Verdazyls as Possible Building Blocks for Multifunctional Molecular Materials: A Case Study on 1,5-Diphenyl-3-(p-iodophenyl)-verdazyl Focusing on Magnetism, Electron Transfer and the Applicability of the Sonogashira-Hagihara Reaction


Abstract
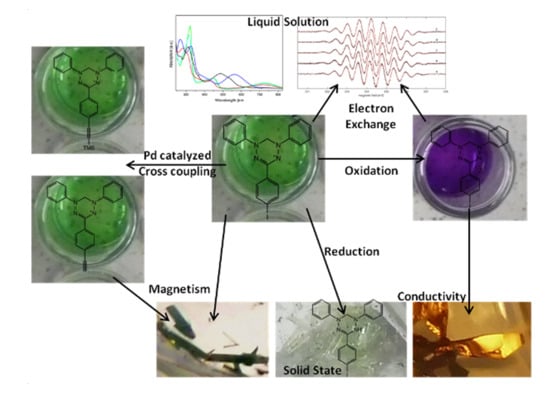
:
1. Introduction
2. Results
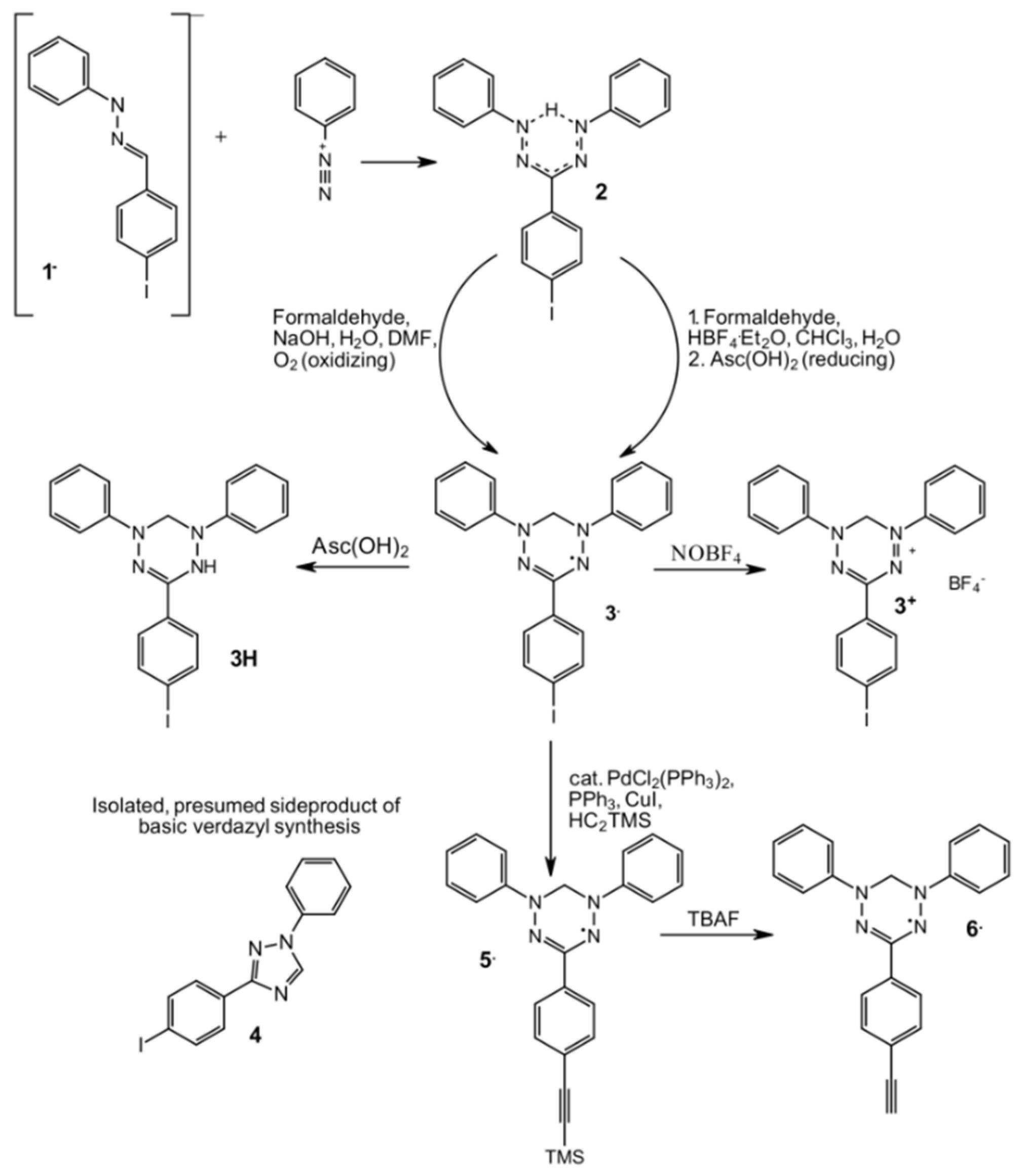
2.1. Syntheses
2.2. Crystallography
2.2.1. General Remarks and Crystallization Procedures
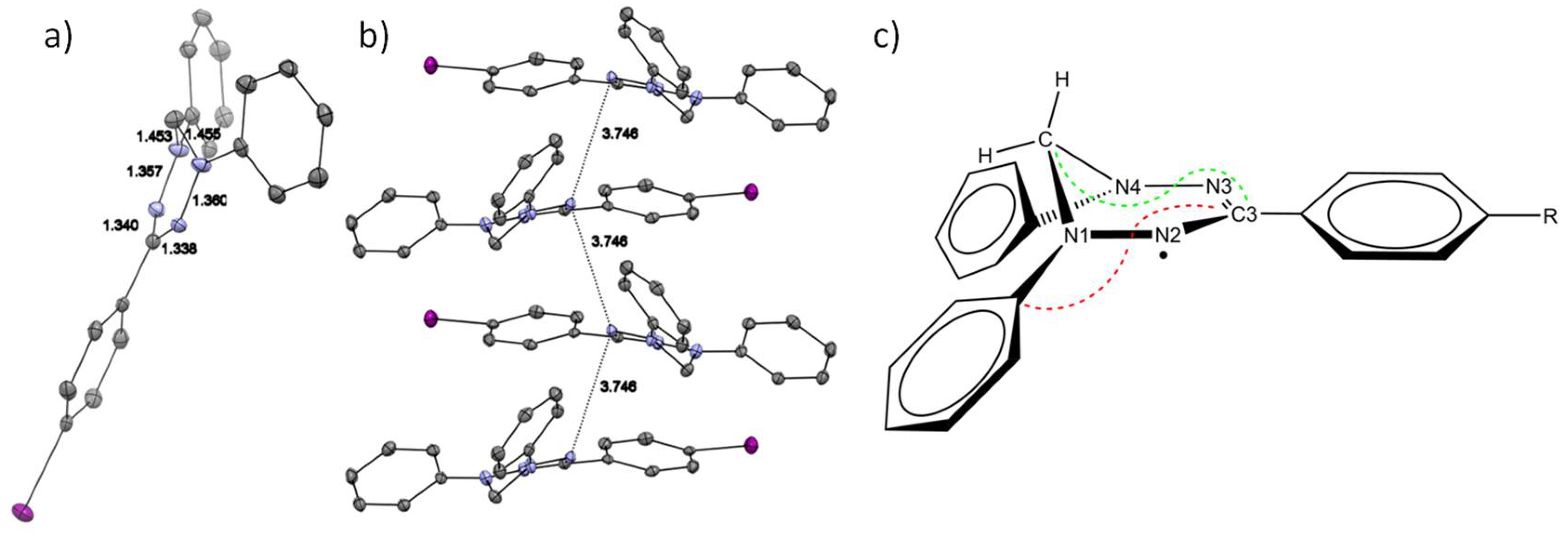
2.2.2. Discussion of the Crystal Structures
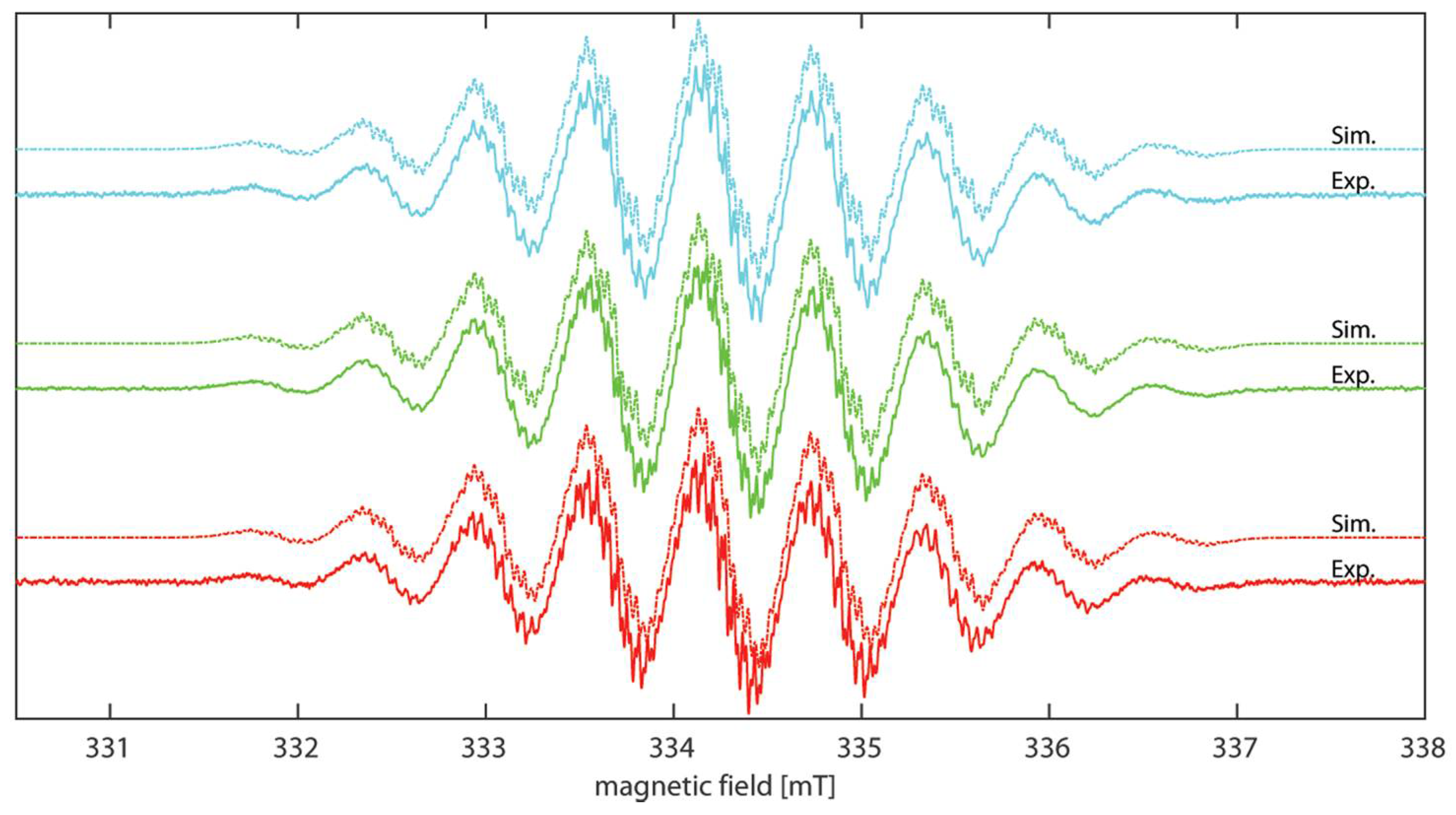
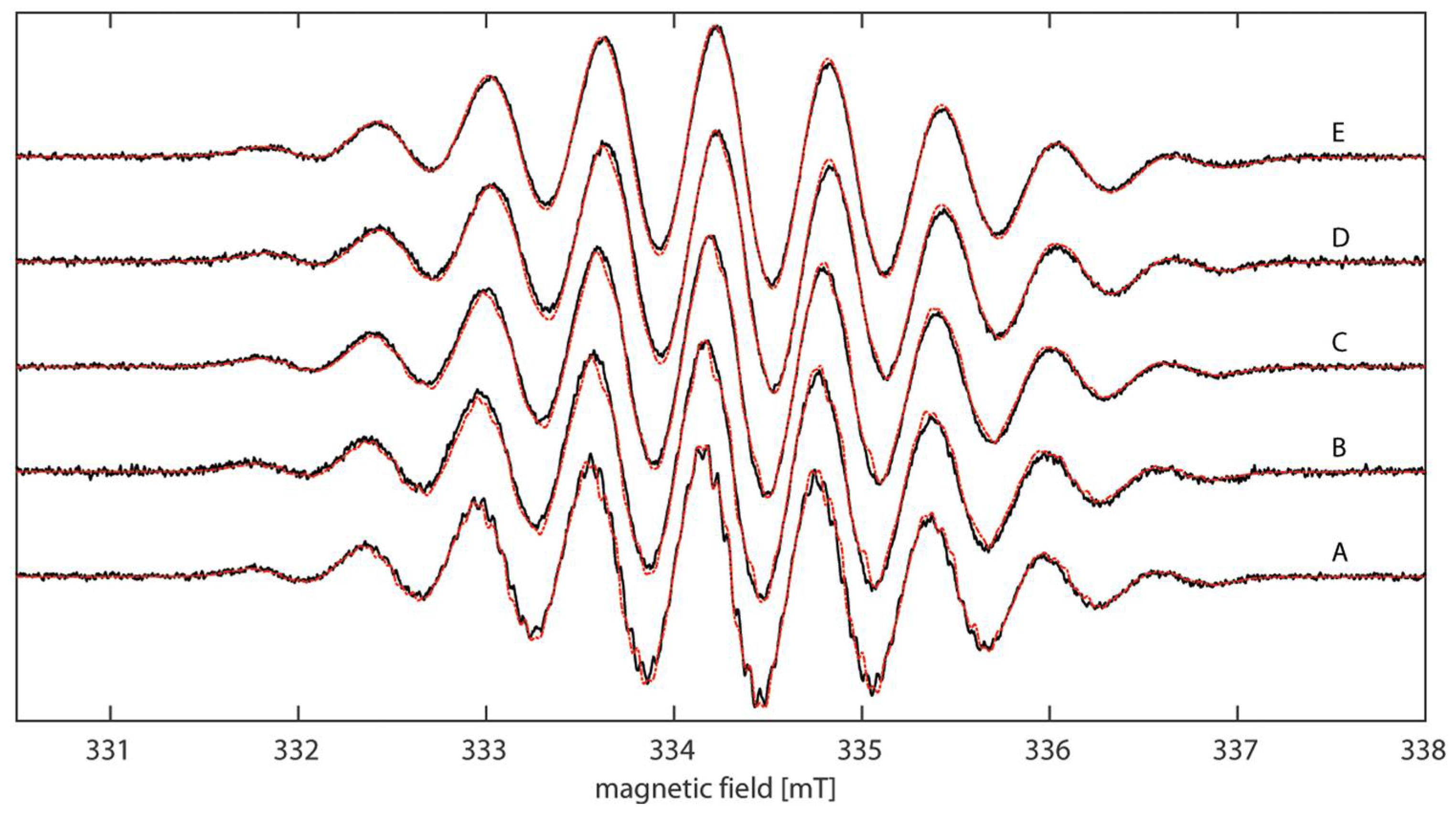
2.3. EPR Spectroscopy
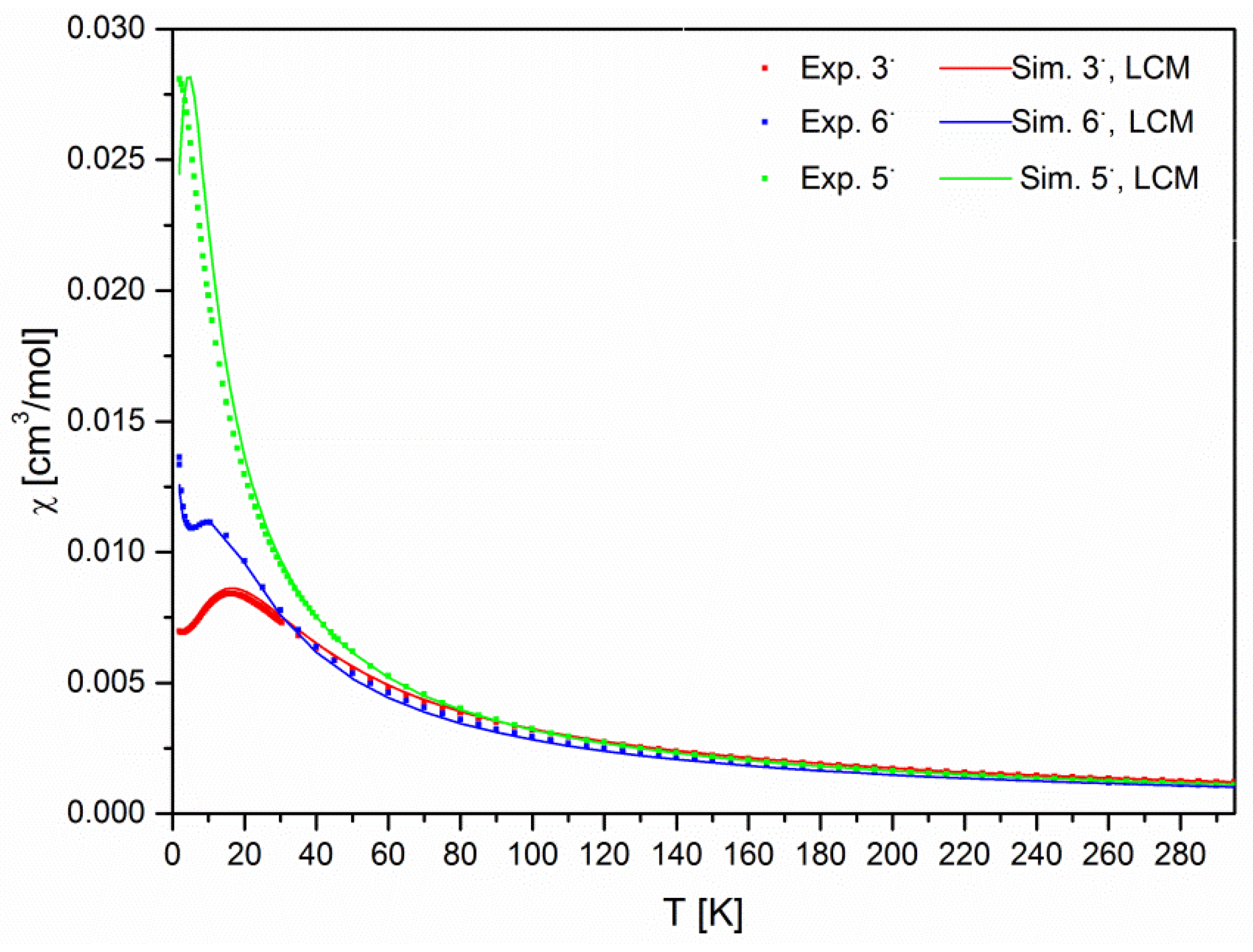
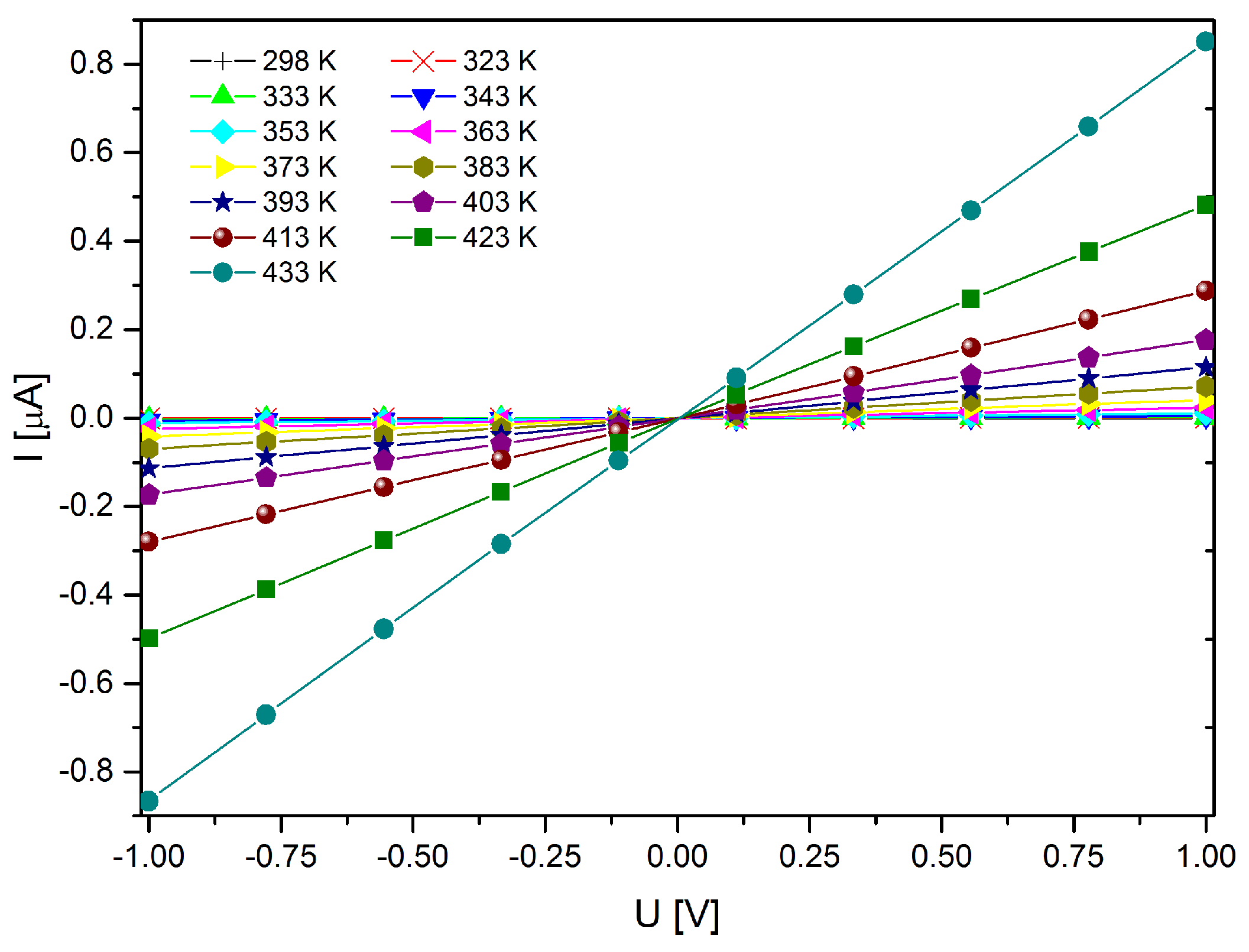
2.4. Magnetometry and Conductivity
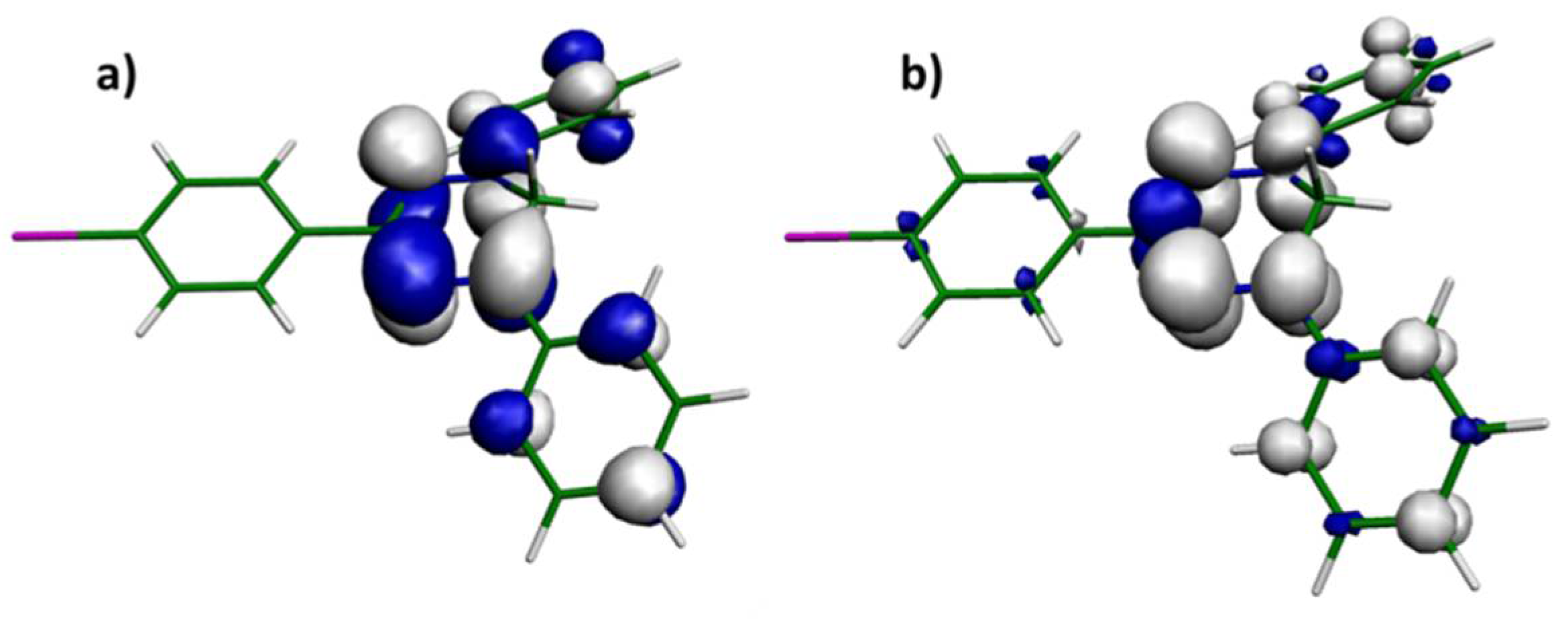
2.5. DFT Calculations
3. Materials and Methods
3.1. Syntheses
3.2. Magnetometry and Conductivity Measurements
3.3. DFT Calculations
3.4. EPR Sample Preparation
3.5. EPR Measurements
3.6. X-ray Diffractometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ET | electron transfer |
| MMM | multifunctional molecular materials |
| AF | antiferromagnetic |
| J | isotropic exchange coupling constant |
| TTF | tetrathiafulvalene |
| (BS)-DFT | (broken-symmetry) density functional theory |
| (cw) EPR | (continuous wave) electron paramagnetic resonance |
| TMS | trimethylsilyl |
| DMF | dimethylformamide |
| Asc(OH)2 | ascorbic acid |
| TBAF | tetrabutylammonium fluoride |
| DCM | dichloromethane |
| HOMO | highest occupied molecular orbital |
| LUMO | lowest unoccupied molecular orbital |
| Aiso | isotropic hyperfine coupling constant |
| LWPP | peak-to-peak linewidth |
| gyromagnetic ratio of the electron | |
| kdiff | diffusion rate constant |
| kET | rate constant of ET |
| α | proportionality constant to calculate kET |
| η | solvent viscosity |
| R | universal gas constant |
| T | temperature |
| TEMPO | tetramethylpiperidine-N-oxide |
| Hexc | exchange spin-Hamiltonian operator |
| HFX | Hartree-Fock Exchange |
| χ | magnetic susceptibility constant |
| LCM | linear chain model |
| IR | infrared |
| TLC | thin layer chromatography |
| I | electrical current |
| U | voltage |
| σ | conductivity |
| λ | reorganization energy |
| TMI | transition metal ions |
References
- Ratera, I.; Veciana, J. Playing with organic radicals as building blocks for functional molecular materials. Chem. Soc. Rev. 2012, 41, 303–349. [Google Scholar] [CrossRef] [PubMed]
- Train, C.; Norel, L.; Baumgarten, M. Organic radicals, a promising route towards original Molecule-Based magnetic materials. Coord. Chem. Rev. 2009, 253, 2342–2351. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, Y.; Keshri, S.K.; Mukhopadhyay, P. Recent advances in organic radicals and their magnetism. Magnetochemistry 2016, 2, 42. [Google Scholar] [CrossRef]
- Oyaizu, K.; Nishide, H. Radical polymers for organic electronic devices: A radical departure from conjugated polymers? Adv. Mater. 2009, 21, 2339–2344. [Google Scholar] [CrossRef]
- Train, C.; Gruselle, M.; Verdaguer, M. The fruitful introduction of chirality and control of absolute configurations in molecular magnets. Chem. Soc. Rev. 2011, 40, 3297–3312. [Google Scholar] [CrossRef] [PubMed]
- Kirk, M.L.; Shultz, D.A. Transition metal complexes of donor–acceptor biradicals. Coord. Chem. Rev. 2013, 257, 218–233. [Google Scholar] [CrossRef]
- Gallagher, N.M.; Olankitwanit, A.; Rajca, A. High-spin organic molecules. J. Org. Chem. 2015, 80, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.K.; Henderson, A.T. A new stable free radical: DI-t-Butylnitroxide. J. Am. Chem. Soc. 1961, 83, 4671–4672. [Google Scholar] [CrossRef]
