
Visible-Light, Iodine-Promoted Formation of N-Sulfonyl Imines and N-Alkylsulfonamides from Aldehydes and Hypervalent Iodine Reagents


Abstract
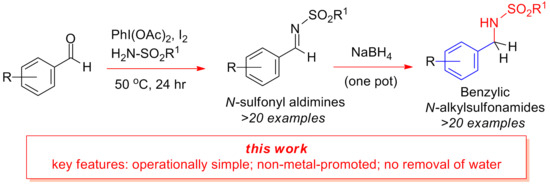
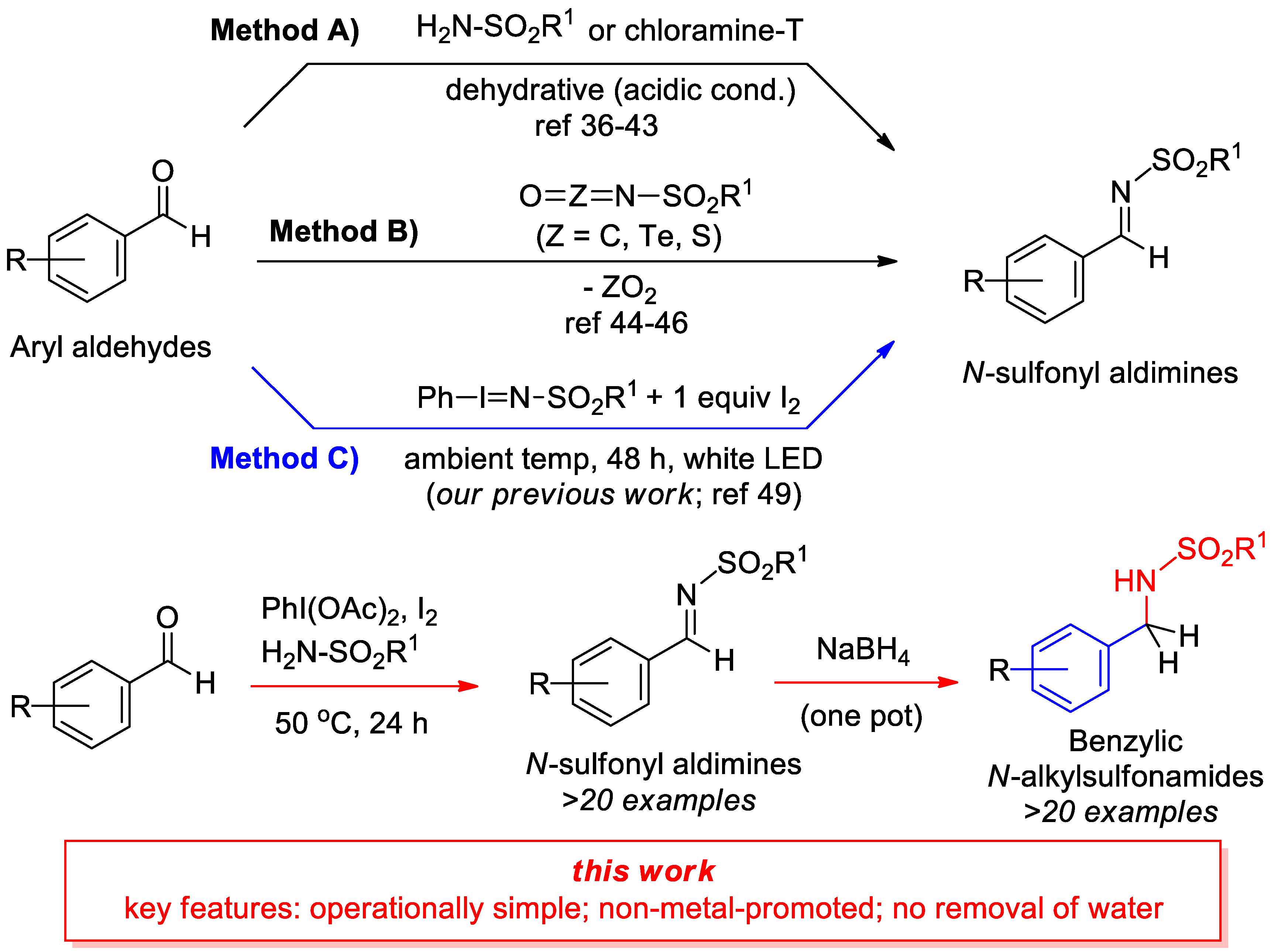
:
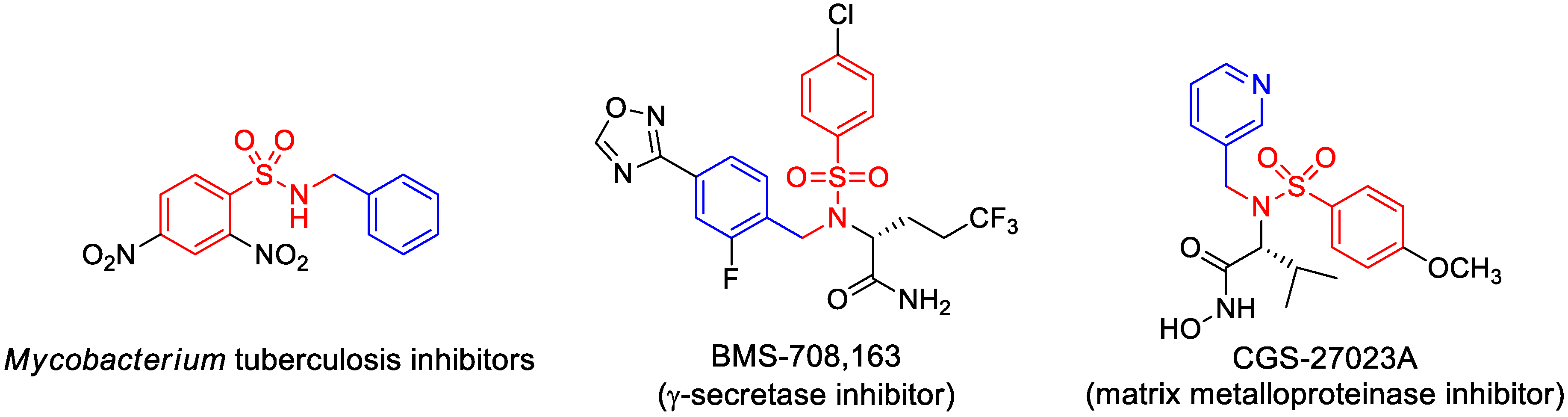
1. Introduction
2. Results
2.1. Optimization of Reaction Conditions and Control Reactions
2.2. Substrate Scope
2.3. Investigation of Mechanism
2.3.1. Deuterium-Labeling Experiment
2.3.2. Isolation of Mechanistic Intermediates
3. Discussion
3.1. Trends in Reactivity
3.2. Proposed Mechanism
4. Materials and Methods
4.1. Chemicals and Instruments
4.2. Synthetic Procedures
4.2.1. General Procedure for the Preparation of N-Sulfonyl Imines (2) (1H-NMR Yields)
4.2.2. General Procedure for the Preparation of N-Alkylsulfonamides (3)
4.2.3. Isotopically Labeled Experiment
4.2.4. Observation of Carboxylic Acid Intermediate
4.2.5. Carboxylic Acid-Promoted Condensation
4.3. Characterization Data
4.3.1. 4-Chloro-N-[(4-methylphenyl)methyl]-benzenesulfonamide (3bc)
4.3.2. 2-Chloro-N-[(4-methylphenyl)methyl]-benzenesulfonamide (3bd)
4.3.3. N-[(4-Bromophenyl)methyl]-4-chlorobenzenesulfonamide (3dc)
4.3.4. 4-Methyl-N-[(4-nitrophenyl)methyl]-benzenesulfonamide (3fa)
4.3.5. N-[(4-Cyanophenyl)methyl]-4-methylbenzenesulfonamide (3ga)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Carlo, B.; Donna, M.H.; Amos, B.S. Carboxylic acid (bio) isosteres in drug design. Chem. Med. Chem. 2013, 8, 385–395. [Google Scholar]
- Ballatore, C.; Soper, J.H.; Piscitelli, F.; James, M.; Huang, L.; Atasoylu, O.; Huryn, D.M.; Trojanowski, J.Q.; Lee, V.M.; Brunden, K.R.; et al. Cyclopentane-1,3-dione: A novel isostere for the carboxylic acid functional group. Application to the design of potent thromboxane (A2) receptor antagonists. J. Med. Chem. 2011, 54, 6969–6983. [Google Scholar] [CrossRef] [PubMed]
- Malwal, S.R.; Sriram, D.; Yogeeswari, P.; Konkimalla, V.B.; Chakrapani, H. Design, synthesis and evaluation of thiol-activated sources of sulfur dioxide (SO2) as antimycobacterial agents. J. Med. Chem. 2012, 55, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Mukherjee, A.; Saha, A.; Jha, T. Arylsulfonamides and selectivity of matrix metalloproteinase-2: An overview. Eur. J. Med. Chem. 2017, 129, 72–109. [Google Scholar] [CrossRef] [PubMed]
- Bloch, R. Additions of organometallic reagents to CN bonds: Reactivity and selectivity. Chem. Rev. 1998, 98, 1407–1438. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Ishitani, H. Catalytic enantioselective addition to imines. Chem. Rev. 1999, 99, 1069–1094. [Google Scholar] [CrossRef] [PubMed]
