Synthesis and Antioxidant Activity of Caffeic Acid Derivatives


Abstract
:
1. Introduction
2. Results and Discussion
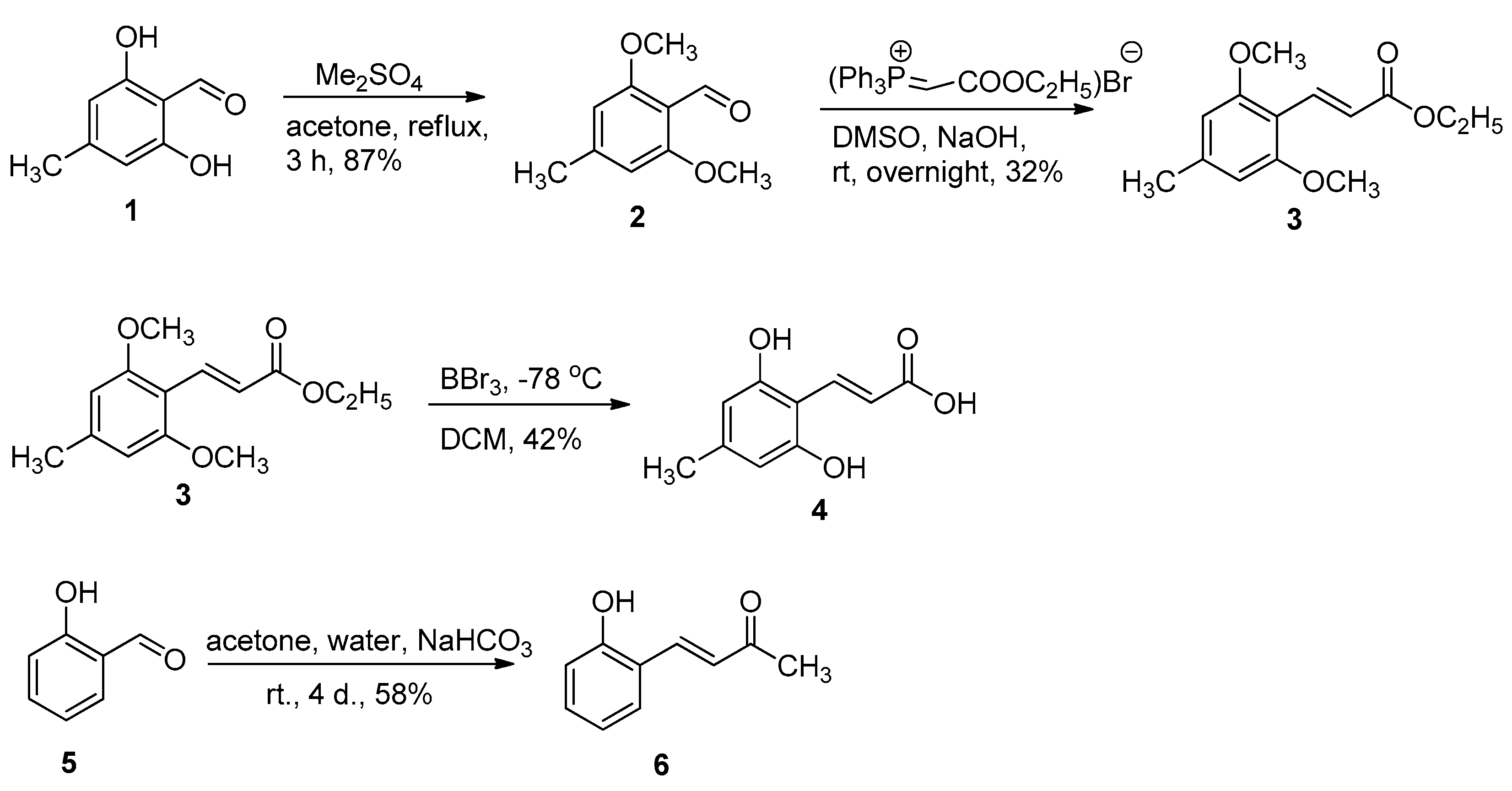
2.1. Chemistry
2.2. Biological Activity
2.2.1. Antioxidant Activity of CA Derivatives
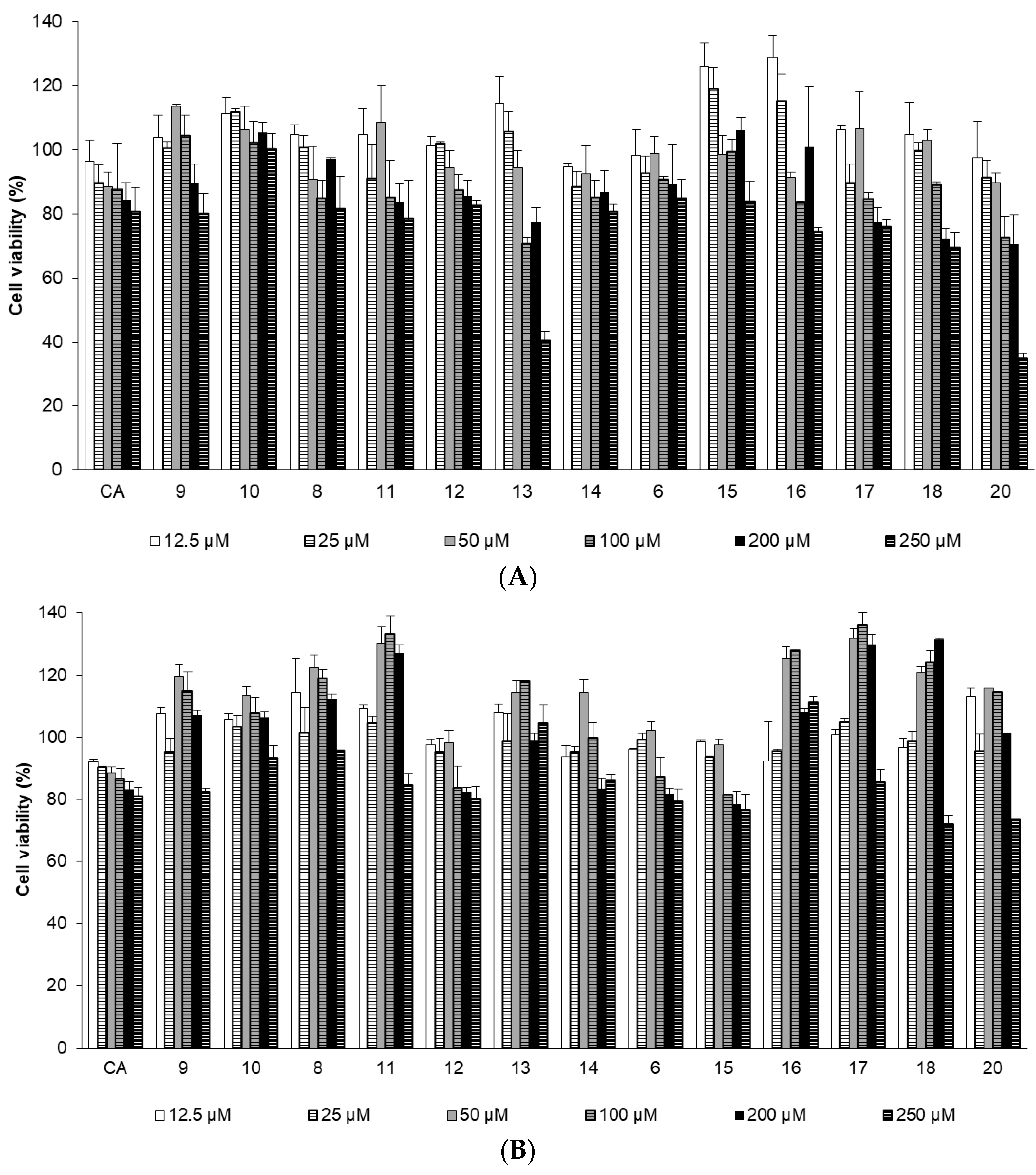
2.2.2. Effects of CA Derivatives on NHDF Cell Viability
3. Experimental
3.1. Chemistry
3.1.1. General Materials and Methods
3.1.2. General Method for the HWE Reaction in Water
3.2. Biological Activities
3.2.1. Calculation of log Po/w
3.2.2. Determination of the Oxidation Inhibition of the o/w Emulsion
3.2.3. DPPH Assay
3.2.4. Cell Viability Assay in the Normal Human Dermal Fibroblast (NHDF) Cell Line
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CA | caffeic acid |
| HWE | Horner–Wadsworth–Emmons |
| DCM | dichloromethane |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
| EtOAc | ethyl acetate |
| DMSO | dimethyl sulfoxide |
| log Po/w | partition coefficient between n-octanol and water |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| NHDF | normal human dermal fibroblasts |
| TBARS | thiobarbituric acid reactive substances |
References
- Shahidi, F.; Yeo, J. Bioactivities of phenolics by focusing on suppression of chronic diseases: A review. Int. J. Mol. Sci. 2018, 19, 1573. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Gilardini Montani, M.S.; Merendino, N.; Romani, A.; Velotti, F. Hydroxytyrosol-derived compounds: A basis for the creation of new pharmacological agents for cancer prevention and therapy. J. Med. Chem. 2015, 58, 9089–9107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tang, Y.; Li, N.G.; Zhu, Y.; Duan, J.A. Bioactivity and chemical synthesis of caffeic acid phenethyl ester and its derivatives. Molecules 2014, 19, 16458–16476. [Google Scholar] [CrossRef] [PubMed]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouysegu, L. Plant polyphenols: Chemical properties, biological activities, and synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.A.; Girao da Cruz, T.; Andrade, J.L.; Milhazes, N.; Borges, F.; Marques, M.P.M. Anticancer activity of phenolic acids of natural or synthetic origin: A structure-activity study. J. Med. Chem. 2003, 46, 5395–5401. [Google Scholar] [CrossRef] [PubMed]