- Osiecki, J.H.; Ullman, E.F. Studies of free radicals. I. α-Nitronyl nitroxides, a new class of stable radicals. J. Am. Chem. Soc. 1968, 90, 1078–1079. [Google Scholar] [CrossRef]
- Likhtenshtein, G.I.; Yamauchi, J.; Nakatsuji, S.; Smirnov, A.I.; Tamura, R. Nitroxides: Applications in Chemistry, Biomedicine, and Materials Science, 1st ed.; Wiley-VCH: Weinheim, Germany, 2008; ISBN 978-3527318896. [Google Scholar]
- Tebben, L.; Studer, A. nitroxides: Applications in synthesis and in polymer chemistry. Angew. Chem. Int. Ed. 2011, 50, 5034–5068. [Google Scholar] [CrossRef] [PubMed]
- Haugland, M.M.; Lovett, J.E.; Anderson, E.A. Advances in the synthesis of nitroxide radicals for use in biomolecule spin labelling. Chem. Soc. Rev. 2018, 47, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Trischmann, H. surprisingly stable nitrogenous free radicals. Angew. Chem. Int. Ed. 1963, 2, 155. [Google Scholar] [CrossRef]
- Neugebauer, F.A.; Fischer, H. 6-Oxoverdazyls. Angew. Chem. Int. Ed. 1980, 19, 724–725. [Google Scholar] [CrossRef]
- Koivisto, B.D.; Hicks, R.G. The magnetochemistry of verdazyl radical-based materials. Coord. Chem. Rev. 2005, 249, 2612–2630. [Google Scholar] [CrossRef]
- Koivisto, B.D.; Ichimura, A.S.; McDonald, R.; Lemaire, M.T.; Thompson, L.K.; Hicks, R.G. Intramolecular π-Dimerization in a 1,1′-Bis (Verdazyl) ferrocene diradical. J. Am. Chem. Soc. 2006, 128, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Norel, L.; Rota, J.; Chamoreau, L.; Pilet, G.; Robert, V.; Train, C. Spin transition and exchange interaction: janus visions of supramolecular spin coupling between face-to-face verdazyl radicals. Angew. Chem. Int. Ed. 2011, 50, 7128–7131. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.G.; Lemaire, M.T.; Öhrström, L.; Richardson, J.F.; Thompson, L.K.; Xu, Z. Strong supramolecular-based magnetic exchange in π-stacked radicals. structure and magnetism of a hydrogen-bonded verdazyl radical: hydroquinone molecular solid. J. Am. Chem. Soc. 2001, 123, 7154–7159. [Google Scholar] [CrossRef] [PubMed]
- Norel, L.; Chamoreau, L.-M.; Train, C. Modulation of intermolecular exchange interaction in organic radical based compounds: magneto-structural analysis of phenol and imidazolium substituted oxoverdazyl radicals. Polyhedron 2010, 29, 342–348. [Google Scholar] [CrossRef]
- Kumar, V.; Shova, S.; Maurel, V.; Novitchi, G.; Train, C. Crystallographic insights into the synthesis and magnetic properties of oxoverdazyl radicals functionalized by benzoic acid. Eur. J. Inorg. Chem. 2018, 2018, 517–524. [Google Scholar] [CrossRef]
- Miyashiro, S.; Ishii, T.; Miura, Y.; Yoshioka, N. Synthesis and magnetic properties of stable radical derivatives carrying a phenylacetylene unit. Molecules 2018, 23, 371. [Google Scholar] [CrossRef] [PubMed]
- Eusterwiemann, S.; Doerenkamp, C.; Dresselhaus, T.; Janka, O.; de Oliveira, M., Jr.; Daniliuc, C.; Eckert, H.; Neugebauer, J.; Pöttgen, R.; Studer, A. Strong intermolecular antiferromagnetic verdazyl–verdazyl coupling in the solid state. Phys. Chem. Chem. Phys. 2017, 19, 15681–15685. [Google Scholar] [CrossRef] [PubMed]
- Merhi, A.; Roisnel, T.; Rigaut, S.; Train, C.; Norel, L. Ferromagnetic intermolecular exchange interaction in ethynyl-verdazyl radical crystals. CrystEngComm 2014, 16, 9783–9787. [Google Scholar] [CrossRef]