- Otomaru, Y.; Tokunaga, N.; Shintani, R.; Hayashi, T. C2-symmetric bicyclo[3.3.1]nonadiene as a chiral ligand for rhodium-catalyzed asymmetric arylation of N-(4-Nitrobenzenesulfonyl)arylimines. Org. Lett. 2005, 7, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Yu, H.-J.; Wu, N.-Y.; Feng, C.-G.; Lin, G.-Q. C1-symmetric dicyclopentadienes as new chiral diene ligands for asymmetric rhodium-catalyzed arylation of N-Tosylarylimines. Org. Lett. 2010, 12, 3820–3823. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Hepburn, H.B.; Chotsaeng, N.; Lam, H.W. Enantioselective rhodium-catalyzed nucleophilic allylation of cyclic imines with allylboron reagents. Angew. Chem. Int. Ed. 2012, 51, 8309–8313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensel, A.; Nagura, K.; Delvos, L.B.; Oestreich, M. Enantioselective addition of silicon nucleophiles to aldimines using a preformed NHC-copper(I) complex as the catalyst. Angew. Chem. 2014, 53, 4964–4967. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.R.; Kelly, C.B.; Siegenfeld, A.P.; Molander, G.A. Mild, redox-neutral alkylation of imines enabled by an organic photocatalyst. ACS Catal. 2017, 7, 1766–1770. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Silverman, S.M. Enantioselective construction of pyrrolidines by palladium-catalyzed asymmetric [3 + 2] cycloaddition of trimethylenemethane with imines. J. Am. Chem. Soc. 2012, 134, 4941–4954. [Google Scholar] [CrossRef] [PubMed]
- Illa, O.; Namutebi, M.; Saha, C.; Ostovar, M.; Chen, C.C.; Haddow, M.F.; Nocquet-Thibault, S.; Lusi, M.; McGarrigle, E.M.; Aggarwal, V.K. Practical and highly selective sulfur ylide-mediated asymmetric epoxidations and aziridinations using a cheap and readily available chiral sulfide: Extensive studies to map out scope, limitations, and rationalization of diastereo- and enantioselectivities. J. Am. Chem. Soc. 2013, 135, 11951–11966. [Google Scholar] [CrossRef] [PubMed]
- Lykke, L.; Halskov, K.S.; Carlsen, B.D.; Chen, V.X.; Jorgensen, K.A. Catalytic asymmetric diaziridination. J. Am. Chem. Soc. 2013, 135, 4692–4695. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Hisakuni, D.; Lin, C.H.; Minakata, S. 2-Halogenoimidazolium salt catalyzed aza-Diels-Alder reaction through halogen-bond formation. Org. Lett. 2015, 17, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Esquivias, J.; Gómez Arrayás, R.; Carretero, J.C. A copper(II)-catalyzed aza-friedel–crafts reaction of N-(2-pyridyl)sulfonyl aldimines: Synthesis of unsymmetrical diaryl amines and triaryl methanes. Angew. Chem. Int. Ed. 2006, 45, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.-X.; Xie, J.-H.; Duan, H.