
Zinc (II)-Mediated Selective O-Benzylation of 2-Oxo-1,2-Dihydropyridines Systems

Abstract
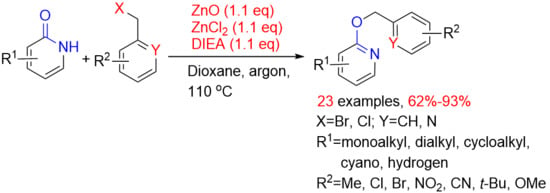
:
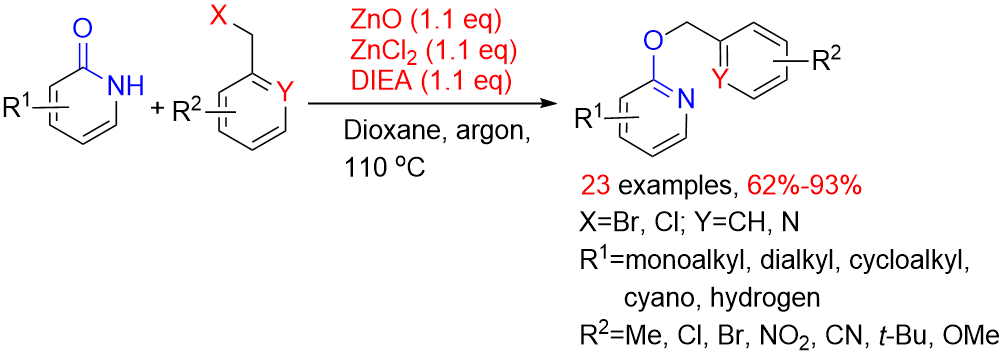
1. Introduction
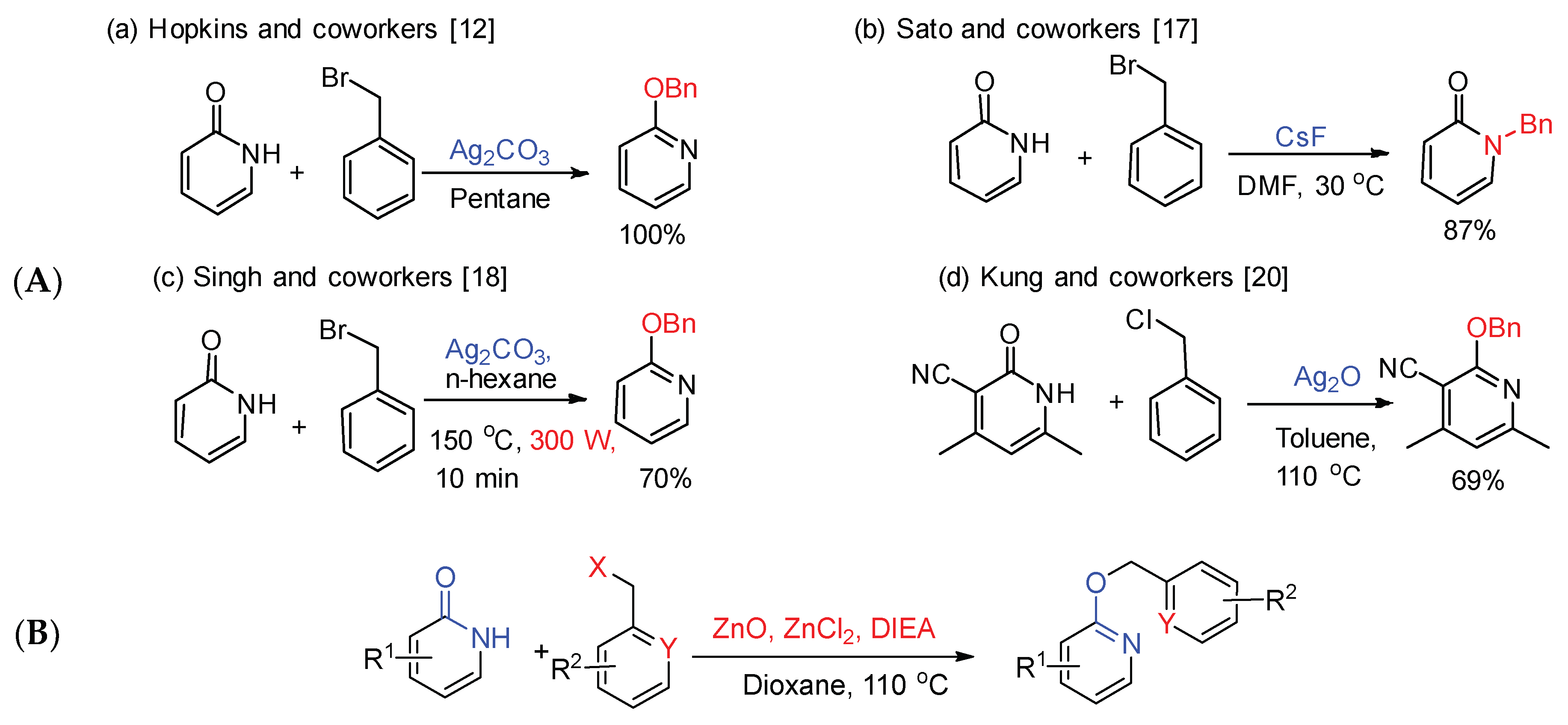
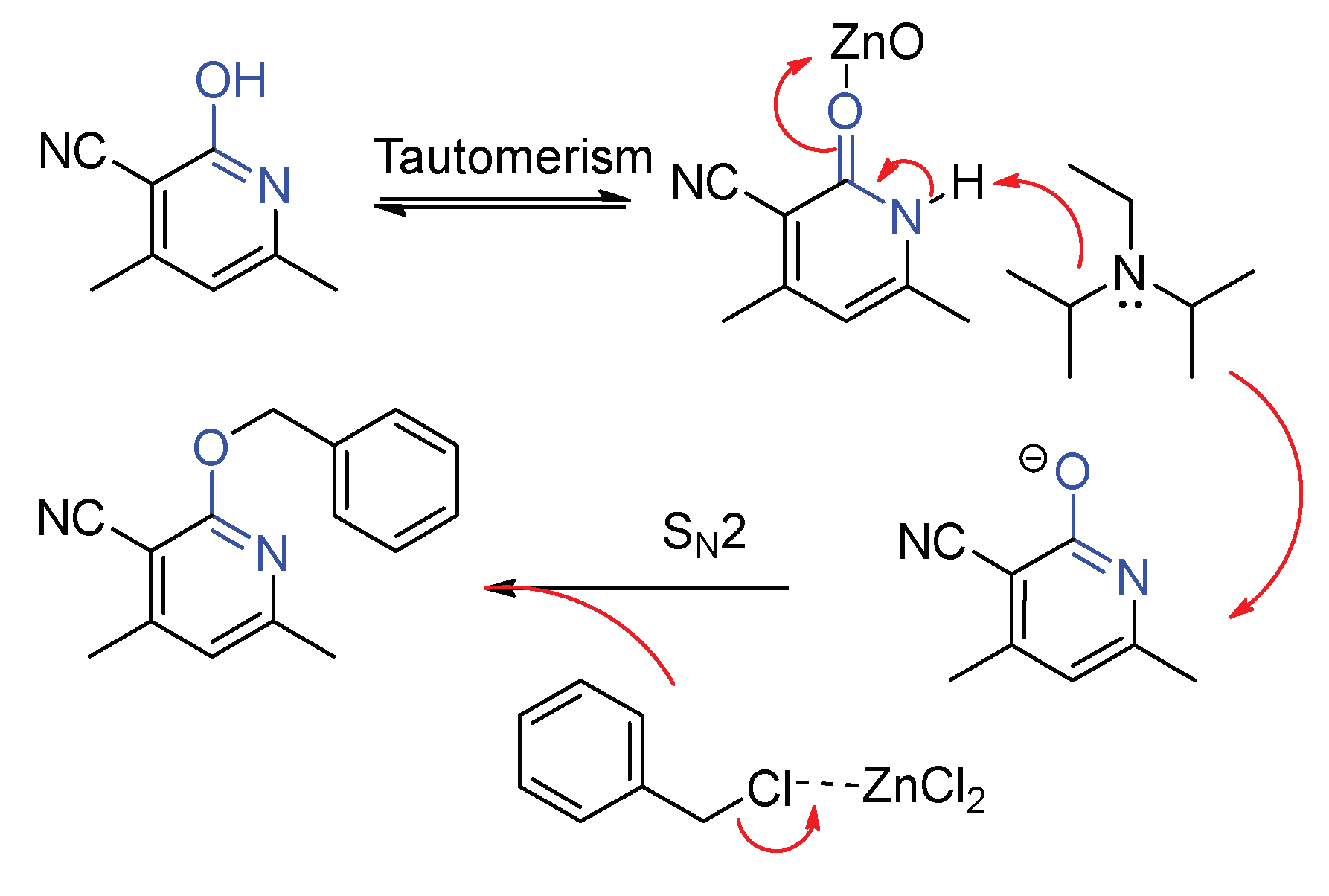
2. Results and Discussion
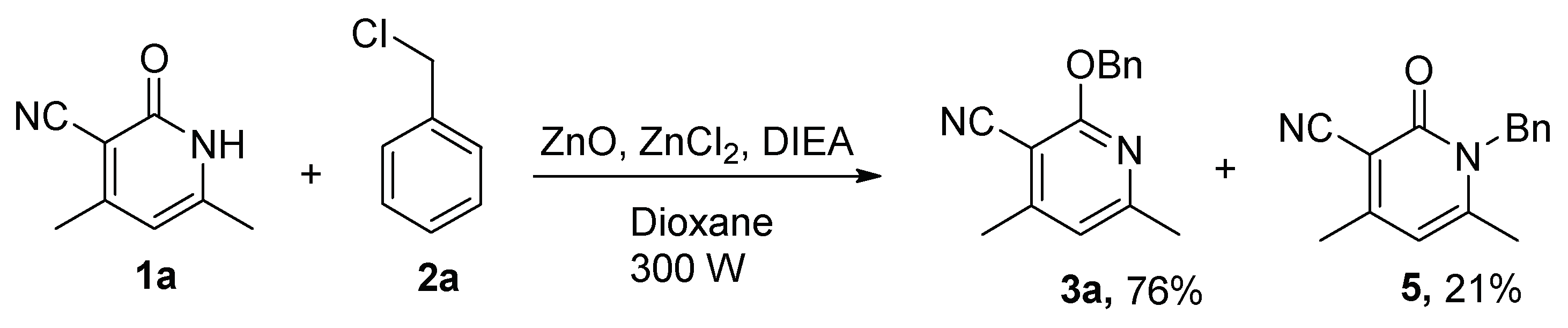
2.1. Optimization of the Reaction Conditions
2.2. Scope with Respect to Substituted 2-Oxo-1,2-Dihydropyridines
2.3. Scope with Respect to Substituted Benzyl Halides
3. Materials and Methods
3.1. Materials
3.2. Microwave-Assisted Synthesis of 3a
3.3. General Procedures for the Synthesis of Substituted 2-Oxo-1,2-dihydropyridines of 1a–1k
3.4. General Procedures for the Synthesis of 3a–3m
3.5. General Procedures for the Synthesis of 4a–4k
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mercedes, T.; Salvador, G.; Margarita, P. New synthetic methods to 2-pyridone rings. Curr. Org. Chem. 2005, 9, 1757–1779. [Google Scholar]
- Verissimo, E.; Berry, N.; Gibbons, P.; Cristiano, M.L.S.; Rosenthal, P.J.; Gut, J.; Ward, S.A.; O’Neill, P.M. Design and synthesis of novel 2-pyridone peptidomimetic falcipain 2/3 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4210–4214. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, P.E.J.; Dyer, D.L.; Naylor-Olsen, A.M.; Vacca, J.P.; Gardell, S.J.; Lewis, S.D.; Lucas, B.J., Jr.; Lyle, E.A.; Lynch, J.J., Jr.; Mulichak, A.M. L-373,890, an achiral, noncovalent, subnanomolar thrombin inhibitor. Bioorg. Med. Chem. Lett. 1997, 7, 1497–1500. [Google Scholar] [CrossRef]
- Reiner, J.E.; Lim-Wilby, M.S.; Brunck, T.K.; Ha-Uong, T.; Goldman, E.A.; Abelman, M.A.; Nutt, R.F.; Semple, J.E.; Tamura, S.Y. Investigation of the S3 site of thrombin: Design, synthesis and biological activity of 4-substituted 3-amino-2-pyridones incorporating P1-argininals. Bioorg. Med. Chem. Lett. 1999, 9, 895–900. [Google Scholar] [CrossRef]