- Rota, J.-B.; Le Guennic, B.; Robert, V. Toward verdazyl radical-based materials: Ab initio inspection of potential organic candidates for spin-crossover phenomenon. Inorg. Chem. 2010, 49, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Jornet, J.; Deumal, M.; Ribas-Ariño, J.; Bearpark, M.J.; Robb, M.A.; Hicks, R.G.; Novoa, J.J. Direct versus mediated through-space magnetic interactions: A first principles, bottom-up reinvestigation of the magnetism of the pyridyl-verdazyl: Hydroquinone molecular co-crystal. Chem. Eur. J. 2006, 12, 3995–4005. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, T.; Eusterwiemann, S.; Matuschek, D.R.; Daniliuc, C.G.; Janka, O.; Pöttgen, R.; Studer, A.; Neugebauer, J. Black-box determination of temperature-dependent susceptibilities for crystalline organic radicals with complex magnetic topologies. Phys. Chem. Chem. Phys. 2016, 18, 28262–28273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas-Torrent, M.; Crivillers, N.; Mugnaini, V.; Ratera, I.; Rovira, C.; Veciana, J. Organic radicals on surfaces: Towards molecular spintronics. J. Mater. Chem. 2009, 19, 1691–1695. [Google Scholar] [CrossRef]
- Sanvito, S. Molecular spintronics. Chem. Soc. Rev. 2011, 40, 3336–3355. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Komatsu, H.; Suzuki, K. Interplay between magnetism and conductivity derived from spin-polarized donor radicals. Chem. Soc. Rev. 2011, 40, 3105–3118. [Google Scholar] [CrossRef] [PubMed]
- Chahma, M.; Wang, X.; van der Est, A.; Pilkington, M. Synthesis and characterization of a new family of spin bearing TTF ligands. J. Org. Chem. 2006, 71, 2750–2755. [Google Scholar] [CrossRef] [PubMed]
- Chahma, M.; Macnamara, K.; Van der Est, A.; Alberola, A.; Polo, V.; Pilkington, M. Synthesis and characterization of a TTF-π-Verdazyl radical—A new building block for conducting and/or magnetic Systems. New J. Chem. 2007, 31, 1973–1978. [Google Scholar] [CrossRef]
- Polo, V.; Alberola, A.; Andres, J.; Anthony, J.; Pilkington, M. Towards understanding of magnetic interactions within a series of tetrathiafulvalene–π conjugated-verdazyl diradical cation system: A density functional theory study. Phys. Chem. Chem. Phys. 2008, 10, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Venneri, S.; Wilson, J.; Rawson, J.M.; Pilkington, M. Structural, magnetic and DFT studies on a charge-transfer salt of a Tetrathiafulvalenepyridyl-(1,5-diisopropyl) verdazyl diradical cation. ChemPlusChem 2015, 80, 1624–1633. [Google Scholar] [CrossRef]
- Gilroy, J.B.; McKinnon, S.D.; Koivisto, B.D.; Hicks, R.G. Electrochemical studies of verdazyl radicals. Org. Lett. 2007, 9, 4837–4840. [Google Scholar] [CrossRef] [PubMed]
- Haller, B.C.; Chambers, D.; Cheng, R.; Chemistruck, V.; Hom, T.F.; Li, Z.; Nguyen, J.; Ichimura, A.; Brook, D.J. Oxidation of electron donor-substituted verdazyls: Building blocks for molecular switches. J. Phys. Chem. A 2015, 119, 10750–10760. [Google Scholar] [CrossRef] [PubMed]
- Bancerz, M.; Youn, B.; DaCosta, M.V.; Georges, M.K. A hydrazine-and phosgene-free synthesis of tetrazinanones, precursors to 1,5-Dialkyl-6-Oxoverdazyl radicals. J. Org. Chem. 2012, 77, 2415–2421. [Google Scholar] [CrossRef] [PubMed]
- Paré, E.C.; Brook, D.J.; Brieger, A.; Badik, M.; Schinke, M. Synthesis of 1,5-Diisopropyl substituted 6-Oxoverdazyls. Org. Biomol. Chem. 2005, 3, 4258–4261. [Google Scholar] [CrossRef] [PubMed]
- Matuschek, D.; Eusterwiemann, S.; Stegemann, L.; Doerenkamp, C.; Wibbeling, B.; Daniliuc, C.G.; Doltsinis, N.L.; Strassert, C.A.; Eckert, H.; Studer, A. Profluorescent verdazyl radicals—Synthesis and characterization. Chem. Sci. 2015, 6, 4712–4716. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Trevisan, T.; Lieu, E.; Brook, D.J. Suzuki–miyaura coupling of verdazyl radicals. Eur. J. Org. Chem. 2017, 2017, 1125–1131. [Google Scholar] [CrossRef]
