounds, dihydroartemisinin and artemisone: Implications for embryotoxicity. Toxicology 2007, 241, 66–74. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | D10 (CQ-S) Mean ± S.D. IC50, nM a | Ratio IC50 cpd/IC50 DHA | SI IC50WEHI-164/IC50 cpd b | W-2 (CQ-R) Mean ± S.D. IC50, nM a | Ratio IC50 cpd/IC50 DHA | SI IC50WEHI-164/IC50 cpd b | WEHI-164 Mean ± S.D. IC50, nM c |
|---|---|---|---|---|---|---|---|
| 10 | 1.4 ± 0.5 | 0.41 | 107 | 1.1 ± 0.4 | 0.73 | 136 | 150 ± 10 |
| 18 | 1.3 ± 0.1 | 0.38 | 215 | 1.2 ± 0.3 | 0.8 | 233 | 280 ± 80 |
| 19 | 0.87 ± 0.25 | 0.26 | 195 | 0.71 ± 0.2 | 0.5 | 239 | 170 ± 30 |
| 20 | 5.7 ± 2.9 | 1.7 | 596 | 4.4 ± 2.3 | 2.9 | 772 | 3,400 ± 400 |
| 21 | 1.9 ± 1.1 | 0.56 | 131 | 1.5 ± 0.5 | 1.0 | 166 | 250 ± 90 |
| 22 | 2.3 ± 1.0 | 0.68 | 222 | 1.1 ± 0.4 | 0.73 | 463 | 510 ± 100 |
| 23 | 0.74 ± 0.34 | 0.22 | 878 | 0.74 ± 0.3 | 0.49 | 878 | 650 ± 60 |
| 24 | 4.8 ± 2.9 | 1.4 | 102 | 4.1 ± 3.2 | 2.7 | 119 | 490 ± 200 |
| 25 | 0.66 ± 0.2 | 0.19 | 242 | 0.58 ± 0.3 | 0.39 | 275 | 160 ± 40 |
| 26 | 1.3 ± 0.8 | 0.38 | 161 | 0.82 ± 0.25 | 0.55 | 256 | 210 ± 10 |
| DHA 2 | 3.4 ± 1.6 | 211 | 1.5 ± 0.3 | 480 | 720 ± 140 | ||
| Artemisone 7 | 1.1 ± 0.4 | 0.32 | 269 | 0.9 ± 0.4 | 0.6 | 328 | 296 ± 59 |
| Chloroquine | 23.4 ± 10.1 | 263.6 ± 142.9 | >10,000 |
| Compound | Mean (±S.D.) IC50, µM a | ||
|---|---|---|---|
| K562b | HMEC b | HDF b | |
| 18 | 0.81 ± 0.24 | 4.41 ± 2.29 | 382.5 ± 31.2 |
| 21 | 0.78 ± 0.15 | 2.50 ± 0.80 | 533.6 ± 9.9 |
| 23 | 1.95 ± 0.48 | nd | 313.6 ± 85.6 |
| 24 | 0.87 ± 0.05 | 2.39 ± 1.51 | 389.1 ± 8.6 |
| DHA 2 | 2.76 ± 0.66 | 1.16 ± 0.40 | 88.7 ± 13.3 |
| Artemisone 7 | 0.26 ± 0.04 | nd | 88.3 ± 33.8 |
| Camptothecin | 0.03 ± 0.01 | nd | 2.62 ± 0.84 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Parapini, S.; Williams, I.D.; Misiano, P.; Wong, H.N.; Taramelli, D.; Basilico, N.; Haynes, R.K. Facile Preparation of N-Glycosylated 10-Piperazinyl Artemisinin Derivatives and Evaluation of Their Antimalarial and Cytotoxic Activities. Molecules 2018, 23, 1713. https://doi.org/10.3390/molecules23071713
Wu Y, Parapini S, Williams ID, Misiano P, Wong HN, Taramelli D, Basilico N, Haynes RK. Facile Preparation of N-Glycosylated 10-Piperazinyl Artemisinin Derivatives and Evaluation of Their Antimalarial and Cytotoxic Activities. Molecules. 2018; 23(7):1713. https://doi.org/10.3390/molecules23071713
Chicago/Turabian StyleWu, Yuet, Silvia Parapini, Ian D. Williams, Paola Misiano, Ho Ning Wong, Donatella Taramelli, Nicoletta Basilico, and Richard K. Haynes. 2018. "Facile Preparation of N-Glycosylated 10-Piperazinyl Artemisinin Derivatives and Evaluation of Their Antimalarial and Cytotoxic Activities" Molecules 23, no. 7: 1713. https://doi.org/10.3390/molecules23071713
APA StyleWu, Y., Parapini, S., Williams, I. D., Misiano, P., Wong, H. N., Taramelli, D., Basilico, N., & Haynes, R. K. (2018). Facile Preparation of N-Glycosylated 10-Piperazinyl Artemisinin Derivatives and Evaluation of Their Antimalarial and Cytotoxic Activities. Molecules, 23(7), 1713. https://doi.org/10.3390/molecules23071713