- Sanderson, P.E.; Cutrona, K.J.; Dorsey, B.D.; Dyer, D.L.; McDonough, C.M.; Naylor-Olsen, A.M.; Chen, I.W.; Chen, Z.; Cook, J.J.; Gardell, S.J.; et al. L-374,087, an efficacious, orally bioavailable, pyridinone acetamide thrombin inhibitor. Bioorg. Med. Chem. Lett. 1998, 8, 817–822. [Google Scholar] [CrossRef]
- Veale, C.A.; Bernstein, P.R.; Bryant, C.; Ceccarelli, C.; Damewood, J.R., Jr.; Earley, R.; Feeney, S.W.; Gomes, B.; Kosmider, B.J.; Steelman, G.B.; et al. Nonpeptidic inhibitors of human leukocyte elastase. 5. design, synthesis, and X-ray crystallography of a series of orally active 5-aminopyrimidin-6-one-containing trifluoromethyl ketones. J. Med. Chem. 1995, 38, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Brown, F.J.; Andisik, D.W.; Bernstein, P.R.; Bryant, C.B.; Ceccarelli, C.; Damewood, J.R., Jr.; Edwards, P.D.; Earley, R.A.; Feeney, S.; Green, R.C.; et al. Design of orally active, non-peptidic inhibitors of human leukocyte elastase. J. Med. Chem. 1994, 37, 1259–1261. [Google Scholar] [CrossRef] [PubMed]
- Semple, G.; Ashworth, D.M.; Batt, A.R.; Baxter, A.J.; Benzies, D.W.; Elliot, L.H.; Evans, D.M.; Franklin, R.J.; Hudson, P.; Jenkins, P.D.; et al. Peptidomimetic aminomethylene ketone inhibitors of interleukin-i[3-converting enzyme (ice). Bioorg. Med. Chem. Lett. 1998, 8, 959–964. [Google Scholar] [CrossRef]
- Semple, G.; Ashworth, D.M.; Batt, A.R.; Baxter, A.J.; Benzies, D.W.; Elliot, L.H.; Evans, D.M.; Franklin, R.J.; Hudson, P.; Jenkins, P.D.; et al. Pyridone-based peptidomimetic inhibitors of interleukin-lβ-converting enzyme (ice). Bioorg. Med. Chem. Lett. 1997, 7, 1337–1342. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Adlington, R.M.; Conte, A.; Irlapati, N.R.; Marquez, R.; Pritchard, G. Total synthesis of pyridovericin: Studies toward the biomimetic synthesis of pyridomacrolidin. J. Org. Lett. 2002, 4, 2125–2127. [Google Scholar] [CrossRef]