-F.; Wang, L.X.; Zhou, Q.L. Asymmetric friedel-crafts addition of indoles to N-sulfonyl aldimines: A simple approach to optically active 3-indolyl-methanamine derivatives. Org. Lett. 2006, 8, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, S.; Fu, X.; Yan, C.H. Metal-free aerobic oxidative coupling of amines to imines. Chem. Commun. 2011, 47, 10148–10150. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Liao, Y.; Lin, L.; Luo, W.; Liu, X.; Feng, X. N,N′-dioxide-scandium(III)-catalyzed asymmetric Aza-Friedel-Crafts reaction of sesamol with aldimines. J. Org. Chem. 2014, 79, 10662–10668. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Lv, H.; Guan, Y.H.; Zhu, H.B.; Cui, X.M.; Guo, K. Assembly of indenamine derivatives through in situ formed N-sulfonyliminium ion initiated cyclization. Chem. Commun. 2014, 50, 4119–4122. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Nishida, A.; Nakagawa, M. Ytterbium(III) Triflate/TMSCl: Efficient catalyst for imino ene reaction. Org. Lett. 2000, 2, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Nishida, A.; Nakagawa, M. Imino ene reaction catalyzed by ytterbium(III) triflate and TMSCl or TMSOTf. J. Org. Chem. 2003, 68, 3112–3120. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Bisai, A.; Pandey, A.; Singh, V.K. Imino-ene reaction of N-tosyl arylaldimines with α-methylstyrene: Application in the synthesis of important amines. Tetrahedron Lett. 2005, 46, 5039–5041. [Google Scholar] [CrossRef]
- Oliver, L.H.; Puls, L.A.; Tobey, S.L. Brønsted acid promoted imino-ene reactions. Tetrahedron Lett. 2008, 49, 4636–4639. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kiyohara, H.; Yamaguchi, M. Catalytic Silicon-mediated carbon-carbon bond-forming reactions of unactivated amides. J. Am. Chem. Soc. 2011, 133, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-H.; Huang, F.-P.; Zhu, P.; Dong, Z.-W.; Hui, X.-P. Synergistic chiral ion pair catalysts for asymmetric catalytic synthesis of quaternary α,β-diamino acids. Org. Lett. 2012, 14, 2010–2013. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, M.K.; Ghosh, K.; Yadav, A.K.; Nanaji, Y.; Halder, S.; Sayyad, M. Memory of chirality (MOC) concept in imino-aldol reaction: Enantioselective synthesis of α,β-diamino esters and aziridines. J. Org. Chem. 2013, 78, 2311–2326. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhao, J.C.G. Highly enantioselective three-component direct mannich reactions of unfunctionalized ketones catalyzed by bifunctional organocatalysts. Org. Lett. 2013, 15, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.S.; Tauchert, M.E.; Bergman, R.G.; Ellman, J.A. Rhodium(III)-catalyzed arylation of boc-imines via C−H bond functionalization. J. Am. Chem. Soc. 2011, 133, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Hesp, K.D.; Bergman, R.G.; Ellman, J.A. Rhodium-catalyzed synthesis of branched amines by direct addition of benzamides to imines. Org. Lett. 2012, 14, 2304–2307. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.A.; Zhao, B.; Shi, Y. Recent advances in transition metal-catalyzed sp3 C–H amination adjacent to double bonds and carbonyl groups. Chem. Soc. Rev. 2012, 41, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, L.; Li, X. Rh(III)-catalyzed olefination of N-sulfonyl imines: Synthesis of ortho-olefinated benzaldehydes. Org. Lett. 2013, 15, 6294–6297. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, K.; Azcargorta, A.R.; Cheng, Y.; Bolm, C. Directed additions of 2-arylpyridines and related substrates to cyclic imines through rhodium-catalyzed C–H functionalization. Org. Lett. 2014, 16, 2538–2541. [Google Scholar] [CrossRef] [PubMed]
- Wynne, J.H.; Price, S.E.; Rorer, J.R.; Stalick, W.M. Synthesis of novel functionalized N-tosylaldimines. Synth. Commun. 2003, 33, 341–352. [Google Scholar] [CrossRef]
- Khalafi-Nezhad, A.; Parhami, A.; Zare, A.; Shirazi, A.N.; Zare, A.R.M.; Hassaninejad, A. Triarylmethyl chlorides as novel, efficient, and mild organic catalysts for the synthesis of N-sulfonyl imines under neutral conditions. Can. J. Chem. 2008, 86, 456–461. [Google Scholar] [CrossRef]
- Wu, X.-F.; Vovard-Le Bray, C.; Bechki, L.; Darcel, C. Iron-catalyzed sulfonylimine synthesis under neutral conditions. Tetrahedron 2009, 65, 7380–7384. [Google Scholar] [CrossRef]
- Chang, J.W.W.; Ton, T.M.U.; Tania, S.; Taylor, P.C.; Chan, P.W.H. Practical copper(i)-catalysed amidation of aldehydes. Chem. Commun. 2010, 46, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Chawla, R.; Singh, A.K.; Yadav, L.D.S. An organocatalytic synthesis of N-sulfonyl imines using chloramine-T in aqueous medium. Tetrahedron Lett. 2014, 55, 3553–3556. [Google Scholar] [CrossRef]
- Morales, S.; Guijarro, F.G.; Garcia Ruano, J.L.; Cid, M.B. A general aminocatalytic method for the synthesis of aldimines. J. Am. Chem. Soc. 2014, 136, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.T.; Visco, M.D.; Marsini, M.A.; Grinberg, N.; Busacca, C.A.; Mattson, A.E.; Senanayake, C.H. A general method for imine formation using B(OCH2CF3)3. Org. Lett. 2015, 17, 2442–2445. [Google Scholar] [CrossRef] [PubMed]
- Sharghi, H.; Hosseini-Sarvari, M.; Ebrahimpourmoghaddam, S. A novel method for the synthesis of N-sulfonyl aldimines using AlCl3 under solvent-free conditions (SFC). Arkivoc 2007, xv, 255–264. [Google Scholar]