- Eid, H.M.; Vallerand, D.; Muhammad, A.; Durst, T.; Haddad, P.S.; Martineau, L.C. Structural constraints and the importance of lipophilicity for the mitochondrial uncoupling activity of naturally occurring caffeic acid esters with potential for the treatment of insulin resistance. Biochem. Pharmacol. 2010, 79, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yoshioka, Y.; Kato, E.; Katsuki, S.; Iida, O.; Hosokawa, K.; Kawabata, J. Methyl caffeate as an α-glucosidase inhibitor from Solanum torvum fruits and the activity of related compounds. Biosci. Biotechnol. Biochem. 2010, 74, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Ganugapati, J.; Swarna, S. Molecular docking studies of antidiabetic activity of cinnamon compounds. Asian J. Pharm. Clin. Res. 2014, 7, 31–34. [Google Scholar]
- Fiuza, S.M.; Gomes, C.; Teixeira, L.J.; Girao da Cruz, M.T.; Cordeiro, M.N.D.S.; Milhazes, N.; Borgesa, F.; Marques, M.P.M. Phenolic acid derivatives with potential anticancer properties––A structure–activity relationship study. Part 1: Methyl, propyl and octyl esters of caffeic and gallic acids. Bioorg. Med. Chem. 2004, 12, 3581–3589. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.S.; Oliveira, S.; Paiva-Martins, F.; Ferreira, P.M.T.; Pereira, D.M.; Andrade, P.B.; Valentao, P. Synthesis and preliminary biological evaluation of new phenolic and catecholic dehydroamino acid derivatives. Tetrahedron 2017, 73, 6199–6209. [Google Scholar] [CrossRef]
- Chen, J.H.; Chi-Tang, H. Antioxidant activities of caffeic acid and its related hydroxycinnamic acid compounds. J. Agric. Food Chem. 1997, 7, 2374–2378. [Google Scholar] [CrossRef]
- Sato, Y.; Itagaki, S.; Kurokawa, T.; Ogura, J.; Kobayashi, M.; Hirano, T.; Sugawara, M.; Iseki, K. In vitro and in vivo antioxidant properties of chlorogenic acid and caffeic acid. Int. J. Pharm. 2010, 403, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Losada-Barreiro, S.; Bravo-Díaz, C. Free radicals and polyphenols: The redox chemistry of neurodegenerative diseases. Eur. J. Med. Chem. 2017, 133, 379–402. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Darokar, M.P.; Fatima, A.; Kumar, J.K.; Chowdhury, C.; Saxena, H.O.; Dwivedi, G.R.; Shrivastava, K.; Gupta, V.; Chattopadhyay, S.K.; et al. Synthesis of diverse analogues of Oenostacin and their antibacterial activities. Bioorg. Med. Chem. 2007, 15, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Siquet, C.; Paiva-Martins, F.; Lima, J.L.F.C.; Reis, S.; Borges, F. Antioxidant profile of dihydroxy- and trihydroxyphenolic acids- A structure–activity relationship study. Free Radic. Res. 2006, 40, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.C.; Damu, A.G.; Cherng, C.Y.; Jeng, J.F.; Teng, C.M.; Lee, E.J.; Wu, T.S. Isolation of a natural antioxidant, dehydrozingerone from Zingiber officinale and synthesis of its analogues for recognition of effective antioxidant and antityrosinase agents. Arch. Pharm. Res. 2005, 28, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, I.E.; Navarro, D.; Rocord, E.; Asther, M. Fungal biotransformation of p-coumaric acid into caffeic acid by Pycnoporus cinnabarinus: An alternative for producing a strong natural oxidant. World J. Microbiol. Biotechnol. 2003, 19, 157–160. [Google Scholar] [CrossRef]
- Kroon, P.A.; Williamson, G. Hydroxycinnamates in plants and food: Current and future perspectives. J. Sci. Food Agric. 1999, 79, 355–361. [Google Scholar] [CrossRef]
- Touaibia, M.; Jean-François, J.; Doiron, J. Caffeic acid, a versatile pharmacophore: An overview. Mini Rev. Med. Chem. 2011, 11, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Arai, Y.; Kino, K. Biotechnological production of caffeic acid by bacterial cytochrome P450 CYP199A2. Appl. Environ. Microbiol. 2012, 78, 6087–6094. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.A.; Ghadami, M. Efficient synthesis of α,β-unsaturated ketones with trans-selective Horner–Wadsworth–Emmons reaction in water. Environ. Chem. Lett. 2016, 14, 223–228. [Google Scholar] [CrossRef]
- Wadsworth, W.S. Synthetic applications of phosphoryl-stabilized anions. Org. React. 2005, 25, 73–253. [Google Scholar]
- Wadsworth, W.S.; Emmons, W.D. The utility of phosphonate carbanions in olefin synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. [Google Scholar] [CrossRef]