- Youngdale, G.A.; Oglia, T.F. 1,2-Dihydro-2-oxo-6-(2,2-dimethylpropyl)-3-pyridinecarboxylic acid, analogues, and derivatives. A new class of oral hypoglycemic agents. J. Med. Chem. 1985, 28, 1790–1796. [Google Scholar] [CrossRef] [PubMed]
- Kornblum, N.; Smiley, R.A.; Blackwood, R.K.; Iffland, D.C. The mechanism of the reaction of silver nitrite with alkyl halides. The contrasting reactions of silver and alkali metal salts with alkyl halides. The alkylation of ambident anions. J. Am. Chem. Soc. 1955, 77, 6269–6280. [Google Scholar] [CrossRef]
- Kornblum, N.; Paul, J.B.; William, J.L.N. Solvation as a factor in the alkylation of ambident anions: The importance of the hydrogen bonding capacity of the solvent. J. Am. Chem. Soc. 1963, 85, 1141–1147. [Google Scholar] [CrossRef]
- Kornblum, N.; Seltzer, R.; Haberfield, P. Solvation as a factor in the alkylation of ambident anions: The importance of the dielectric factor. J. Am. Chem. Soc. 1963, 85, 1148–1154. [Google Scholar] [CrossRef]
- Hopkins, G.C.; Jonak, J.P.; Tieckelmann, H.; Minnemeyer, H.J. Alkylations of heterocyclic ambident anions. I. 2-hydroxypyrimidines. J. Org. Chem. 1966, 31, 3969–3973. [Google Scholar] [CrossRef]
- Hopkins, G.; Jonak, J.; Minnemeyer, H.; Tieckelmann, H. Alkylations of heterocyclic ambident anions. 11. alkylation of 2-pyridone salts. J. Org. Chem. 1967, 32, 4040–4044. [Google Scholar] [CrossRef]
- Sato, T.; Yoshimatsu, K.; Otera, J. CsF in organic synthesis. tuning of n- or o-alkylation of 2-pyridone. Synlett 1995, 1995, 845–846. [Google Scholar] [CrossRef]
