Machine Learning for Drug-Target Interaction Prediction


Abstract
:
1. Introduction
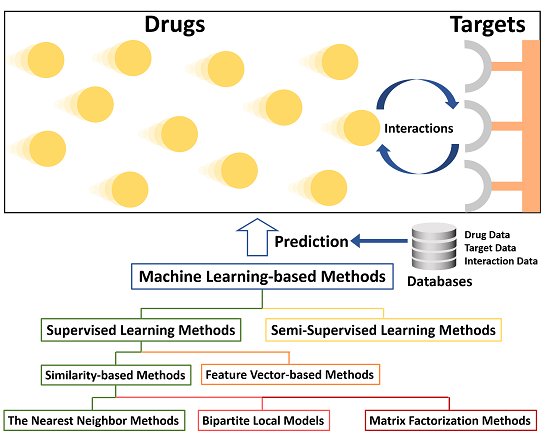
- Supervised Learning MethodsBoth positive labels and negative labels are required in the training set. Then these labeled samples are used to train the learning models for subsequent DTI prediction.
- Similarity-based methodsThe similarities among drugs or among targets are calculated via various similarity measurement strategies. Similarity matrices can be utilized in various types of kernel functions:
- (i)
- The nearest neighbor methods: The nearest neighbor methods make predictions based on the information of the nearest neighbors.
- (ii)
- Bipartite local models: Two local models are firstly trained for drugs and targets respectively. The final prediction result for each drug-target pair is computed based on the operation of the two independent prediction scores.
- (iii)
- Matrix factorization methods: Drug-target interaction matrix is factorized into two latent feature matrices that when multiplied together can approximate the original matrix.
- Feature vector-based methodsThe training data is represented as feature vectors. Then some machine learning models, like Random Forest, can be utilized for prediction based on these vectors.
- Semi-Supervised Learning MethodsSemi-supervised learning methods make predictions only based on a small amount of labeled data and a large amount of unlabeled data.To our best knowledge, there are already some excellent reviews on chemogenomic approaches for DTI prediction [6,15,16,17,18,19]. Compared to previous works, we focus on the special topic of machine learning methods used in DTI prediction. Besides, we utilize a hierarchical classification scheme and summarize several latest prediction methods such as [20,21,22,23] which are hardly mentioned in any previous review. In particular, review [17] is written only from a narrow viewpoint, namely similarity-based approaches, which are a subclass of machine learning methods. Surveys [6,15,18,19] all provide a more general and comprehensive overview of chemogenomic approaches rather than emphasizing machine learning. In recent years, machine learning has made breakthroughs and attracted a lot of public attention. Discussing state-of-the-art DTI prediction strategies from this special perspective can demonstrate more methodology details. Although review [16] also focuses on learning-based methods, its emphasis is only on supervised learning. In comparison, we provide more detailed sub-classes and introduce newly developed methods after review [16] was published.The rest of this article is organized as follows: The “Databases” section describes current available data sources for DTI prediction research. The “Methods” section briefly introduces several representative machine learning methods via a hierarchical classification scheme. Then we discuss advantages and limitations of methods in each category as well as remaining challenges. Finally, the “Conclusions and Outlook” section makes a future perspective for machine leaning in DTI prediction.
2. Databases
3. Methods
3.1. Supervised Learning Methods
3.1.1. Similarity-Based Methods
3.1.2. Feature Vector-Based Methods
3.2. Semi-Supervised Learning Methods
3.3. Discussion
4. Conclusions and Outlook
5. Key Points
- Identifying drug-target interactions is the vital first step in drug discovery research.
- A number of existing professional databases serve known data resources for DTI prediction and thus promote the drug discovery.
- Machine learning-base methods are generally effective and reliable for DTI prediction.
- Different machine learning methods have their merits and demerits. Hence, it is essential to choose appropriate methods or assemble models for special prediction tasks.
- A more effective prediction model can be established by integrating more heterogeneous data sources of drugs and targets.