- Sisko, J.; Weinreb, S.M. Addition of grignard and organolithium reagents to N-sulfonyl aldimines generated in situ from aldehydes and N-sulfinylsulfonamides. J. Org. Chem. 1990, 55, 393–395. [Google Scholar] [CrossRef]
- Trost, B.M.; Marrs, C. A convenient synthesis of N-tosylimines. J. Org. Chem. 1991, 56, 6468–6470. [Google Scholar] [CrossRef]
- Huang, D.; Wang, X.; Wang, X.; Chen, W.; Wang, X.; Hu, Y. Synthesis of N-sulfonyl arylaldimines developed by retesting an old process. Org. Lett. 2016, 18, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Lamar, A.A.; Nicholas, K.M. Iodine-catalyzed aminosulfonation of hydrocarbons by imidoiodinanes: A synthetic and mechanistic investigation. J. Org. Chem. 2010, 75, 7644–7650. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, A.C.; Hancock, E.N.; Anders, E.J.; Tierney, M.M.; Morgan, H.R.; Scott, K.A.; Lamar, A.A. Visible-light-mediated, nitrogen-centered radical amination of tertiary alkyl halides under metal-free conditions to form α-tertiary amines. Org. Biomol. Chem. 2016, 14, 4387–4392. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.D.; Scott, K.A.; DeMier, B.C.; Morgan, H.R.; Macgruder, J.A.; Lamar, A.A. Formation of N-sulfonyl imines from iminoiodinanes by iodine-promoted, N-centered radical sulfonamidation of aldehydes. Org. Biomol. Chem. 2017, 15, 9209–9216. [Google Scholar] [CrossRef] [PubMed]
- Zard, S.Z. Recent progress in the generation and use of nitrogen-centred radicals. Chem. Soc. Rev. 2008, 37, 1603–1618. [Google Scholar] [CrossRef] [PubMed]
- Höfling, S.B.; Heinrich, M.R. Nitrogen-centered radical scavengers. Synthesis 2011, 2011, 173–189. [Google Scholar]
- Chen, J.R.; Hu, X.Q.; Lu, L.Q.; Xiao, W.J. Visible light photoredox-controlled reactions of N-radicals and radical ions. Chem. Soc. Rev. 2016, 45, 2044–2056. [Google Scholar] [CrossRef] [PubMed]
- Achar, T.K.; Mal, P. Radical-induced metal and solvent-free cross-coupling using TBAI-TBHP: Oxidative amidation of aldehydes and alcohols with N-chloramines via C–H activation. J. Org. Chem. 2015, 80, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.M.; Lu, H.; Cui, Y.; Lizardi, C.L.; Arzua, T.N.; Wojtas, L.; Cui, X.; Zhang, X.P. Selective radical amination of aldehydic C(sp 2)-H bonds with fluoroaryl azides via Co(II)-based metalloradical catalysis: Synthesis of fluoroaryl amides from aldehydes under neutral and nonoxidative conditions. Chem. Sci. 2014, 5, 2422–2427. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.; Chen, S.; Shi, E.; Xu, Y.; Wan, X. Cross coupling of acyl and aminyl radicals: Direct synthesis of amides catalyzed by Bu4NI with TBHP as an oxidant. Angew. Chem. 2012, 51, 3231–3235. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Courtneidge, J.L.; Lusztyk, J.; Pagé, D. Alkoxyl radicals from alcohols. spectroscopic detection of intermediate alkyl and acyl hypoiodites in the Suárez and Beebe reactions. Tetrahedron Lett. 1994, 35, 1003–1006. [Google Scholar] [CrossRef]
- Wang, D.H.; Hao, X.S.; Wu, D.F.; Yu, J.Q. Palladium-catalyzed oxidation of Boc-protected N-methylamines with IOAc as the Oxidant: A Boc-directed sp3 C–H bond activation. Org. Lett. 2006, 8, 3387–3390. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Jones, R.M. The sulfonamide group as a structural alert: A distorted story? Curr. Opin. Drug Discov. Devel. 2008, 11, 72–79. [Google Scholar] [PubMed]
- Davies, P.W.; Martin, N.; Spencer, N. Isotopic labelling studies for a gold-catalysed skeletal rearrangement of alkynyl aziridines. Beilstein J. Org. Chem. 2011, 7, 839–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Pu, D.; Wen, F.; Wu, J. δ and α SP3 C−H bond oxidation of sulfonamides with PhI(OAc)2/I2 under metal-free conditions. J. Org. Chem. 2007, 72, 8994–8997. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Li, W.; Pu, D.; Zhang, L. Transition-metal-free intermolecular amination of sp3 C−H bonds with sulfonamides. Org. Lett. 2009, 11, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Martín, A.; Pérez-Martín, I.; Suárez, E. Intramolecular hydrogen abstraction promoted by amidyl radicals. evidence for electronic factors in the nucleophilic cyclization of ambident amides to oxocarbenium ions. Org. Lett. 2005, 7, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Togo, H.; Hoshina, Y.; Muraki, T.; Nakayama, H.; Yokoyama, M. Study on radical amidation onto aromatic rings with (diacyloxyiodo) arenes. J. Org. Chem. 1998, 63, 5193–5200. [Google Scholar] [CrossRef]
- Yang, H.T.; Ren, W.L.; Dong, C.P.; Yang, Y.; Sun, X.Q.; Miao, C.B. PhI(OAc)2/I2-mediated [3 + 2] reaction of [60]fullerene with amides for the preparation of fullerooxazoles. Tetrahedron Lett. 2013, 54, 6799–6803. [Google Scholar] [CrossRef]
- Minakata, S. Utilization of N–X bonds in the synthesis of N-heterocycles. Accounts Chem. Res. 2009, 42, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Martínez, C.; Muñiz, K. An iodine-catalyzed Hofmann–Löffler reaction. Angew. Chem. Int. Ed. 2015, 54, 8287–8291. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Deng, M.; Qian, C. The effect of coordination on the reaction of N-tosyl imines with diethylzinc. Tetrahedron 2005, 61, 12238–12243. [Google Scholar] [CrossRef]