- Singh, B.K.; Cavalluzzo, C.; Maeyer, M.D.; Debyser, Z.; Parmarb, V.S.; Eycken, E.V. Microwave-assisted silver(I)-mediated selective o-alkylation of aromatic imidate systems. Synthesis 2009, 16, 2725–2728. [Google Scholar] [CrossRef]
- Vavilina, G.; Zicmanis, A.; Mekss, P.; Klavins, M. Alkylation of the 2-hydroxypyridine anion in ionic liquid media. Chem. Heterocyclic. Compd. 2008, 44, 549–558. [Google Scholar] [CrossRef]
- Kung, P.P.; Rui, E.; Bergqvist, S.; Bingham, P.; Braganza, J.; Collins, M.; Cui, M.; Diehl, W.; Dinh, D.; Fan, C.; et al. Design and synthesis of pyridone-containing 3,4-dihydroisoquinoline-1(2H)-ones as a novel class of enhancer of zeste homolog 2 (EZH2) inhibitors. J. Med. Chem. 2016, 59, 8306–8325. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.N.; Mallarrddy, G.; Nagender, P.; Rao, P.S.; Poorinachandra, Y.; Ranjithreddy, P.; Kumar, C.G.; Narsaiah, B. Synthesis of novel triazole functionalized pyridine derivatives as potential antimicrobial and anti-biofilm agents. Indian J. Chem. Sect. B 2016, 55B, 1361–1375. [Google Scholar]
- Almena, I.; Diaz-Ortiz, A.; Diez-Barra, E.; de la Hoz, A.; Loupy, A. Solvent-free benzylation of 2-pyridone. Regiospecific N- or C-alkylation. Chem. Lett. 1996, 27, 333–334. [Google Scholar] [CrossRef]
- De la Hoz, A.; Prieto, M.P.; Rajzmann, M.; de Cózar, A.; Díaz-Ortiz, A.; Moreno, A.; Cossíoc, F.P. Selectivity under microwave irradiation. Benzylation of 2-pyridone: An experimental and theoretical study. Tetrahedron 2008, 64, 8169–8176. [Google Scholar] [CrossRef]
- Elassar, A.Z.A. Synthesis and reactions of 3-cyano-4,6-dimethyl-2-pyridone. J. Heterocycl. Chem. 2011, 48, 272–278. [Google Scholar] [CrossRef]
- Azuma, Y.; Morone, M.; Nagayama, K.; Kawamata, Y.; Sato, A. Synthesis and reactions of 4-chloro-1,2-dihydro-6-methyl-2-oxo-3-pyridinecarbonitrile. Heterocycles 2003, 60, 1461–1468. [Google Scholar] [CrossRef]