- In reality, DTI prediction is a regression problem with quantitative bioactivity data.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Masoudi-Nejad, A.; Mousavian, Z.; Bozorgmehr, J.H. Drug-target and disease networks: Polypharmacology in the post-genomic era. In Silico Pharmacol. 2013, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.; Gagnon, J.P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004, 3, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. Pubchem bioassay: 2017 update. Nucleic Acids Res. 2017, 45, D955–D963. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Z. A semi-supervised method for drug-target interaction prediction with consistency in networks. PLoS ONE 2013, 8, e62975. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2016, 17, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Liu, L.; Lu, L.; Zou, Q. Prediction of potential disease-associated micrornas using structural perturbation method. Bioinformatics 2018, 34, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Q.; Rodríguez-Patón, A.; Zeng, X. Meta-path methods for prioritizing candidate disease mirnas. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef]
- Hua, S.; Yun, W.; Zhiqiang, Z.; Zou, Q. A discussion of micrornas in cancers. Curr. Bioinform. 2014, 9, 453–462. [Google Scholar] [CrossRef]
- Zeng, X.; Liao, Y.; Liu, Y.; Zou, Q. Prediction and validation of disease genes using hetesim scores. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Li, D.; Wu, Y.; Zou, Q.; Liu, X. An empirical study of features fusion techniques for protein-protein interaction prediction. Curr. Bioinform. 2016, 11, 4–12. [Google Scholar] [CrossRef]
- Wang, Z.; Zou, Q.; Jiang, Y.; Ju, Y.; Zeng, X. Review of protein subcellular localization prediction. Curr. Bioinform. 2014, 9, 331–342. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Arola, L.; Fernandez-Larrea, J.; Blay, M.; Salvado, M.J.; Blade, C.; Ardevol, A.; Vaque, M.; Pujadas, G. Protein-ligand docking: A review of recent advances and future perspectives. Curr. Pharm. Anal. 2008, 4, 1–19. [Google Scholar] [CrossRef]
- Yamanishi, Y. Chemogenomic approaches to infer drug–target interaction networks. In Data Mining for Systems Biology: Methods and Protocols; Mamitsuka, H., DeLisi, C., Kanehisa, M., Eds.; Humana Press: Totowa, NJ, USA, 2013; Volume 939, pp. 97–113. ISBN 978-1-62703-107-3. [Google Scholar]
- Mousavian, Z.; Masoudi-Nejad, A. Drug-target interaction prediction via chemogenomic space: Learning-based methods. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Takigawa, I.; Mamitsuka, H.; Zhu, S. Similarity-based machine learning methods for predicting drug-target interactions: A brief review. Brief. Bioinform. 2014, 15, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; Zhang, X.; Dai, F.; Yin, J.; Zhang, Y. Drug-target interaction prediction: Databases, web servers and computational models. Brief. Bioinform. 2016, 17, 696–712. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, A.; Wu, M.; Li, X.L.; Kwoh, C.K. Computational prediction of drug-target interactions using chemogenomic approaches: An empirical survey. Brief. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Hong, L.; Xiao, A.; Jiang, T.; Zeng, J. Neodti: Neural integration of neighbor information from a heterogeneous network for discovering new drug-target interactions. Bioinformatics 2018. [Google Scholar] [CrossRef]
- Ma, T.; Xiao, C.; Zhou, J.; Wang, F. Drug similarity integration through attentive multi-view graph auto-encoders. arXiv, 2018; arXiv:1804.10850. [Google Scholar]
- Zhang, W.; Chen, Y.; Liu, F.; Luo, F.; Tian, G.; Li, X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinform. 2017, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Pahikkala, T.; Airola, A.; Pietila, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward more realistic drug-target interaction predictions. Brief. Bioinform. 2015, 16, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microrna function and prioritizing disease-related microrna using biological interaction networks. Brief. Bioinform. 2016, 17, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Ju, Y.; Li, D. Protein folds prediction with hierarchical structured SVM. Curr. Proteom. 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, X.; Ju, Y.; Jiang, Y.; Zhang, Z.; Chen, W. A classification method for microarrays based on diversity. Curr. Bioinform. 2016, 11, 590–597. [Google Scholar] [CrossRef]
