High-Resolution PTP1B Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Miconia albicans

 ,
,  , and
, and 
Abstract
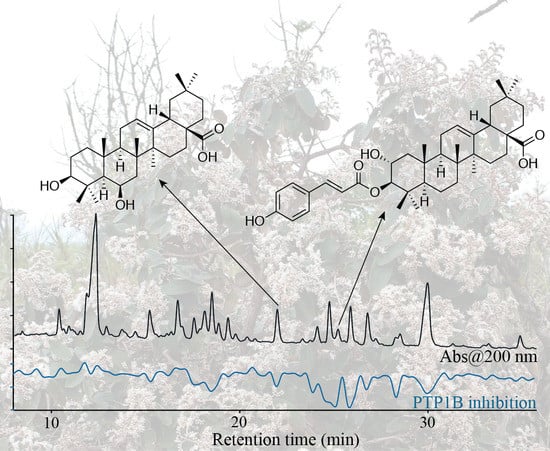
:
1. Introduction
2. Results
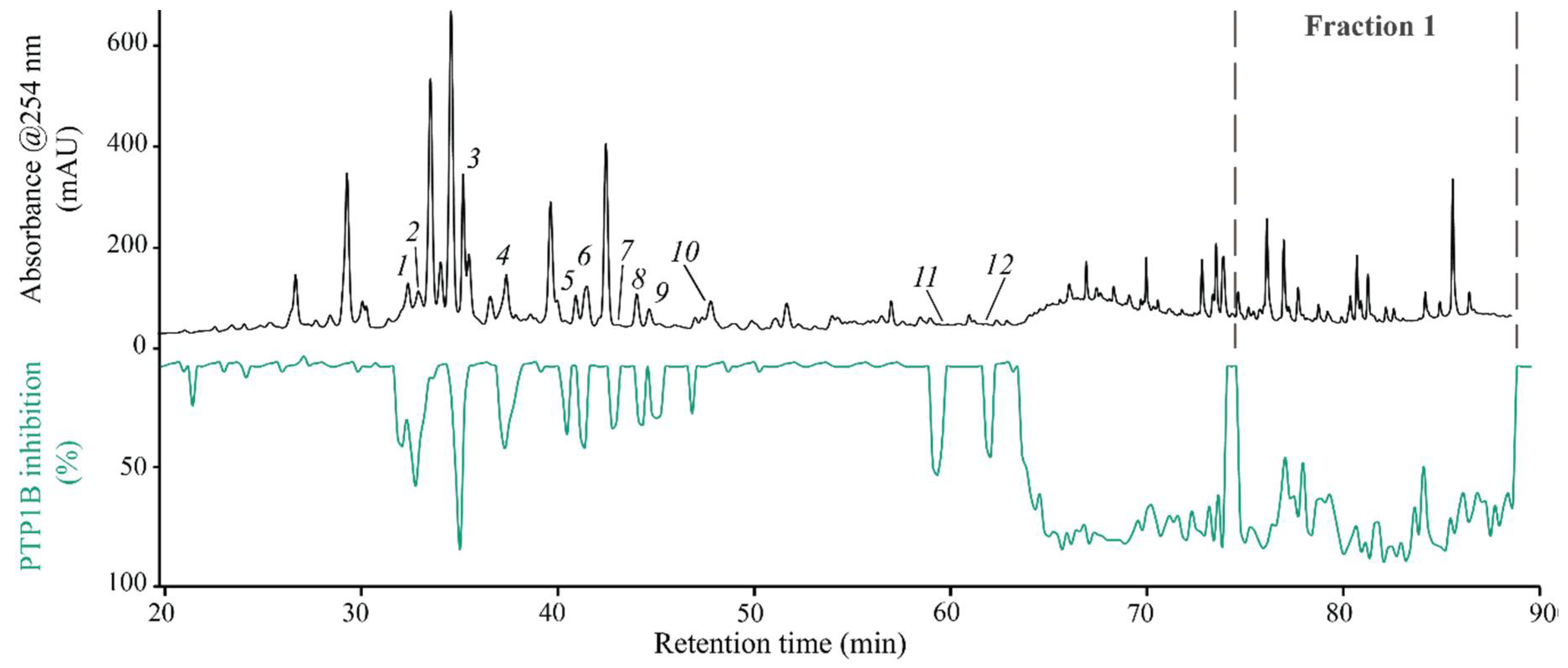
2.1. High-Resolution PTP1B Inhibition Profiling and Identification of Active Compounds from Crude Extract of M. albicans
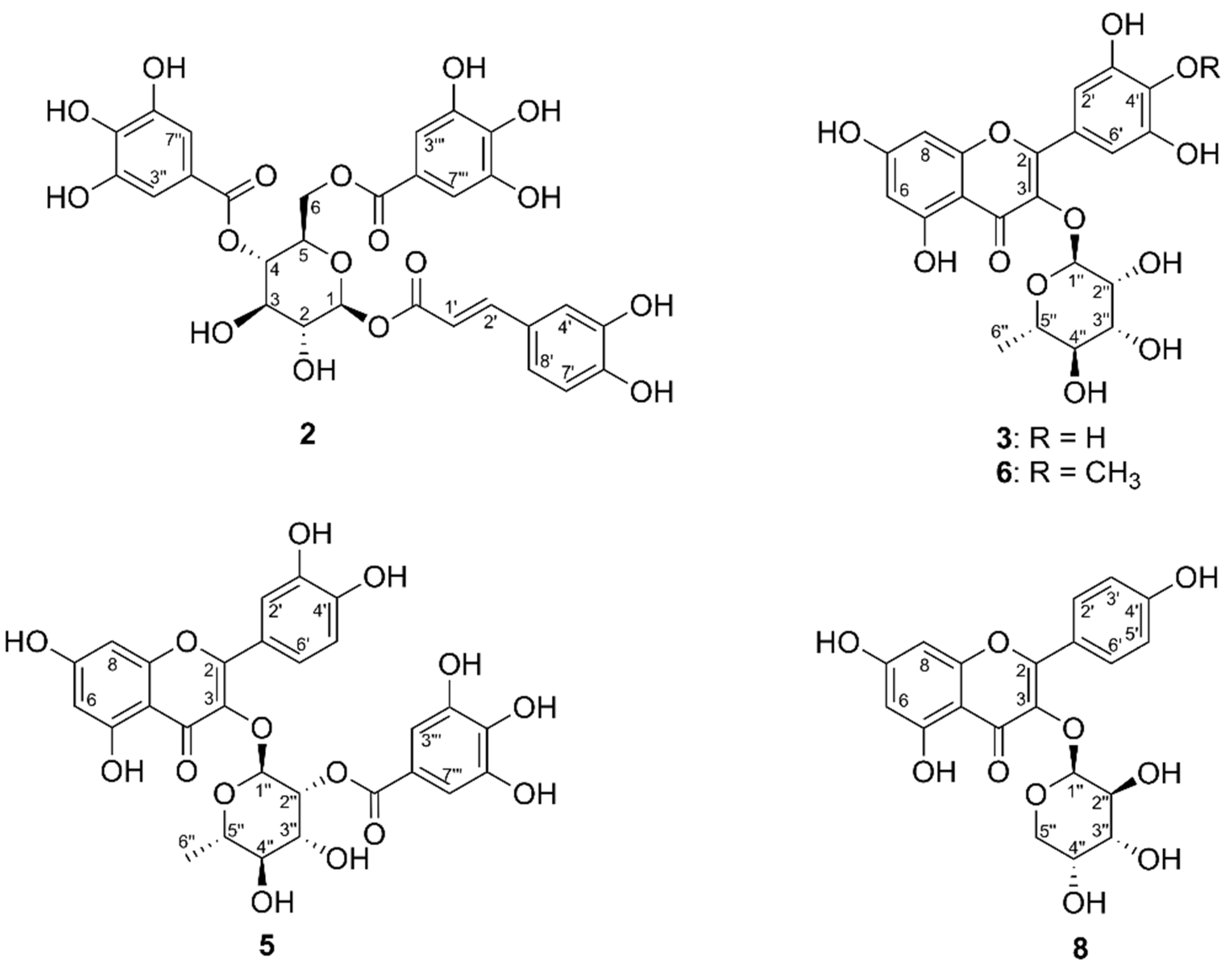
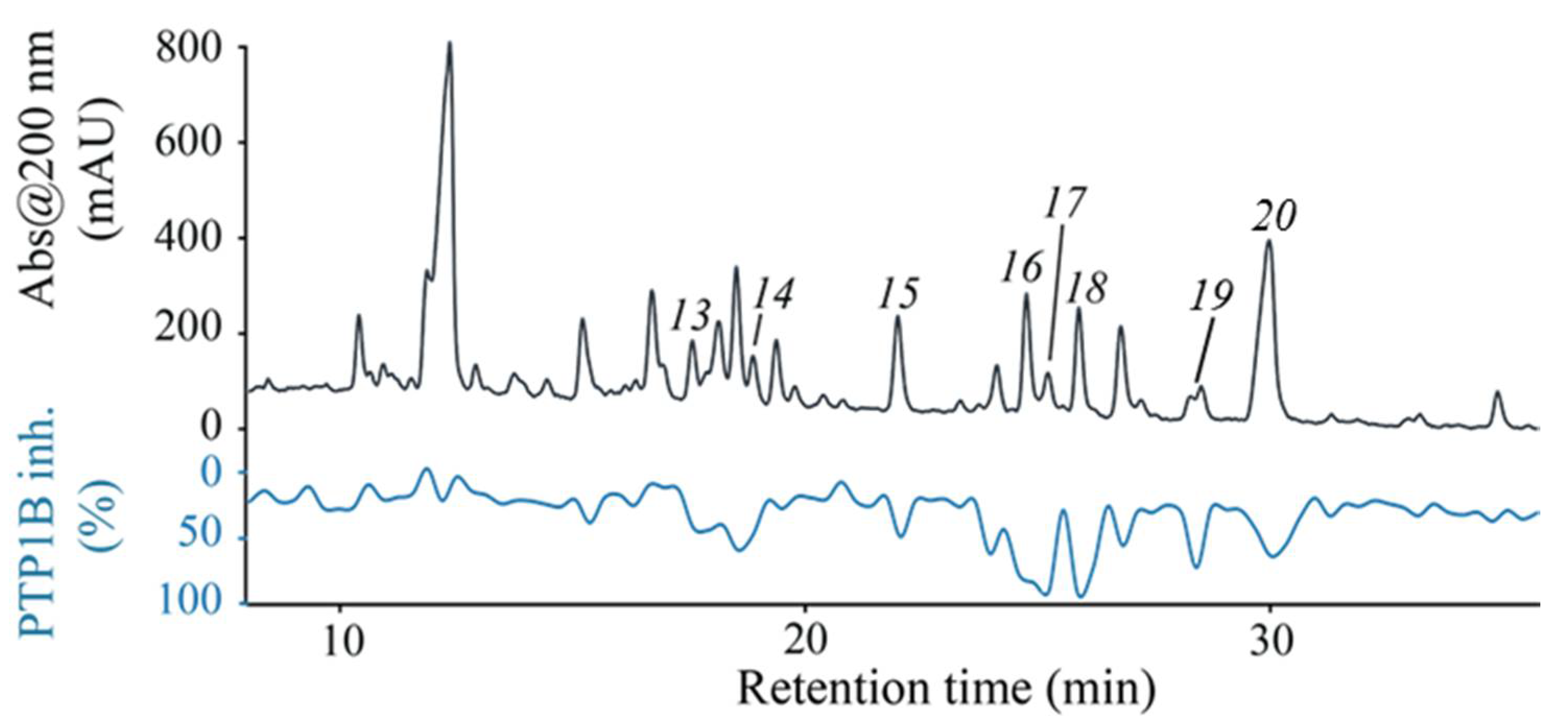
2.2. High-Resolution PTP1B Inhibition Profiling and Identification of Active Compounds from F1
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Preparation of Crude Ethyl Acetate Extracts of Leaves of M. albicans
3.3. Microplate-Based PTP1B Inhibition Assay
3.4. Microfractionation
3.5. Preparative-Scale HPLC for Fractionation of the Crude Extract of M. albicans
3.6. Isolation of PTP1B Inhibitors from the Extract of M. albicans by Analytical-Scale HPLC
3.7. HPLC-HRMS-SPE-NMR Analyses of Triterpenoids from F1
3.8. NMR Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alberti, G.; Zimmet, P.; Shaw, J.; Bloomgarden, Z.; Kaufman, F.; Silink, M. Type 2 diabetes in the young: The evolving epidemic. Diabetes Care 2004, 27, 1798–1811. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.N. Postprandial physiology and the pathogenesis of type 2 diabetes mellitus. Insulin 2008, 3, 132–140. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, D.E. New drug targets for type 2 diabetes and the metabolic syndrome. Nature 2001, 14, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Lee, S.-Y. PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. Expert Opin. Investig. Drugs 2003, 12, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Z.Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Cunha, W.R.; Martins, C.; Ferreira, D.S.; Crotti, A.E.M.; Lopes, N.P.; Albuquerque, S. In vitro trypanocidal activity of triterpenes from Miconia species. Planta Med. 2003, 69, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Martinez, D.M.; Rivas-Morales, C.; Garza-Ramos, M.A.; Verde-Star, M.J.; Nuñez-Gonzalez, M.A.; Leos-Rivas, C. Miconia sp. increases mRNA levels of PPAR gamma and inhibits alpha amylase and alpha glucosidase. Evid.-Based Complement. Altern. Med. 2016, 2016, 5123519. [Google Scholar] [CrossRef] [PubMed]
- Alves, T.M.A.; Silva, A.F.; Brandão, M.; Grandi, T.S.M.; Smânia, E.F.A.; Júnior, A.S.; Zani, C.L. Biological screening of Brazilian medicinal plants. Memórias do Instituto Oswaldo Cruz 2000, 95, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Serpeloni, J.M.; Barcelos, G.R.M.; Mri, M.P.; Yanagui, K.; Vilegas, W.; Varanda, E.A.; Cólus, I.M.S. Cytotoxic and mutagenic evaluation of extracts from plant species of the Miconia genus and their influence on doxorubicin-induced mutagenicity: An in vitro analysis. Exp. Toxicol. Pathol. 2011, 63, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Crevelin, E.J.; Turatti, I.C.C.; Croti, A.E.M.; Veneziani, R.C.S.; Lopes, J.L.C.; Lopes, N.P.; Cunha, W.R. Identification of biologically active triterpenes and sterols present in hexane extracts from Miconia species using high-resolution gas chromatography. Biomed. Chromatogr. 2006, 20, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Kongstad, K.T.; Özdermir, C.; Barzak, A.; Wubshet, S.G.; Staerk, D. Combined use of high-resolution α-glucosidase inhibition profiling and high-performance liquid chromatography-high-resolution mass spectrometry-solid-phase extraction-nuclear magnetic resonance spectroscopy