- Raymond, M.P. Condensations of unsymmetrical ketones. I. condensations with ethyl formate. J. Am. Chem. Soc. 1947, 69, 2670–2672. [Google Scholar]
- Hideshi, K.; Hiroshi, M. A convenient synthesis of 2-aminopyridine-3-carbonitriies. J. Heterocycl. Chem. 1977, 14, 1077–1079. [Google Scholar]
- Angel, A.; Celia, A.; Alfonso, G.O.; Rafael, P.; Martina, V. The reaction of β-aminoenones with substituted acetonitriles. Regiospecific synthesis of 2(1H)-pyridones. J. Heterocycl. Chem. 1987, 24, 709–713. [Google Scholar]
- Akio, S.; Hiroshi, M.B. The cyclization of ethyl cyanoacetate and ketones by ammonium acetate. Bull. Chem. Soc. Jpn. 1967, 40, 1680–1684. [Google Scholar]
- Luisa, M.; Giulia, M.; Pietro, S.; Paola, D.; Maria, G.R.; Franco, B.; Gabriella, B. Synthesis and cardiotonic activity of esters of 2-substituted 5-cyano1,6-dihydro-6-oxo-3-pyridinecarboxylic acids. Crystal structure of 2-methyl, 2-t-butyl and 2-phenyl esters. Eur. J. Med. Chem. 1989, 24, 517–529. [Google Scholar]
- Hassan, M.A.; Soliman, E.A.; Hamed, A.A. Synthesis and reactions of polysubstituted 2(1H)-pyridones and pyridines. Cheminform 1982, 56, 419–423. [Google Scholar]
- Lott, W.A.; Shaw, E. Analogs of aspergillic acid; various antibacterial heterocyclic hydroxamic acids. J. Am. Chem. Soc. 1949, 71, 70–73. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | ZnO (Equiv) | Lewis Acid (Equiv) | Base (Equiv) | Solvent | Time (h) | Yield (%) 2 | |
|---|---|---|---|---|---|---|---|
| 3a | 5 | ||||||
| Entry 1 | 1.1 | -- | -- | Toluene | 24 | -- 3 | -- 3 |
| Entry 2 | 1.1 | ZnCl2 (1.1) | -- | Toluene | 24 | -- 3 | -- 3 |
| Entry 3 | 1.1 | ZnCl2 (1.1) | TEA (1.1) | Toluene | 24 | 71 | -- 3 |
| Entry 4 | -- | ZnCl2 (1.1) | TEA (1.1) | Toluene | 24 | -- 3 | -- 3 |
| Entry 5 | -- | -- | TEA (1.1) | Toluene | 24 | -- 4 | 15 |