- Nagamine, N.; Sakakibara, Y. Statistical prediction of protein chemical interactions based on chemical structure and mass spectrometry data. Bioinformatics 2007, 23, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Mao, T.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Placzek, S.; Schomburg, I.; Chang, A.; Jeske, L.; Ulbrich, M.; Tillack, J.; Schomburg, D. Brenda in 2017: New perspectives and new tools in brenda. Nucleic Acids Res. 2017, 45, D380–D388. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. Pubchem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhang, C.; Zhu, F.; Xu, F.; Chen, S.Y.; Zhang, P.; Li, Y.H.; Yang, S.Y.; Wei, Y.Q.; Tao, L. Therapeutic target database update 2014: A resource for targeted therapeutics. Nucleic Acids Res. 2014, 42, D1118–D1123. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. Drugbank 5.0: A major update to the drugbank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Hecker, N.; Ahmed, J.; Von, E.J.; Dunkel, M.; Macha, K.; Eckert, A.; Gilson, M.K.; Bourne, P.E.; Preissner, R. Supertarget goes quantitative: Update on drug-target interactions. Nucleic Acids Res. 2012, 40, D1113–D1117. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; Mcglinchey, S.; Michalovich, D.; Allazikani, B. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Santos, A.; Von, M.C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J. Supertarget and matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2008, 36, D919–D922. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. Bindingdb: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Magariños, M.P.; Carmona, S.J.; Crowther, G.J.; Ralph, S.A.; Roos, D.S.; Shanmugam, D.; Voorhis, W.C.V.; Agüero, F. TDR targets: A chemogenomics resource for neglected diseases. Nucleic Acids Res. 2012, 40, D1118–D1127. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Campillos, M.; Letunic, I.; Jensen, L.J.; Bork, P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 2010, 6, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Seiler, K.P.; George, G.A.; Happ, M.P.; Bodycombe, N.E.; Carrinski, H.A.; Norton, S.; Brudz, S.; Sullivan, J.P.; Muhlich, J.; Serrano, M. Chembank: A small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2008, 36, D351–D359. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, Q.; Yu, G.; Gai, W.; Li, Y.; Chen, X. DCDB 2.0: A major update of the drug combination database. Database 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Chaudhary, K.; Gupta, S.; Singh, H.; Kumar, S.; Gautam, A.; Kapoor, P.; Raghava, G.P.S. CancerDR: Cancer drug resistance database. Sci. Rep. 2013, 3, 1445. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ren, B.; Chen, M.; Liu, M.X.; Ren, W.; Wang, Q.X.; Zhang, L.X.; Yan, G.Y. ASDCD: Antifungal synergistic drug combination database. PLoS ONE 2014, 9, e86499. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.; Gohlke, B.O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on drug classification and target prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.P.; Kwoh, C.K.; Yang, P.; Li, X.L.; Zheng, J. Drug-target interaction prediction by learning from local information and neighbors. Bioinformatics 2013, 29, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, T.; Marchiori, E. Predicting drug-target interactions for new drug compounds using a weighted nearest neighbor profile. PLoS ONE 2013, 8, e66952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, K.; Yamanishi, Y. Supervised prediction of drug–target interactions using bipartite local models. Bioinformatics 2009, 25, 2397–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zou, H.; Luo, L.; Liu, Q.; Wu, W.; Xiao, W. Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing 2016, 173, 979–987. [Google Scholar] [CrossRef]