- Yrjölä, S.; Parkkari, T.; Navia-Paldanius, D.; Laitinen, T.; Kaczor, A.A.; Kokkola, T.; Adusei-Mensah, F.; Savinainen, J.R.; Laitinen, J.T.; Poso, A.; et al. Potent and selective N-(4-sulfamoylphenyl)thiourea-based GPR55 agonists. Eur. J. Med. Chem. 2016, 107, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liu, C.; Jiang, L.; Xu, Q. Manganese dioxide catalyzed N-alkylation of sulfonamides and amines with alcohols under air. Org. Lett. 2011, 13, 6184–6187. [Google Scholar] [CrossRef] [PubMed]
- Caddick, S.; Wilden, J.D.; Judd, D.B. Direct synthesis of sulfonamides and activated sulfonate esters from sulfonic acids. J. Am. Chem. Soc. 2004, 126, 1024–1025. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Fujita, K.I.; Yamaguchi, R. Simple and versatile catalytic system for N-alkylation of sulfonamides with various alcohols. Org. Lett. 2010, 12, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Shi, F.; Tse, M.K.; GÃrdes, D.; Thurow, K.; Beller, M.; Deng, Y. Copper-catalyzed N-alkylation of sulfonamides with benzylic alcohols: Catalysis and mechanistic studies. Adv. Synth. Catal. 2009, 351, 2949–2958. [Google Scholar] [CrossRef]
- Li, Z.-L.; Jin, L.K.; Cai, C. Nickel-catalyzed product-controllable amidation and imidation of sp3 C–H bonds in substituted toluenes with sulfonamides. Org. Biomol. Chem. 2017, 15, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.R.; Chisholm, J.D. Alkylation of sulfonamides with trichloroacetimidates under thermal conditions. J. Org. Chem. 2016, 81, 8035–8042. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the N-alkylsulfonamide compounds (3aa–3bf; Table 4) are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
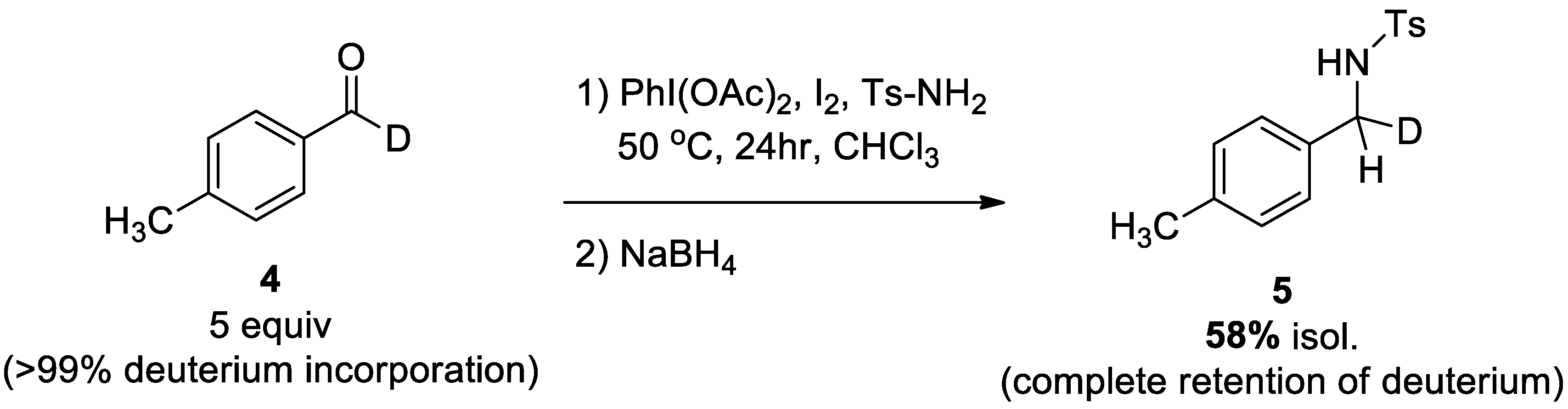{kind=link}
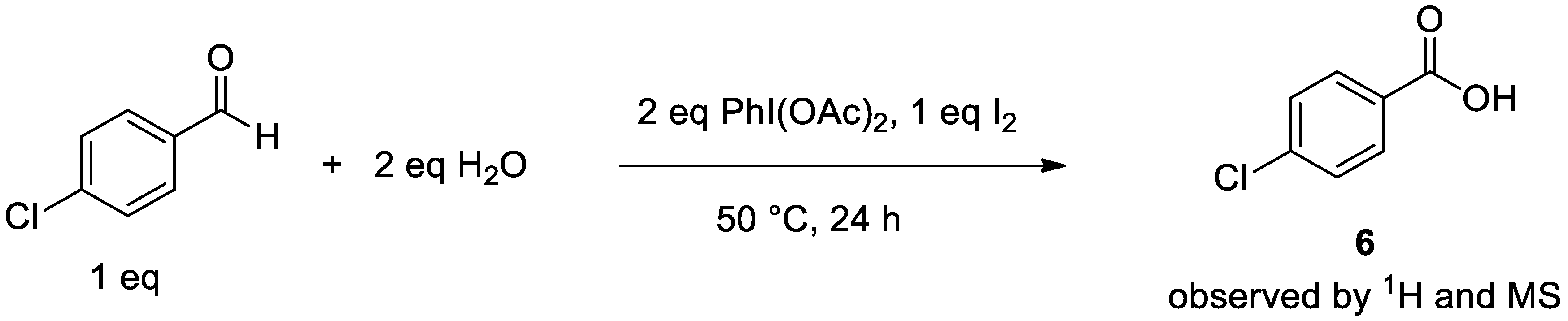{kind=link}
{kind=link}
{kind=link}

| Entry | Equiv. 1 | Equiv. PhI(OAc)2 | Equiv. I2 | Temp (°C) | Time (h) | Light | Additive | Yield 2de (%) 1 |
|---|---|---|---|---|---|---|---|---|
| 1 | 5 | 1 | 1 | 20 | 48 | White LED | None | 34 |
| 2 | 5 | 2 | 1 | 20 | 48 | White LED | None | 42 |
| 3 | 5 | 0 | 1 | 20 | 48 | White LED | None | 0 |
| 4 | 5 | 1 | 0 | 20 | 48 | White LED | None | 0 |
| 5 | 5 | 0 | 0 | 20 | 48 | White LED | None | 0 |
| 6 | 5 | 0 | 0 | 20 | 48 | White LED | 0.55 eq HI | 0 |
| 7 | 5 | 1 | 1 | 50 | 24 | Ambient 2 | None | 57 |
| 8 | 5 | 2 | 1 | 50 | 24 | Ambient | None | 72 |
| 9 | 2 | 2 | 1 | 50 | 24 | Ambient | None | 8 |
| 10 | 1 | 2 | 1 | 50 | 24 | Ambient | None | 1 |
| 11 | 5 | 2 | 1 | 50 | 8 | Ambient | None | 65 |
| 12 | 5 | 2 | 1 | 50 | 4 | Ambient | None | 59 |

| Entry | Variation from Standard Conditions | Yield 2de (%) 1 |