- Petunin, P.V.; Martynko, E.A.; Trusova, M.E.; Kazantsev, M.S.; Rybalova, T.V.; Valiev, R.R.; Uvarov, M.N.; Postnikov, P.S.; Mostovich, E. Verdazyl radical building blocks: Synthesis, structure and sonogashira cross-coupling reactions. Eur. J. Org. Chem. 2018. [Google Scholar] [CrossRef]
- Berry, D.E.; Hicks, R.G.; Gilroy, J.B. The chemistry of formazan dyes. synthesis and characterization of a stable verdazyl radical and a related boron-containing heterocycle. J. Chem. Educ. 2009, 86, 76. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Belyakov, S.A. A direct one-step preparation of triarylverdazylium salts from the corresponding triarylformazans. Synthesis 1997, 1997, 17–19. [Google Scholar] [CrossRef]
- Otting, W.; Neugebauer, F.A. IR-spektroskopische untersuchungen zur frage der isomerie bei formazanen und tetrazoliumsalzen. Eur. J. Inorg. Chem. 1969, 102, 2520–2529. [Google Scholar] [CrossRef]
- Cunningham, C.W.; Burns, G.R.; McKee, V. Photochromic formazans: X-ray crystal structure magnetic resonance and raman spectra of 3-Methyl-1,5-Diphenylformazan and 3-t-Butyl-l,5-Diphenylformazan. J. Chem. Soc. Perkin Trans. 2 1989, 1429–1436. [Google Scholar] [CrossRef]
- Hutton, A.T.; Irving, H.M. Isomers of 3-Methylthio-1,5-Diarylformazans and their interconversion in solution. J. Chem. Soc. Perkin Trans. 2 1982, 1117–1121. [Google Scholar] [CrossRef]
- Schnakenburg, G.; Meyer, A. Syntheses, spectroscopy, and crystal structures of 3-(4-Bromophenyl)-1,5-Diphenylformazan and the 3-(4-Bromophenyl)-1,5-Diphenylverdazyl radical and the crystal structure of the by-product 5-Anilino-3-(4-Bromophenyl)-1-Phenyl-1H-1,2,4-Triazole. Acta Cryst. Sect. E 2018, 74, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, F.A.; Otting, W.; Smith, H.O.; Trischmann, H. Verdazyle, 21. zur Thermischen disproportionierung des 1,3,5-Triphenyl-verdazyls. Eur. J. Inorg. Chem. 1972, 105, 549–553. [Google Scholar]
- Johnston, C.W.; McKinnon, S.D.; Patrick, B.O.; Hicks, R.G. The first “Kuhn verdazyl” ligand and comparative studies of its PdCl2 complex with analogous 6-Oxoverdazyl ligands. Dalton Trans. 2013, 42, 16829–16836. [Google Scholar] [CrossRef] [PubMed]
- Sanz, C.A.; Ferguson, M.J.; McDonald, R.; Patrick, B.O.; Hicks, R.G. Classical and non-classical redox reactions of Pd (II) complexes containing redox-active ligands. Chem. Commun. 2014, 50, 11676–11678. [Google Scholar] [CrossRef] [PubMed]
- Sanz, C.A.; McKay, Z.R.; MacLean, S.W.; Patrick, B.O.; Hicks, R.G. Synthesis and redox reactions of Bis (Verdazyl) palladium complexes. Dalton Trans. 2017, 46, 12636–12644. [Google Scholar] [CrossRef] [PubMed]
- Van Vleck, J. The dipolar broadening of magnetic resonance lines in crystals. Phys. Rev. 1948, 74, 1168–1183. [Google Scholar] [CrossRef]
- Iwase, K.; Yamaguchi, H.; Ono, T.; Shimokawa, T.; Nakano, H.; Matsuo, A.; Kindo, K.; Nojiri, H.; Hosokoshi, Y. Crystal structure and magnetic properties of the Verdazyl Biradical M-Ph-V2 forming a ferromagnetic alternating double chain. J. Phys. Soc. Jpn. 2013, 82, 074719. [Google Scholar] [CrossRef]