for investigation of antidiabetic principles in crude plant extracts. J. Agric. Food Chem. 2015, 63, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kongstad, K.T.; Wiese, S.; Jäger, A.K.; Staerk, D. Edible seaweed as future functional food: Identification of α-glucosidase inhibitors by combined use of high-resolution α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. Food Chem. 2016, 203, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Okutan, L.; Kongstad, K.T.; Jäger, A.K.; Staerk, D. High-resolution α-amylase assay combined with high-performance liquid chromatography–solid-phase extraction–nuclear magnetic resonance spectroscopy for expedited identification of α-amylase inhibitors: Proof of concept and α-amylase inhibitor in cinnamon. J. Agric. Food Chem. 2014, 62, 11465–11471. [Google Scholar] [CrossRef] [PubMed]
- Trinh, B.T.D.; Jäger, A.K.; Staerk, D. High-resolution inhibition profiling combined with HPLC-HRMS-SPE-NMR for identification of PTP1B inhibitors from Vietnamese plants. Molecules 2017, 22, 1228. [Google Scholar] [CrossRef] [PubMed]
- Grosso, C.; Jäger, A.K.; Staerk, D. Coupling of a high-resolution monoamine oxidase-A inhibitor assay and HPLC-SPE-NMR for advanced bioactivity profiling of plant extracts. Phytochem. Anal. 2014, 24, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Agnolet, S.; Wiese, S.; Verpoorte, R.; Staerk, D. Comprehensive analysis of commercial willow bark extracts by new technology platform: Combined use of metabolomics, high-performance liquid chromatography-solid-phase extraction-nuclear magnetic resonance spectroscopy and high-resolution radical scavenging assay. J. Chromatogr. A 2012, 1262, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Wubshet, S.G.; Nielsen, J.; Staerk, D. Coupling