| Entry 6 | 0.1 | ZnCl2 (0.1) | TEA (0.1) | Toluene | 36 | 25 | -- 3 |
| Entry 7 | 1.1 | ZnCl2 (1.1) | TEA (1.1) | Dioxane | 24 | 64 | -- 3 |
| Entry 8 | 0.1 | ZnCl2 (0.1) | DIEA (1.1) | Dioxane | 24 | 13 | -- 3 |
| Entry 9 | 1.1 | ZnCl2(1.1) | DIEA (1.1) | Dioxane | 24 | 90 | -- 3 |
| Entry 10 | 1.1 | ZnCl2 (1.1) | DIEA (1.1) | Ethylene glycol | 24 | -- 3 | -- 3 |
| Entry 11 | 1.1 | ZnCl2 (1.1) | DIEA (1.1) | DMF | 24 | -- 3 | -- 3 |
| Entry 12 | 1.1 | ZnCl2 (1.1) | DIEA (1.1) | DMSO | 24 | -- 3 | -- 3 |
| Entry 13 | 1.1 | ZnCl2 (1.1) | K2CO3 (1.1) | DMF | 24 | 21 | 73 |
| Entry 14 | 1.1 | AlCl3 (1.1) | DIEA (1.1) | Dioxane | 24 | -- 3 | -- 3 |
| Entry 15 | 1.1 | FeCl3 (1.1) | DIEA (1.1) | Dioxane | 24 | -- 3 | -- 3 |
| Entry 16 | 1.1 | CuCl2 (1.1) | DIEA (1.1)) | Dioxane | 24 | -- 3 | -- 3 |
| Entry 17 5 | 1.1 | ZnCl2 (1.1) | DIEA (1.1) | Dioxane | 24 | 86 | -- 3 |

 |

 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Du, F.; Liang, X.; Liu, W.; Fang, T.; Chen, G. Zinc (II)-Mediated Selective O-Benzylation of 2-Oxo-1,2-Dihydropyridines Systems. Molecules 2018, 23, 1784. https://doi.org/10.3390/molecules23071784
Zhou Q, Du F, Liang X, Liu W, Fang T, Chen G. Zinc (II)-Mediated Selective O-Benzylation of 2-Oxo-1,2-Dihydropyridines Systems. Molecules. 2018; 23(7):1784. https://doi.org/10.3390/molecules23071784
Chicago/Turabian StyleZhou, Qifan, Fangyu Du, Xinjie Liang, Wenqiang Liu, Ting Fang, and Guoliang Chen. 2018. "Zinc (II)-Mediated Selective O-Benzylation of 2-Oxo-1,2-Dihydropyridines Systems" Molecules 23, no. 7: 1784. https://doi.org/10.3390/molecules23071784
APA StyleZhou, Q., Du, F., Liang, X., Liu, W., Fang, T., & Chen, G. (2018). Zinc (II)-Mediated Selective O-Benzylation of 2-Oxo-1,2-Dihydropyridines Systems. Molecules, 23(7), 1784. https://doi.org/10.3390/molecules23071784