- Janiak, C. A Critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Marcus, R.A. On the theory of oxidation-reduction reactions involving electron transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Kay, E.R.; Leigh, D.A. Rise of the molecular machines. Angew. Chem. Int. Ed. 2015, 54, 10080–10088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyhach, Y.; Godt, A.; Bauer, C.; Jeschke, G. Spin pair geometry revealed by high-field deer in the presence of conformational distributions. J. Magn. Reson. 2007, 185, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Margraf, D.; Bode, B.E.; Marko, A.; Schiemann, O.; Prisner, T.F. Conformational flexibility of nitroxide biradicals determined by X-band PELDOR experiments. Mol. Phys. 2007, 105, 2153–2160. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Schnakenburg, G.; Schiemann, O. Crystal structure of 4′-{[4-(2,2′:6′,2′′-Terpyridyl-4′-Yl)Phenyl]Ethynyl}biphenyl-4-Yl (2,2,5,5-Tetramethyl-1-Oxyl-3-Pyrrolin-3-Yl)formate benzene 2.5-Solvate. Acta Cryst. Sect. E 2015, 71, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Jassoy, J.J.; Spicher, S.; Berndhäuser, A.; Schiemann, O. Performance of PELDOR, RIDME, SIFTER, and DQC in measuring distances in trityl based Bi-and triradicals: Exchange coupling, pseudosecular coupling and multi-spin effects. Phys. Chem. Chem. Phys. 2018, 20, 13858–13869. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grampp, G.; Rasmussen, K. Solvent dynamical effects on the electron self-exchange rate of the TEMPO/TEMPO+ Couple (TEMPO = 2,2,6,6-Tetramethyl-1-Piperidinyloxy Radical) Part I. ESR-Linebroadening measurements at T = 298 K. Phys. Chem. Chem. Phys. 2002, 4, 5546–5549. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism, 1st ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 1993; ISBN 978-1560815662. [Google Scholar]
- Majumdar, C.K.; Ghosh, D.K. On next-nearest-neighbor interaction in linear chain. I. J. Math. Phys. 1969, 10, 1388–1398. [Google Scholar] [CrossRef]
- Majumdar, C.K.; Ghosh, D.K. On next-nearest-neighbor interaction in linear chain. II. J. Math. Phys. 1969, 10, 1399–1402. [Google Scholar] [CrossRef]
- Heidrich-Meisner, F.; Honecker, A.; Vekua, T. Frustrated ferromagnetic spin-1 2 chain in a magnetic field: The Phase diagram and thermodynamic properties. Phys. Rev. B 2006, 74, 020403. [Google Scholar] [CrossRef]
- Lu, H.; Wang, Y.; Qin, S.; Xiang, T. Zigzag spin chains with antiferromagnetic-ferromagnetic interactions: Transfer-matrix renormalization group study. Phys. Rev. B 2006, 74, 134425. [Google Scholar] [CrossRef]
- Hase, M.; Kuroe, H.; Ozawa, K.; Suzuki, O.; Kitazawa, H.; Kido, G.; Sekine, T. Magnetic properties of Rb2Cu2Mo3O12 including a one-dimensional spin-1/2 heisenberg system with ferromagnetic first-nearest-neighbor and antiferromagnetic second-nearest-neighbor exchange interactions. Phys. Rev. B 2004, 70, 104426. [Google Scholar] [CrossRef]
- Hunklinger, S. Festkörperphysik, 1st ed.; Oldenbourg Verlag: München, Germany, 2007; ISBN 978-3486575620. [Google Scholar]
- Shil, S.; Paul, S.; Misra, A. Charge-transfer-induced magnetism in mixed-stack complexes. J. Phys. Chem. C 2013, 117, 2016–2023. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Blackstock, S.C.; Kim, Y. Estimation of inner shell marcus terms for amino nitrogen compounds by molecular orbital calculations. J. Am. Chem. Soc. 1987, 109, 677–682. [Google Scholar] [CrossRef]
- Rawson, J.M.; Luzon, J.; Palacio, F. Magnetic exchange interactions in perfluorophenyl dithiadiazolyl radicals. Coord. Chem. Rev. 2005, 249, 2631–2641. [Google Scholar] [CrossRef]
- Yan, B.; Cramen, J.; McDonald, R.; Frank, N.L. Ferromagnetic spin-delocalized electron donors for multifunctional materials: π-Conjugated benzotriazinyl radicals. Chem. Commun. 2011, 47, 3201–3203. [Google Scholar] [CrossRef] [PubMed]