|---|---|---|
| 1 | NONE | 72 |
| 2 | 0 equiv. I2 | 40 |
| 3 | 0.2 equiv. I2 | 66 |
| 4 | 4A Molecular sieves | 8 |
| 5 | Reaction in dark 2 | 74 |
| 6 | 1 equiv. TEMPO | 19 |

| Entry | Oil Bath Temperature (°C) | Eq. I2 | Yield 2de (%) 1 |
|---|---|---|---|
| 1 | 20 | 0 | 0 |
| 2 | 20 | 1 | 39 |
| 3 | 30 | 0 | 0 |
| 4 | 30 | 1 | 42 |
| 5 | 40 | 0 | 5 |
| 6 | 40 | 1 | 72 |
| 7 | 50 | 0 | 40 |
| 8 | 50 | 1 | 72 |

| Entry | (Aldehyde) R1 | (Sulfonamide) R2 | Product 2 Yield (%) 1 | Product 3 Yield (%) 2 |
|---|---|---|---|---|
| 1 | 4-OCH3 | 4-CH3 | 2aa 61 | 3aa 61 |
| 2 | 4-CH3 | 4-CH3 | 2ba 71 | 3ba 60 |
| 3 | H | 4-CH3 | 2ca 67 | 3ca 50 |
| 4 | 4-Br | 4-CH3 | 2da 54 | 3da 45 |
| 5 | 4-Cl | 4-CH3 | 2ea 65 | 3ea 74 |
| 6 | 4-NO2 | 4-CH3 | 2fa 50 | 3fa 40 |
| 7 | 4-CN | 4-CH3 | 2ga 52 | 3ga 44 |
| 8 | 3-CH3 | 4-CH3 | 2ha 70 | 3ha 33 |
| 9 | 2-CH3 | 4-CH3 | 2ia 60 | 3ia 33 |
| 10 | 3-OCH3 | 4-CH3 | 2ja 60 | 3ja 40 |
| 11 | 2-OCH3 | 4-CH3 | 2ka 65 | 3ka 40 |
| 12 | 3-Br | 4-CH3 | 2la 50 | 3la 22 |
| 13 | 2-Br | 4-CH3 | 2ma 36 | 3ma 30 |
| 14 | 4-CH3 | H | 2bb 69 | 3bb 40 |
| 15 | 4-Cl | H | 2eb 66 | 3eb 70 |
| 16 | 2-CH3 | H | 2hb 60 | 3hb 38 |
| 17 | 4-CH3 | 4-Cl | 2bc 60 | 3bc 40 |
| 18 | 4-Br | 4-Cl | 2dc 58 | 3dc 54 |
| 19 | 4-CH3 | 2-Cl | 2bd 35 | 3bd 17 |
| 20 | 4-OCH3 | 4-NO2 | 2ae 34 | 3ae 11 |
| 21 | 4-Cl | 4-NO2 | 2ee 27 | 3ee 21 |
| 22 | 4-CH3 | 3-NO2 | 2bf 25 | 3bf 8 |
| 23 | 4-CH3 | 2-NO2 | 2bg 6 | 3bg trace |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hopkins, M.D.; Brandeburg, Z.C.; Hanson, A.J.; Lamar, A.A. Visible-Light, Iodine-Promoted Formation of N-Sulfonyl Imines and N-Alkylsulfonamides from Aldehydes and Hypervalent Iodine Reagents. Molecules 2018, 23, 1838. https://doi.org/10.3390/molecules23081838
Hopkins MD, Brandeburg ZC, Hanson AJ, Lamar AA. Visible-Light, Iodine-Promoted Formation of N-Sulfonyl Imines and N-Alkylsulfonamides from Aldehydes and Hypervalent Iodine Reagents. Molecules. 2018; 23(8):1838. https://doi.org/10.3390/molecules23081838
Chicago/Turabian StyleHopkins, Megan D., Zachary C. Brandeburg, Andrew J. Hanson, and Angus A. Lamar. 2018. "Visible-Light, Iodine-Promoted Formation of N-Sulfonyl Imines and N-Alkylsulfonamides from Aldehydes and Hypervalent Iodine Reagents" Molecules 23, no. 8: 1838. https://doi.org/10.3390/molecules23081838
APA StyleHopkins, M. D., Brandeburg, Z. C., Hanson, A. J., & Lamar, A. A. (2018). Visible-Light, Iodine-Promoted Formation of N-Sulfonyl Imines and N-Alkylsulfonamides from Aldehydes and Hypervalent Iodine Reagents. Molecules, 23(8), 1838. https://doi.org/10.3390/molecules23081838