- Ando, K. Z-Selective Horner–Wadsworth–Emmons reaction of α-substituted ethyl (diarylphosphono)acetates with aldehydes. J. Org. Chem. 1998, 63, 8411–8416. [Google Scholar] [CrossRef]
- Saito, N.; Ryoda, A.; Nakanishi, W.; Kumamoto, T.; Ishikawa, T. Guanidine-catalyzed asymmetric synthesis of 2,2-disubstituted chromane skeletons by intramolecular oxa-Michael addition. Eur. J. Org. Chem. 2008, 2759–2766. [Google Scholar] [CrossRef]
- Patil, V.J.; Mavers, U. Wittig reactions in the presence of Silica gel. Tetrahedron Lett. 1996, 37, 1281–1284. [Google Scholar] [CrossRef]
- Isaacs, N.S.; El-Din, G.N. The application of high pressure to some difficult Wittig reactions. Tetrahedron Lett. 1987, 28, 2191–2192. [Google Scholar] [CrossRef]
- Hooper, D.L.; Garagan, S.; Kayser, M.M. Lithium cation-catalyzed Wittig reactions. J. Org. Chem. 1994, 59, 1126–1128. [Google Scholar] [CrossRef]
- Boulaire, V.L.; Gree, R. Wittig reactions in the ionic solvent [bmim][BF4]. Chem. Commun. 2000, 22, 2195–2196. [Google Scholar] [CrossRef]
- Spinella, A.; Fortunati, T.; Soriente, A. Microwave accelerated Wittig reactions of stabilized phosphorus ylides with ketones under solvent-free conditions. Synlett 1997, 1, 93–94. [Google Scholar] [CrossRef]
- Dambacher, J.; Zhao, W.; El-Batta, A.; Anness, R.; Jiang, C.; Bergdahl, M. Water is an efficient medium for Wittig reactions employing stabilized ylides and aldehydes. Tetrahedron Lett. 2005, 46, 4473–4477. [Google Scholar] [CrossRef]
- El-Batta, A.; Jiang, C.; Zhao, W.; Anness, R.; Cooksy, A.L.; Bergdahl, M. Wittig reactions in water media employing stabilized ylides with aldehydes. Synthesis of α,β-unsaturated esters from mixing aldehydes, α-bromoesters, and Ph3P in aqueous NaHCO3. J. Org. Chem. 2007, 72, 5244–5259. [Google Scholar] [CrossRef] [PubMed]
- Nonnenmacher, A.; Mayer, R.; Plieninger, H. Application of high-pressure on Wittig reactions with resonance stabilized ylides. Liebigs Ann. Chem. 1983, 12, 2135–2140. [Google Scholar] [CrossRef]
- Saija, A.; Tomaino, A.; Lo Cascio, R.; Trombetta, D.; Proteggente, A.; De Pasquale, A.; Uccela, N.; Bonina, F. Ferulic and caffeic acids as potential protective agents against photooxidative skin damage. J. Sci. Food Agric. 1999, 79, 476–480. [Google Scholar] [CrossRef]