HPLC-SPE-NMR with a microplate based high-resolution antioxidant assay for efficient analysis of antioxidants in food—Validation and proof-of-concept study with caper buds. Food Chem. 2013, 141, 4010–4018. [Google Scholar] [CrossRef] [PubMed]
- Wubshet, S.G.; Brighente, I.M.; Moaddel, R.; Staerk, D. Magnetic ligand fishing as a targeting tool for HPLC-HRMS-SPE-NMR: α-glucosidase inhibitory ligands and alkylresorcinol glycosides from Eugenia catharinae. J. Nat. Prod. 2015, 78, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.H.; Hirose, Y.; Iwata, H.; Sakamoto, S.; Tanaka, T.; Kouno, I. Caffeoyl, coumaroyl, galloyl, and hexahydroxydiphenoyl glucoses from Balanophora japonica. Chem. Pharm. Bull. 2001, 49, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jung, E.B.; Seo, S.J.; Park, K.H.; Lee, M.W.; Lee, C.S. Quercetin-3-O-(2″-galloyl)-α-l-rhamnoside prevents TRAIL-induced apoptosis in human keratinocytes by suppressing the caspase-8- and Bid-pathways and mitochondrial pathway. Chem. Biol. Interact. 2013, 204, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, I.I.; Marzouk, M.S.A.; Moharram, F.A.; El-Gindi, M.R.; Hassam, A.M.K. Acylated flavonol glycosides from Eugenia jambolana leaves. Phytochemistry 2001, 58, 1239–1244. [Google Scholar] [CrossRef]
- Bilia, A.R.; Ciampi, L.; Mendez, J.; Morelli, I. Phytochemical investigations of Licania genus. Flavonoids from Licania pyrifolia. Pharmaceutica Acta Helvetiae 1996, 71, 199–204. [Google Scholar] [CrossRef]
