
Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes




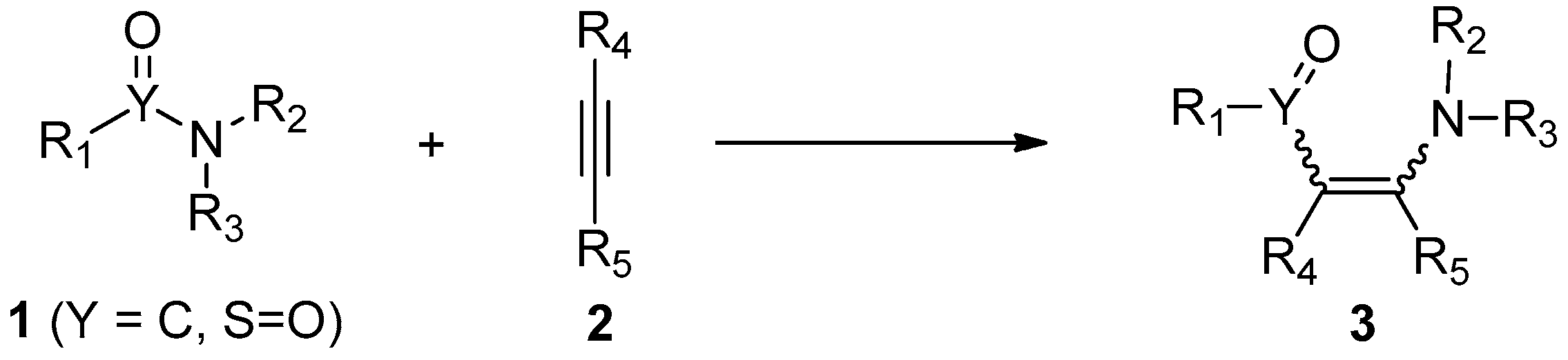