- Saija, A.; Tomaino, A.; Trombetta, D.; de Pasquale, A.; Uccella, N.; Barbuzzi, T.; Paolino, D.; Bonina, F. In vitro and in vivo evaluation of caffeic and ferulic acids as topical photoprotective agents. Int. J. Pharm. 2000, 199, 39–47. [Google Scholar] [CrossRef]
- Pluemsamran, T.; Onkoksoong, T.; Panich, U. Caffeic acid and ferulic acid inhibit UVA-induced matrix metalloproteinase-1 through regulation of antioxidant defense system in keratinocyte HaCaT cells. Photochem. Photobiol. 2012, 88, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Chen, C.C.; Chao, S.W.; Lee, S.S.; Hsu, F.L.; Lu, Y.L.; Hung, M.F.; Chang, C.I. Synthesis of N-hydroxycinnamides capped with a naturally occurring moiety as inhibitors of histone deacetylase. Chem. Med. Chem. 2010, 5, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Viviano, M.; Glasnov, T.N.; Reichart, B.; Tekautz, G.; Kappe, C.O. A scalable two-step continuous flow synthesis of nabumetone and related 4-Aryl-2-butanones. Org. Process Res. Dev. 2011, 15, 858–870. [Google Scholar] [CrossRef]
- Oh, S.; Jang, S.; Kim, D.; Han, I.O.; Jung, J.Ch. Synthesis and evaluation of biological properties of benzylideneacetophenone derivatives. Arch. Pharm. Res. 2006, 29, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Zarnowski, R.; Jaromin, A.; Certik, M.; Czabany, T.; Fontaine, J.; Jakubik, T.; Iqbal, M.C.M.; Grandmougin-Ferjani, A.; Kozubek, A.; Pietr, S.J. The oil of Adenanthera pavonina L. seeds and its emulsions. Z. Naturforsch. C. 2004, 59, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Jaromin, A.; Zarnowski, R.; Kozubek, A. Emulsions of oil from Adenanthera pavonina L. seeds and their protective effect. Cell Mol. Biol. Lett. 2006, 11, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Jaromin, A.; Zarnowski, R.; Piętka-Ottlik, M.; Andes, D.R.; Gubernator, J. Topical delivery of ebselen encapsulated in biopolymeric nanocapsules: Drug repurposing enhanced antifungal activity. Nanomedicine 2018, 13, 1139–1155. [Google Scholar] [CrossRef] [PubMed]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [PubMed]