- Steinmann, D.; Baumgartner, R.R.; Heiss, E.H.; Bartenstein, S.; Atanasov, A.G.; Dirsch, V.M.; Ganzera, M.; Stuppner, H. Bioguided isolation of (9Z)-octadec-9-noic acid from Phellodendron amurense Rupr. and identification of fatty acids as PTP1B inhibitors. Planta Med. 2012, 78, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Shibata, E.; Kanno, T.; Tsuchiya, A.; Kuribayachi, K.; Tabata, C.; Nakano, T.; Nishizaki, T. Free fatty acids inhibit protein tyrosine phosphatase 1B and activate Akt. Cell. Physiol. Biochem. 2013, 32, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Patočka, J. Biologically active pentacyclic triterpenes and their current medicine signification. J. Appl. Biomed. 2003, 1, 7–12. [Google Scholar]
- Wen, X.; Sun, H.; Liu, J.; Ceng, K.; Zheng, P.; Zhang, L.; Hao, J.; Zhang, L.; Ni, P.; Zographos, S.E.; et al. Naturally occurring pentacyclic triterpenes as inhibitors of glycogen phosphorylase: Synthesis, structure-activity relationships, and X-ray christallographic studies. J. Med. Chem. 2008, 51, 3540–3554. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Espinosa, J.J.; Rios, M.Y.; López-Martínez, S.; López-Vallejo, F.; Medina-Franco, J.L.; Paoli, P.; Camici, G.; Navarrete-Vázquez, G.; Ortiz-Andade, R.; Estrada-Soto, S. Antidiabetic activity of some pentacyclic acid triterpenoids, role of PTB-1B: In vitro, in silico, and in vivo approaches. Eur. J. Med. Chem. 2011, 46, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Guinda, A.; Delgado, T.; Rada, M.; Cayuela, J.A. Biochemical basis of the antidiabetic activity of oleanolic acid and related pentacyclic triterpenes. Diabetes 2013, 62, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.W.; Shen, Q.; Yang, F.; Wang, B.; Zou, H.; Li, J.Y.; Li, J.; Tang, J. Synthesis and biological evaluation of heterocyclic ring-substituted maslinic acid derivatives as novel inhibitors of protein tyrosine phosphatase 1B. Bioorg. Med. Chem. Lett. 2009, 19, 6618–6622. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.W.; Cha, J.M.; Choi, S.U.; Lee, K.R. A new triterpene glycoside from stem of Lagerstroemia indica. Arch. Pharm. Res. 2016, 39, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.R.; Sheppard, V.; Medford, K.A.; Tinto, W.F.; Reynolds, W.F.; McLean, S.F. Triterpenes from Miconia stenostachya. J. Nat. Prod. 1992, 55, 963–966. [Google Scholar] [CrossRef]
- Yagi, A.; Okamura, N.; Haraguchi, Y.; Noda, K.; Nishioka, I. Studies on the constituents of Zyziphi fructus II.: Structure of new p-coumaroylates of maslinic acid. Chem. Pharm. Bull. 1978, 26, 3075–3079. [Google Scholar] [CrossRef]
- Gong, K.K.; Li, P.L.; Qiao, D.; Zhang, X.W.; Cu, M.J.; Qin, G.F.; Tang, X.L.; Li, G.Q. Cytotoxic and antiviral triterpenoids from the mangrove plant Sonneratia paracaseolaris. Molecules 2017, 22, 1319. [Google Scholar] [CrossRef] [PubMed]
- Sevindik, H.G.; Ozgen, U.; Atila, A.; Er, H.O.; Kazaz, C.; Duman, H. Phytochemicl studies and quantitative HPLC analysis of rosmarinic acid and luteolin 5-O-β-d-glucopyranoside on Thymus praecox subsp. grossheimii var. grossheimii. Chem. Pharm. Bull. 2015, 63, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Venditti, A.; Sanna, C.; Lorenzetti, L.M.; Ballero, M.; Bainco, A. New coumarinyl ethers in Daphne oleoides Schreb. Collected from Sardinia island. Chem. Biodivers. 2017, 14, e1700072. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Wubshet, S.G.; Tahtah, Y.; Heskes, A.M.; Kongstad, K.T.; Pateraki, I.; Hamberger, B.; Møller, B.M.; Staerk, D. Identification of PTP1B and α-glucosidase inhibitory serrulatanes from Eremophila spp. by combined use of dual high-resolution PTP1B and α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. J. Nat. Prod. 2016, 79, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