{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
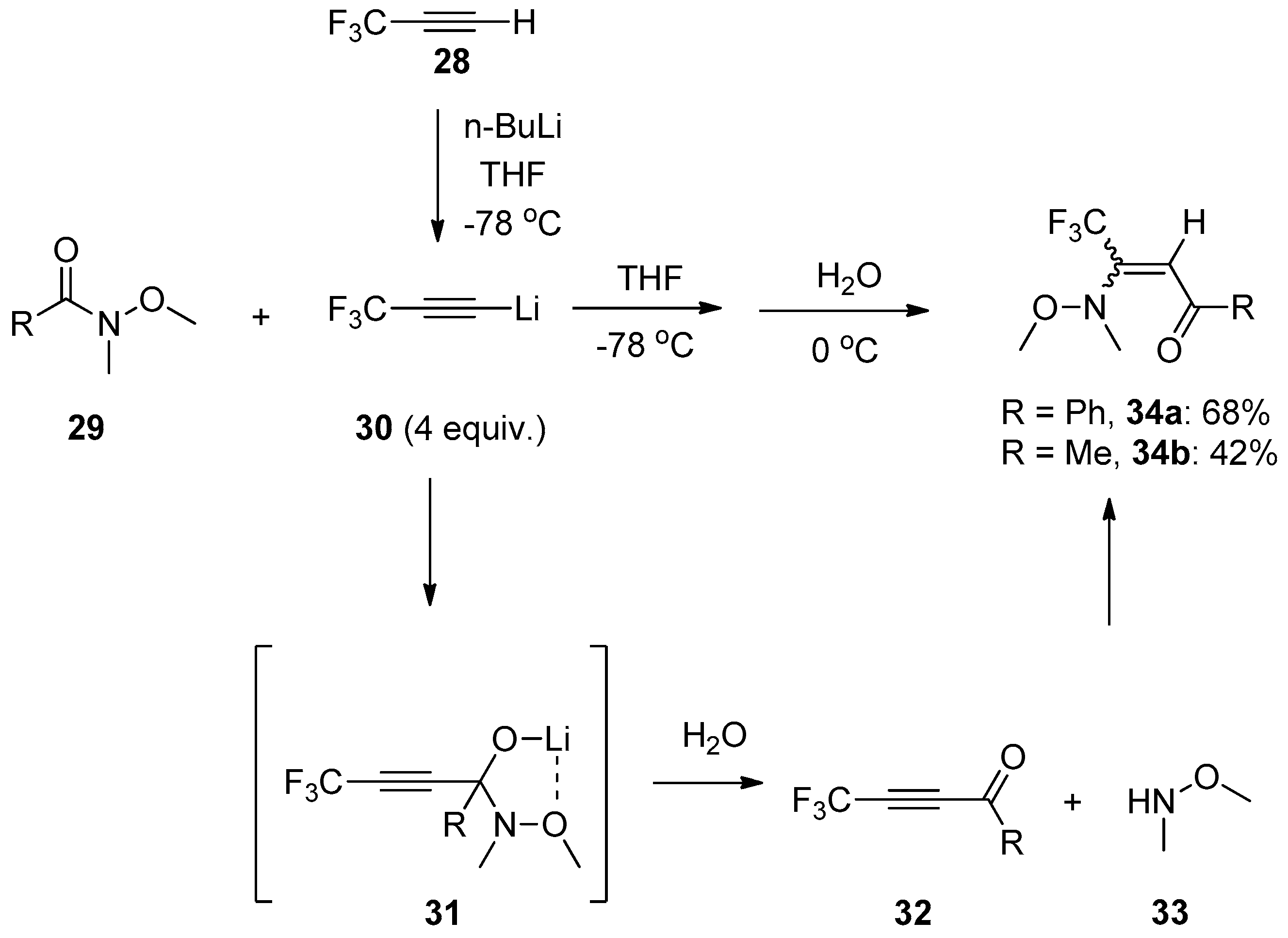{kind=link}