- Shi, J.Y.; Yiu, S.M. SRP: A concise non-parametric similarity-rank-based model for predicting drug-target interactions. In Proceedings of the 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Washington, DC, USA, 9–12 November 2015; IEEE: New York, NY, USA, 2015; pp. 1636–1641. [Google Scholar]
- Zong, N.; Kim, H.; Ngo, V.; Harismendy, O. Deep mining heterogeneous networks of biomedical linked data to predict novel drug-target associations. Bioinformatics 2017, 33, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Liu, C.; Jiang, J.; Lu, W.; Li, W.; Liu, G.; Zhou, W.; Huang, J.; Tang, Y. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 2012, 8, e1002503. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and bi-random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gönen, M. Predicting drug–target interactions from chemical and genomic kernels using bayesian matrix factorization. Bioinformatics 2012, 28, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Cobanoglu, M.C.; Liu, C.; Hu, F.; Oltvai, Z.N.; Bahar, I. Predicting drug–target interactions using probabilistic matrix factorization. J. Chem. Inf. Model. 2013, 53, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ding, H.; Mamitsuka, H.; Zhu, S. Collaborative matrix factorization with multiple similarities for predicting drug-target interactions. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Chicago, IL, USA, 11–14 August 2013; ACM: New York, NY, USA, 2013; pp. 1025–1033. [Google Scholar]
- Ezzat, A.; Zhao, P.; Wu, M.; Li, X.L.; Kwoh, C.K. Drug-target interaction prediction with graph regularized matrix factorization. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 14, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, J.B.; Silva, V.D.; Langford, J.C. A global geometric framework for nonlinear dimensionality reduction. Science 2000, 290, 2319–2323. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, J.; Xu, X.; Li, Y.; Zhao, H.; Fang, Y.; Li, X.; Zhou, W.; Wang, W.; Wang, Y. A systematic prediction of multiple drug-target interactions from chemical, genomic, and pharmacological data. PLoS ONE 2012, 7, e37608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, J. Predicting drug-target interactions using restricted boltzmann machines. Bioinformatics 2013, 29, i126–i134. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Ding, Y.; Seal, A.; Chen, B.; Sun, Y.; Bolton, E. Predicting drug target interactions using meta-path-based semantic network analysis. BMC Bioinform. 2016, 17, 160. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Wu, L.Y.; Zhou, X.; Wong, S.T. Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces. BMC Syst. Biol. 2010, 4, S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpert, D.H.; Macready, W.G. No free lunch theorems for optimization. IEEE Trans Evol. Comput. 1997, 1, 67–82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, F.; Hu, J.; Sorrentino, R. Label propagation prediction of drug-drug interactions based on clinical side effects. Sci. Rep. 2015, 5, 12339. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, A.; Wu, M.; Li, X.L.; Kwoh, C.K. Drug-target interaction prediction using ensemble learning and dimensionality reduction. Methods 2017, 129, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, A.; Wu, M.; Li, X.L.; Kwoh, C.K. Drug-target interaction prediction via class imbalance-aware ensemble learning. BMC Bioinform. 2016, 17, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R. An ensemble learning approach for improving drug–target interactions prediction. In Proceedings of the 4th International Conference on Computer Engineering and Networks, Shanghai, China, 19–20 July 2015; Wong, W.E., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 433–442. [Google Scholar]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-generation machine learning for biological networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular rna detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Goadrich, M. The relationship between Precision-Recall and ROC curves. In Proceedings of the 23rd International Conference on Machine Learning (ICML ’06), Pittsburgh, PA, USA, 25–29 June 2006; ACM Press: New York, NY, USA, 2006; pp. 233–240. [Google Scholar] [Green Version]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Database and URL | Brief Descriptions |