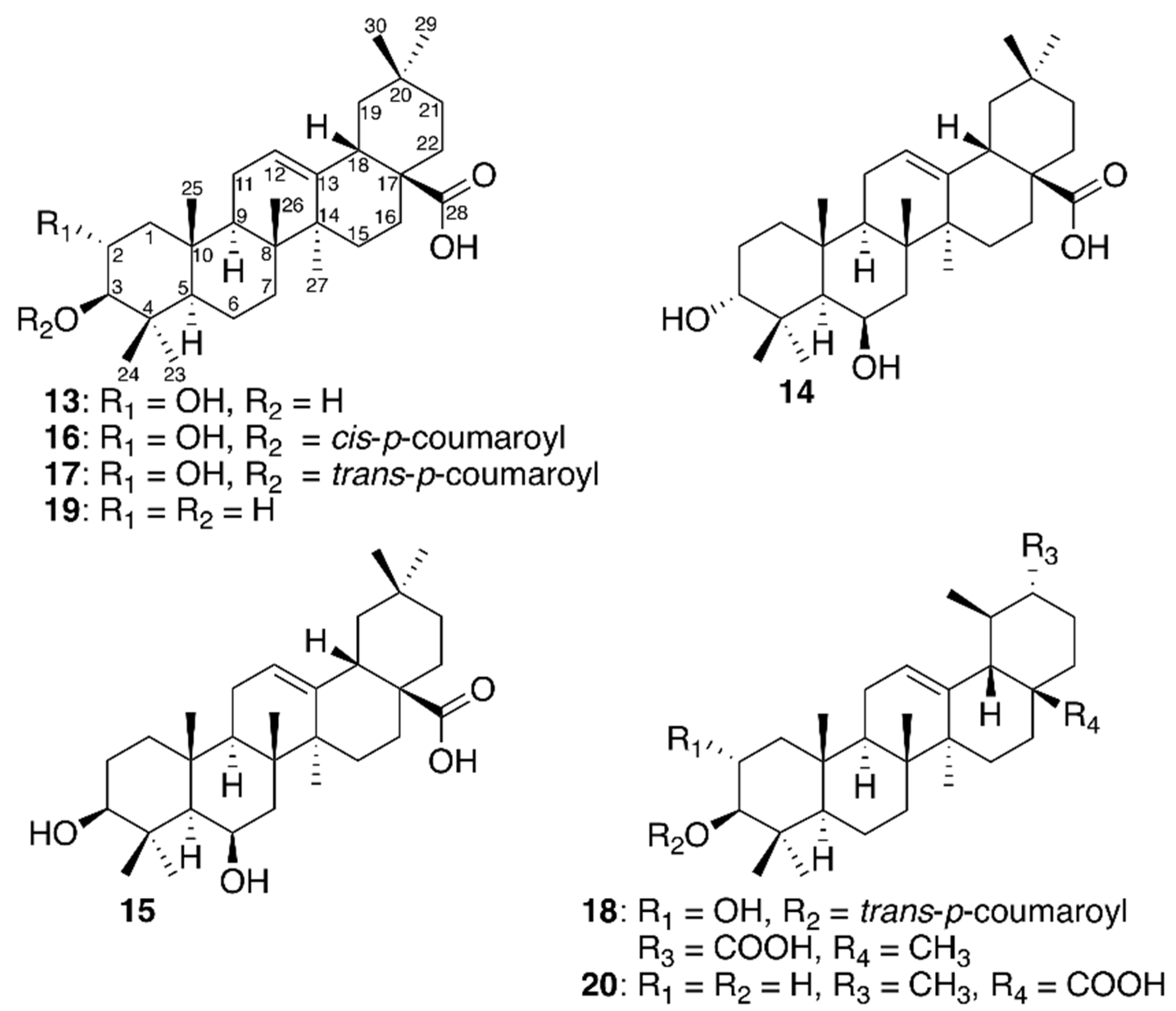{kind=link}
| Compound | Name | IC50 (μM) a |
|---|---|---|
| 13 | Maslinic acid | 3.21 ± 0.8 |
| 14 | 3-epi-sumaresinolic acid | 2.87 ± 0.4 |
| 15 | Sumaresinolic acid | 1.84 ± 0.3 |
| 16 | 3-O-cis-p-coumaroyl-maslinic acid | 0.46 ± 0.07 |
| 17 | 3-O-trans-p-coumaroyl-maslinic acid | 1.08 ± 0.3 |
| 18 | 3-O-trans-p-coumaroyl-2α-hydroxydulcioic acid | 1.6 ± 0.5 |
| 19 | Oleanolic acid | 2.88 ± 0.6 |
| 20 | Ursolic acid | 2.18 ± 0.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Cássia Lemos Lima, R.; T. Kongstad, K.; Kato, L.; José das Silva, M.; Franzyk, H.; Staerk, D. High-Resolution PTP1B Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Miconia albicans. Molecules 2018, 23, 1755. https://doi.org/10.3390/molecules23071755
De Cássia Lemos Lima R, T. Kongstad K, Kato L, José das Silva M, Franzyk H, Staerk D. High-Resolution PTP1B Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Miconia albicans. Molecules. 2018; 23(7):1755. https://doi.org/10.3390/molecules23071755
Chicago/Turabian StyleDe Cássia Lemos Lima, Rita, Kenneth T. Kongstad, Lucília Kato, Marcos José das Silva, Henrik Franzyk, and Dan Staerk. 2018. "High-Resolution PTP1B Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Miconia albicans" Molecules 23, no. 7: 1755. https://doi.org/10.3390/molecules23071755
APA StyleDe Cássia Lemos Lima, R., T. Kongstad, K., Kato, L., José das Silva, M., Franzyk, H., & Staerk, D. (2018). High-Resolution PTP1B Inhibition Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of PTP1B Inhibitors from Miconia albicans. Molecules, 23(7), 1755. https://doi.org/10.3390/molecules23071755