{kind=link}
{kind=link}
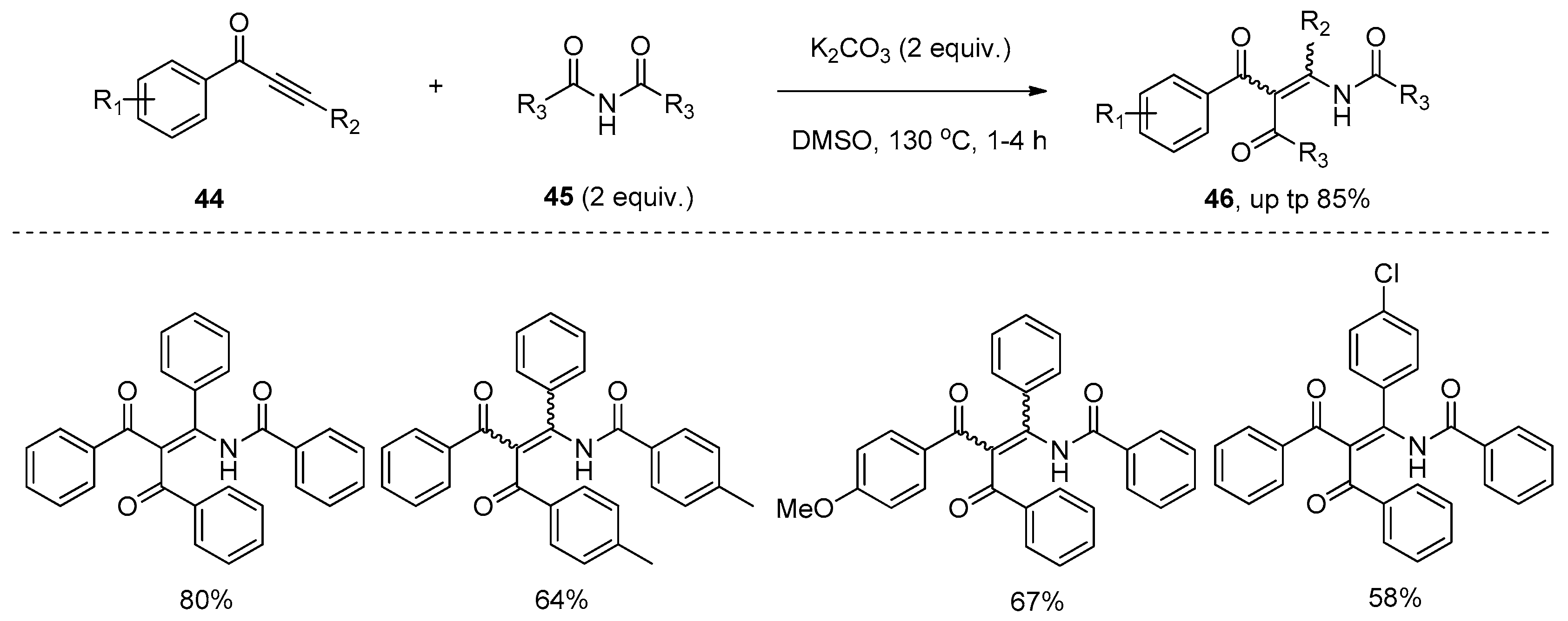{kind=link}