|---|---|
| KEGG [29] http://www.genome.jp/kegg | An encyclopedia of genes and genomes for both functional interpretation and practical application of genomic information. |
| BRENDA [30] http://www.brenda-enzymes.org/ | The main enzyme and enzyme-ligand information system. |
| PubChem [31] https://pubchem.ncbi.nlm.nih.gov/ | A database for information on chemical substances and their biological activities involving three inter-linked databases, i.e., Substance, Compound and BioAssay. |
| TTD [32] http://bidd.nus.edu.sg/group/ttd/ttd.asp | Therapeutic Target Database providing comprehensive information about the drug resistance mutations, gene expressions and target combinations data. |
| DrugBank [33] http://www.drugbank.ca | Consisting of two parts information involving detailed drug data (i.e., chemical, pharmacological and pharmaceutical) and drug target information (i.e., sequence, structure, and pathway) respectively. |
| SuperTarget [34] http://bioinf-apache.charite.de/supertarget | A database integrating drug-related information with more than 330,000 compound-target protein relations. |
| ChEMBL [35] https://www.ebi.ac.uk/chembldb | Data resource for molecule structures and molecule-protein interactions collected from the primary published literature on a regular basis. |
| STITCH [36] http://stitch.embl.de/ | Repository of known and predicted chemical-protein interactions. |
| MATADOR [37] http://matador.embl.de/ | A database of protein-chemical interactions including as many direct and indirect interactions as possible. |
| BindingDB [38] http://www.bindingdb.org/bind | A public database of protein-ligand binding affinities. |
| TDR targets [39] http://tdrtargets.org/ | A chemogenomics resource for neglected tropical diseases. |
| SIDER [40] http://sideeffects.embl.de/ | Serving information on marketed medicines and their recorded adverse drug reactions. |
| ChemBank [41] http://chembank.broad.harvard.edu/ | Collections of available data derived from small molecules and small-molecule screens and resources for studying their properties. |
| DCDB [42] http://www.cls.zju.edu.cn/dcdb/ | The Drug Combination Database for collecting and organizing known examples of drug combinations. |
| CancerDR [43] http://crdd.osdd.net/raghava/cancerdr/ | Cancer Drug Resistance Database of 148 anticancer drugs and their effectiveness against around 1000 cancer cell lines. |
| ASDCD [44] http://asdcd.amss.ac.cn/ | The first Antifungal Synergistic Drug Combination Database including published synergistic antifungal drug combinations, targets, indications, and other pertinent data. |
| SuperPred [45] http://prediction.charite.de/ | Resource of compound-target interactions. |
| Databases | The Number of Compounds | The Number of Targets | The Number of Compound-Target Interactions |
|---|---|---|---|
| KEGG | 18,380 | 26,885,475 | |
| BRENDA | 7341 | ||
| PubChem | 96,479,316 | 68,868 | |
| TTD | 34,019 | 3101 | |
| DrugBank | 11,682 | 26,889 | 131,724 |
| SuperTarget | 195,770 | 6219 | 332,828 |
| ChEMBL | 2,275,906 | 12,091 | |
| STITCH | 500,000 | 9,600,000 | 1,600,000,000 |
| MATADOR | 775 | ||
| BindingDB | 652,068 | 7082 | 1,454,892 |
| TDR targets | 2,000,000 | 5300 | |
| SIDER | 5868 | 1430 | 139,756 |
| ChemBank | 1,700,000 | ||
| DCDB | 904 | 805 | |
| CancerDR | 148 | 116 | |
| ASDCD | 105 | 1225 | 210 |
| SuperPred | 341,000 | 1800 | 665,000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.; Liu, X.; Jin, S.; Lin, J.; Liu, J. Machine Learning for Drug-Target Interaction Prediction. Molecules 2018, 23, 2208. https://doi.org/10.3390/molecules23092208
Chen R, Liu X, Jin S, Lin J, Liu J. Machine Learning for Drug-Target Interaction Prediction. Molecules. 2018; 23(9):2208. https://doi.org/10.3390/molecules23092208
Chicago/Turabian StyleChen, Ruolan, Xiangrong Liu, Shuting Jin, Jiawei Lin, and Juan Liu. 2018. "Machine Learning for Drug-Target Interaction Prediction" Molecules 23, no. 9: 2208. https://doi.org/10.3390/molecules23092208
APA StyleChen, R., Liu, X., Jin, S., Lin, J., & Liu, J. (2018). Machine Learning for Drug-Target Interaction Prediction. Molecules, 23(9), 2208. https://doi.org/10.3390/molecules23092208