- Zhang, Z.; Xiao, B.; Chen, Q.; Lian, X.Y. Synthesis and biological evaluation of caffeic acid 3,4-dihydroxyphenethyl ester. J. Nat. Prod. 2010, 73, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
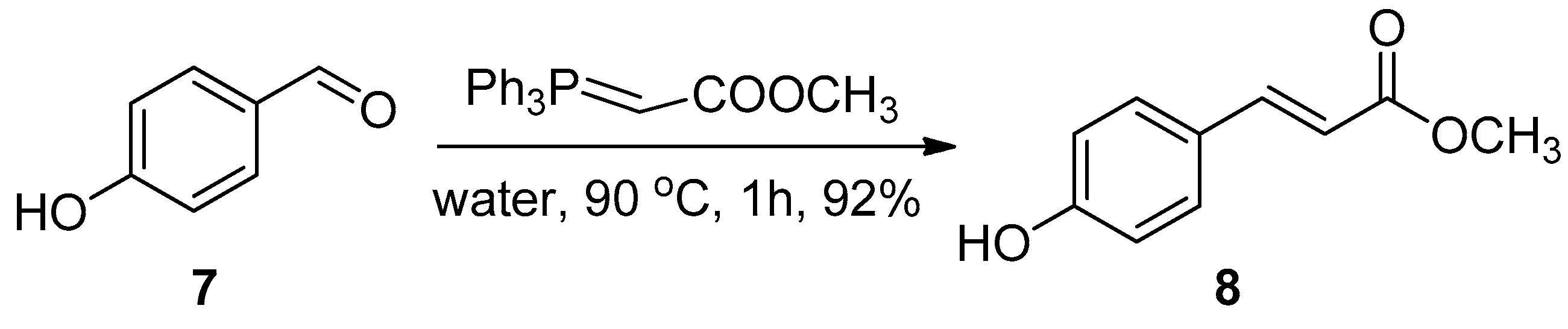{kind=link}

| Entry | Aldehyde | Ylide | Product (E Isomer) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1. |  |  |  | 0.5 | 82 |
| 2. |  | |  | 1.0 | 86 |
| 3. |  | |  | 1.0 | 91 |
| 4. |  | |  | 5.0 | 86 |
| 5. |  | |  | 1.0 | 74 |
| 6. |  | |  | 1.0 | 37 |
| 7. |  | |  | 0.5 | 99 |
| 8. |  |  |  | 2.5 | 85 |
| 9. |  | |  | 3.0 | 97 |
| 10. |  | |  | 2.0 | 83 |
| 11. |  | |  | 2.0 | 78 |
| 12. |  | |  | 0.5 | 70 |
| 13. |  | |  | 5.0 | 0 |
| 14. |  | |  | 0.5 | 98 |
| Compound | log Po/w a | Inhibition of Oxidation of o/w Emulsion b | DPPH IC50 (µM) |
|---|---|---|---|
| CA | 0.93 | 26.1 ± 2.9 | 32.2 |
| 9 | 1.82 | 2.6 ± 1.5 | n.a.c |
| 10 | 1.78 | 18.4 ± 2.2 | n.a.c |
| 8 | 1.81 | 22.7 ± 6.4 | n.a.c |
| 11 | 1.31 | 33.7 ± 6.5 | 1015.9 |
| 12 | 0.99 | 55.5 ± 2 | 18.6 |
| 13 | 0.95 | 38.6 ± 2.3 | 53.3 |
| 14 | 1.76 | 14.8 ± 2 | 58.9 |
| 6 | 1.94 | n.a.c | n.a.d |
| 15 | 1.82 | n.a.c | n.a.d |
| 16 | 1.85 | 6.8 ± 4.4 | n.a.d |
| 17 | 1.49 | 4.6 ± 1.3 | 688.2 |
| 18 | 1.05 | 28.6 ±4 | 16.8 |
| 20 | 1.86 | 7.9 ± 2.1 | 61.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidoryk, K.; Jaromin, A.; Filipczak, N.; Cmoch, P.; Cybulski, M. Synthesis and Antioxidant Activity of Caffeic Acid Derivatives. Molecules 2018, 23, 2199. https://doi.org/10.3390/molecules23092199
Sidoryk K, Jaromin A, Filipczak N, Cmoch P, Cybulski M. Synthesis and Antioxidant Activity of Caffeic Acid Derivatives. Molecules. 2018; 23(9):2199. https://doi.org/10.3390/molecules23092199
Chicago/Turabian StyleSidoryk, Katarzyna, Anna Jaromin, Nina Filipczak, Piotr Cmoch, and Marcin Cybulski. 2018. "Synthesis and Antioxidant Activity of Caffeic Acid Derivatives" Molecules 23, no. 9: 2199. https://doi.org/10.3390/molecules23092199
APA StyleSidoryk, K., Jaromin, A., Filipczak, N., Cmoch, P., & Cybulski, M. (2018). Synthesis and Antioxidant Activity of Caffeic Acid Derivatives. Molecules, 23(9), 2199. https://doi.org/10.3390/molecules23092199