- Novoa, J.; Deumal, M.; Jornet-Somoza, J. Calculation of microscopic exchange interactions and modelling of macroscopic magnetic properties in molecule-based magnets. Chem. Soc. Rev. 2011, 40, 3182–3212. [Google Scholar] [CrossRef] [PubMed]
- Autschbach, J.; Srebro, M. Delocalization error and “functional tuning” in Kohn–Sham calculations of molecular properties. Acc. Chem. Res. 2014, 47, 2592–2602. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zheng, X.; Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Local scaling correction for reducing delocalization error in density functional approximations. Phys. Rev. Lett. 2015, 114, 053001. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Autschbach, J. Influence of the delocalization error and applicability of optimal functional tuning in density functional calculations of nonlinear optical properties of organic donor–acceptor chromophores. ChemPhysChem 2013, 14, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Mori-Sánchez, P.; Cohen, A.J.; Yang, W. Delocalization errors in density functionals and implications for main-group thermochemistry. J. Chem. Phys. 2008, 129, 204112. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Cho, D.; Ko, C.K.; Ikabata, Y.; Wakayama, K.; Yoshikawa, T.; Nakai, H.; Lee, J.Y. Effect of hartree-fock exact exchange on intramolecular magnetic coupling constants of organic diradicals. J. Chem. Phys. 2015, 142, 024318. [Google Scholar] [CrossRef] [PubMed]
- Shil, S.; Herrmann, C. Performance of range-separated hybrid exchange-correlation functionals for the calculation of magnetic exchange coupling constants of organic diradicals. J. Comput. Chem. 2018, 39, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.; Devlin, F.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Noodleman, L.; Case, D.A. Density-functional theory of spin polarization and spin coupling in iron—Sulfur clusters. Adv. Inorg. Chem. 1992, 38, 423–470. [Google Scholar]
- Caballol, R.; Castell, O.; Illas, F.; de PR Moreira, I.; Malrieu, J. Remarks on the proper use of the broken symmetry approach to magnetic coupling. J. Phys. Chem. A 1997, 101, 7860–7866. [Google Scholar] [CrossRef]
- Sheldrick, G.M. ShelxT—Integrated space-group and crystal structure determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with Shelxl. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Fielding, A.J.; Concilio, M.G.; Heaven, G.; Hollas, M.A. New developments in spin labels for pulsed dipolar EPR. Molecules 2014, 19, 16998–17025. [Google Scholar] [CrossRef] [PubMed]
- Shevelev, G.Y.; Krumkacheva, O.A.; Lomzov, A.A.; Kuzhelev, A.A.; Rogozhnikova, O.Y.; Trukhin, D.V.; Troitskaya, T.I.; Tormyshev, V.M.; Fedin, M.V.; Pyshnyi, D.V. Physiological-temperature distance measurement in nucleic acid using triarylmethyl-based spin labels and pulsed dipolar EPR spectroscopy. J. Am. Chem. Soc. 2014, 136, 9874–9877. [Google Scholar] [CrossRef] [PubMed]
- Jassoy, J.J.; Berndhäuser, A.; Duthie, F.; Kühn, S.P.; Hagelueken, G.; Schiemann, O. Versatile trityl spin labels for nanometer distance measurements on biomolecules in vitro and within cells. Angew. Chem. Int. Ed. 2017, 56, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Jassoy, J.J.; Meyer, A.; Spicher, S.; Wübben, C.; Schiemann, O. Synthesis of nanometer sized bis- and tris-trityl model compounds with different extent of spin-spin coupling. Molecules 2018, 23, 682. [Google Scholar] [CrossRef] [PubMed]
- Bobko, A.A.; Dhimitruka, I.; Zweier, J.L.; Khramtsov, V.V. Trityl radicals as persistent dual function pH and oxygen probes for in vivo electron paramagnetic resonance spectroscopy and imaging: Concept and experiment. J. Am. Chem. Soc. 2007, 129, 7240–7241. [Google Scholar] [CrossRef] [PubMed]
- Herrling, T.; Fuchs, J.; Rehberg, J.; Groth, N. UV-induced free radicals in the skin detected by ESR spectroscopy and imaging using nitroxides. Free Radic. Biol. Med. 2003, 35, 59–67. [Google Scholar] [CrossRef]
- Bobko, A.A.; Dhimitruka, I.; Eubank, T.D.; Marsh, C.B.; Zweier, J.L.; Khramtsov, V.V. Trityl-based EPR probe with enhanced sensitivity to Oxygen. Free Radic. Biol. Med. 2009, 47, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Sauvée, C.; Rosay, M.; Casano, G.; Aussenac, F.; Weber, R.T.; Ouari, O.; Tordo, P. Highly efficient, water-soluble polarizing agents for dynamic nuclear polarization at high frequency. Angew. Chem. Int. Ed. 2013, 52, 10858–10861. [Google Scholar] [CrossRef] [PubMed]
- Dane, E.L.; Corzilius, B.; Rizzato, E.; Stocker, P.; Maly, T.; Smith, A.A.; Griffin, R.G.; Ouari, O.; Tordo, P.; Swager, T.M. Rigid orthogonal Bis-TEMPO biradicals with improved solubility for dynamic nuclear polarization. J. Org. Chem. 2012, 77, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Gómez, J.L.; Monteagudo, E.; Lloveras, V.; Parella, T.; Veciana, J.; Vidal-Gancedo, J. Optimized polarization build-up times in dissolution DNP-NMR using a benzyl amino derivative of BDPA. RSC Adv. 2016, 6, 27077–27082. [Google Scholar] [CrossRef] [Green Version]
- Kubicki, D.J.; Casano, G.; Schwarzwälder, M.; Abel, S.; Sauvée, C.; Ganesan, K.; Yulikov, M.; Rossini, A.J.; Jeschke, G.; Copéret, C.; et al. Rational design of dinitroxide biradicals for efficient cross-effect dynamic nuclear polarization. Chem. Sci. 2016, 7, 550–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathies, G.; Caporini, M.A.; Michaelis, V.K.; Liu, Y.; Hu, K.-N.; Mance, D.; Zweier, J.L.; Rosay, M.; Baldus, M.; Griffin, R.G. Efficient dynamic nuclear polarization at 800 MHz/527 GHz with trityl-nitroxide biradicals. Angew. Chem. Int. Ed. 2015, 54, 11770–11774. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.-N.; Bajaj, V.S.; Rosay, M.; Baldus, M.; Griffin, R.G. High-frequency dynamic nuclear polarization using TEMPO and trityl radicals. J. Chem. Phys. 2007, 126, 044512. [Google Scholar] [CrossRef] [PubMed]
- De Nooy, A.E.J.; Besemer, A.C.; Van Bekkum, H. Highly selective nitroxyl radical-mediated oxidation of primary alcohol groups in water-soluble glucan. Carbohyd. Res. 1995, 269, 89–98. [Google Scholar] [CrossRef]
- Ryland, B.L.; Stahl, S.S. Practical aerobic oxidations of alcohols and amines with homogenous copper/TEMPO and related catalyst systems. Angew. Chem. Int. Ed. 2014, 53, 8804–8838. [Google Scholar] [CrossRef] [PubMed]
- Blasi, D.; Nikolaidou, D.M.; Terenziani, F.; Ratera, I.; Veciana, J. Excimers from stable and persistent supramolecular radical-pairs in Red/NIR-emitting organic nanoparticles and polymeric films. Phys. Chem. Chem. Phys. 2017, 19, 9313–9319. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, T.; Miyamoto, D.; Nagasaki, Y. Design of core-shell-type nanoparticles carrying stable radicals in the core. Biomacromolecules 2009, 10, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Le, T.-N.; Grewal, H.; Changcoco, V.; Truong, V.; Brook, D.J.R. Water soluble, chiral, verdazyl radicals derived from aldoses. Tetrahedron 2016, 72, 6368–6374. [Google Scholar] [CrossRef] [Green Version]
- Thurber, K.R.; Le, T.-N.; Changcoco, V.; Truong, V.; Brook, D.J.R. Verdazyl-ribose: A new radical for solid-state dynamic nuclear polarization at high magnetic field. J. Magn. Res. 2018, 289, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kashara, T.; Chen, E.K.Y.; Hamer, G.K.; Georges, M.K. 1,3-Dipolar cycloaddition reactions initiated with the 1,5-Dimethyl-3-phenyl-6-oxoverdazyl radical. Eur. J. Org. Chem. 2008, 2008, 4571–4574. [Google Scholar] [CrossRef]
- Bancerz, M.; Georges, M.K. Verdazyl radicals as substrates for organic synthesis: A synthesis of 3-Methyl-5-aryl-1,3,4-oxadiazolones. J. Org. Chem. 2011, 76, 6377–6382. [Google Scholar] [CrossRef] [PubMed]
- Cumaraswamy, A.A.; Hamer, G.K.; Georges, M.K. Verdazyl radicals as substrates for organic synthesis: The synthesis and characterization of [12]-, [13]-, and [21]-Paraheteraphanes. Eur. J. Org. Chem. 2012, 2012, 1717–1722. [Google Scholar] [CrossRef]
- Bancerz, M.; Prack, E.; Georges, M.K. Triphenyl verdazyl radicals’ reactivity with alkyne carboxylates as a synthetic route to 1-(Phenyldiazenyl) Isoquinoline-3,4-dicarboxylates. Tetrahedron Lett. 2012, 53, 4026–4029. [Google Scholar] [CrossRef]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | giso | Aiso(N1/N2) a [MHz] | Aiso(HN-Phenyl) b [MHz] | LWPPc [MHz] |
|---|---|---|---|---|
| 3 | 2.00365 | 16.81/16.62 | 3.05, 1.30, 3.30, 0.76 | 0.22, 0.60 |
| 5 | 2.00366 | 16.81/16.62 | 3.05, 1.25, 3.30, 0.72 | 0.22, 0.60 |
| 6 | 2.00366 | 16.81/16.62 | 3.05, 1.25, 3.30, 0.72 | 0.22, 0.60 |
| Cmpd | C3NiNjCH2 a [°] | C3NNCPh a [°] | d(C3-N) b [Å] | d(N-N) b [Å] | d(N-CH2) b [Å] |
|---|---|---|---|---|---|
| 3· exp. | 22.30 | 168.70 | 1.339 | 1.359 | 1.454 |
| 3· DFT | 21.55 | 166.77 | 1.334 | 1.342 | 1.448 |
| 3+ exp. | 18.38 | 160.50 | 1.351 | 1.305 | 1.461 |
| 3+ DFT | 17.11 | 154.73 | 1.345 | 1.296 | 1.458 |
| 3H exp. | 32.61/19.54 | 95.69/161.11 | 1.396/1.290 | 1.410/1.399 | 1.454/1.456 |
| 3H DFT | 34.27/16.15 | 96.10/174.96 | 1.398/1.289 | 1.401/1.362 | 1.465/1.441 |
| 5· exp. | 20.97 | 171.14 | 1.340 | 1.342 | 1.459 |
| 5· DFT | 21.48 | 166.48 | 1.335 | 1.341 | 1.448 |
| 6· exp. | 23.34 | 169.76 | 1.337 | 1.366 | 1.461 |
| 6· DFT | 21.51 | 166.06 | 1.335 | 1.341 | 1.448 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jobelius, H.; Wagner, N.; Schnakenburg, G.; Meyer, A. Verdazyls as Possible Building Blocks for Multifunctional Molecular Materials: A Case Study on 1,5-Diphenyl-3-(p-iodophenyl)-verdazyl Focusing on Magnetism, Electron Transfer and the Applicability of the Sonogashira-Hagihara Reaction. Molecules 2018, 23, 1758. https://doi.org/10.3390/molecules23071758
Jobelius H, Wagner N, Schnakenburg G, Meyer A. Verdazyls as Possible Building Blocks for Multifunctional Molecular Materials: A Case Study on 1,5-Diphenyl-3-(p-iodophenyl)-verdazyl Focusing on Magnetism, Electron Transfer and the Applicability of the Sonogashira-Hagihara Reaction. Molecules. 2018; 23(7):1758. https://doi.org/10.3390/molecules23071758
Chicago/Turabian StyleJobelius, Hannah, Norbert Wagner, Gregor Schnakenburg, and Andreas Meyer. 2018. "Verdazyls as Possible Building Blocks for Multifunctional Molecular Materials: A Case Study on 1,5-Diphenyl-3-(p-iodophenyl)-verdazyl Focusing on Magnetism, Electron Transfer and the Applicability of the Sonogashira-Hagihara Reaction" Molecules 23, no. 7: 1758. https://doi.org/10.3390/molecules23071758
APA StyleJobelius, H., Wagner, N., Schnakenburg, G., & Meyer, A. (2018). Verdazyls as Possible Building Blocks for Multifunctional Molecular Materials: A Case Study on 1,5-Diphenyl-3-(p-iodophenyl)-verdazyl Focusing on Magnetism, Electron Transfer and the Applicability of the Sonogashira-Hagihara Reaction. Molecules, 23(7), 1758. https://doi.org/10.3390/molecules23071758