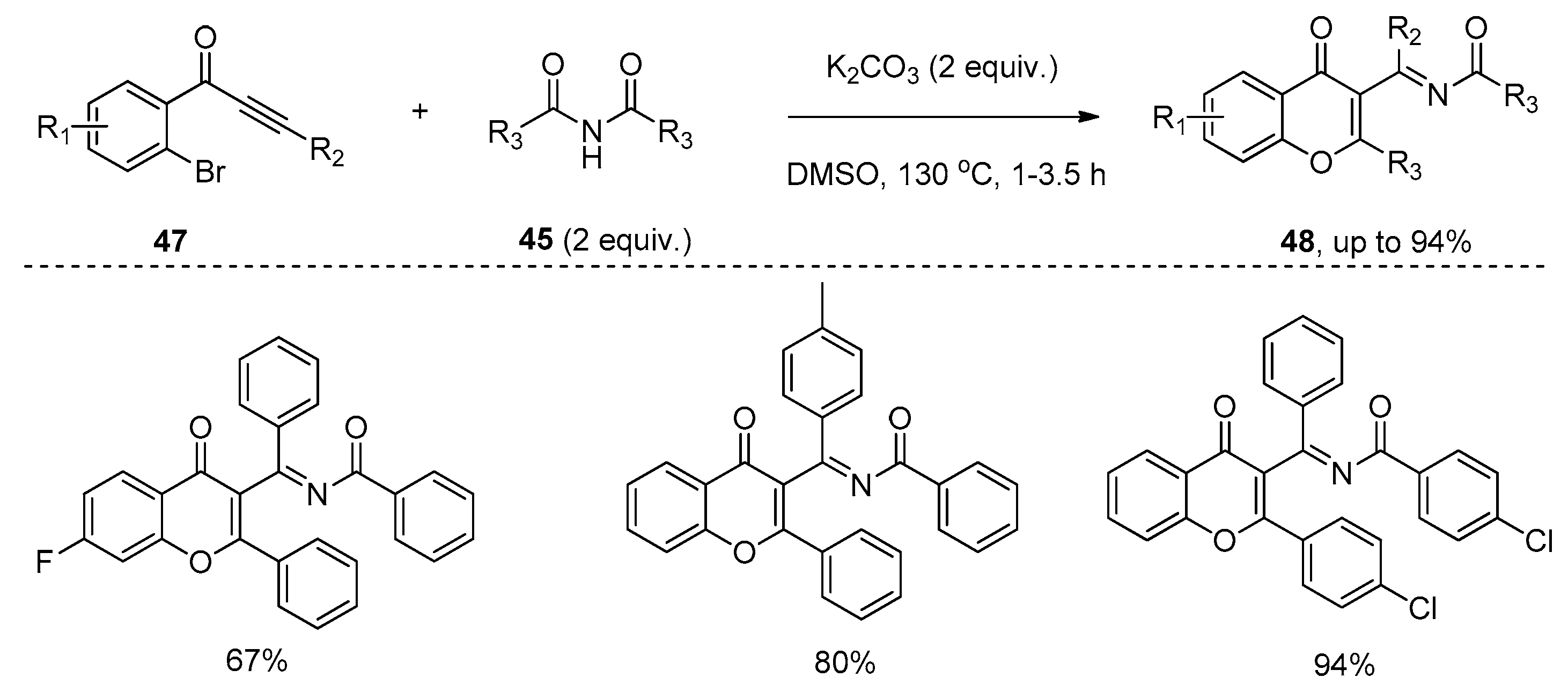{kind=link}
{kind=link}
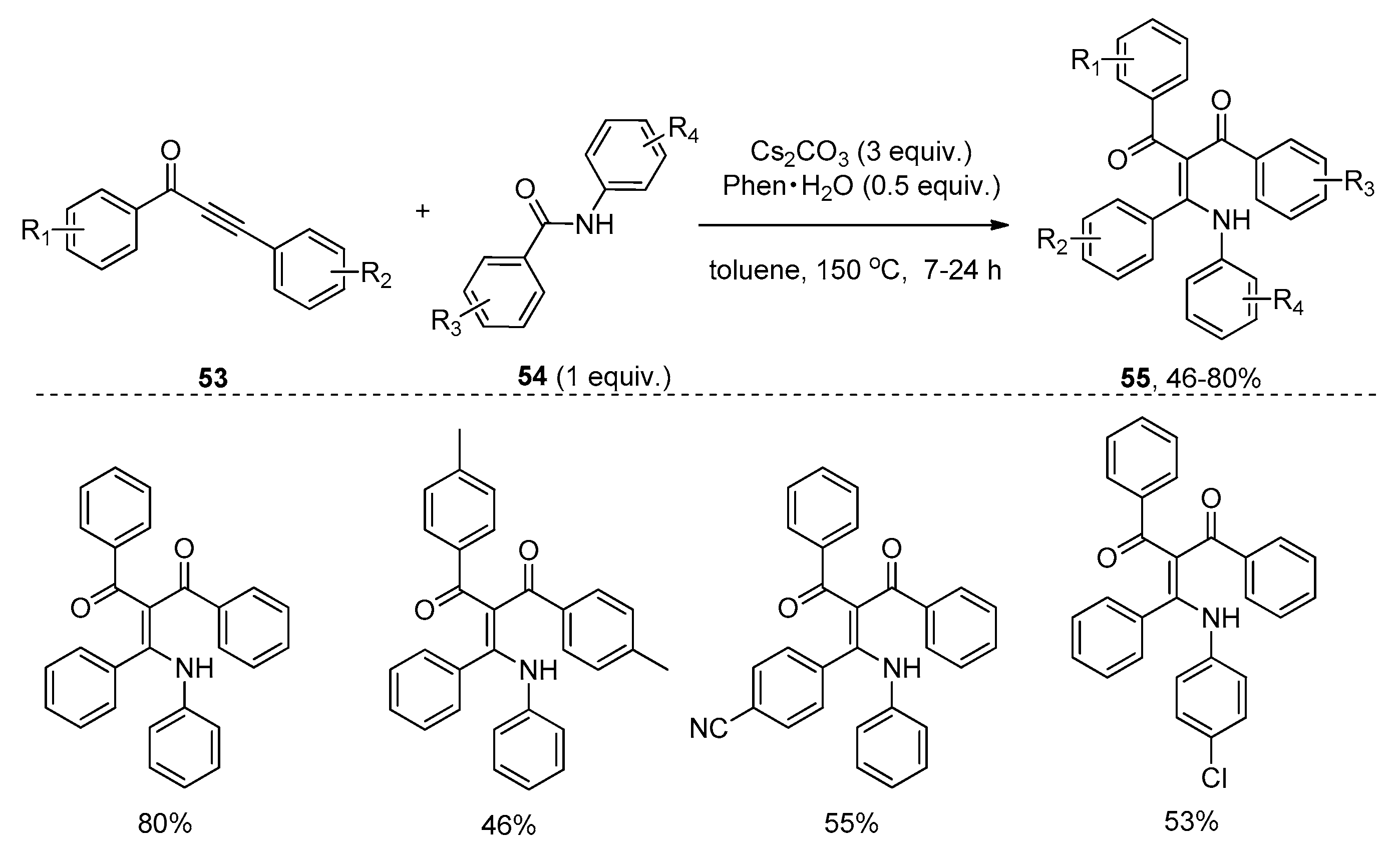{kind=link}
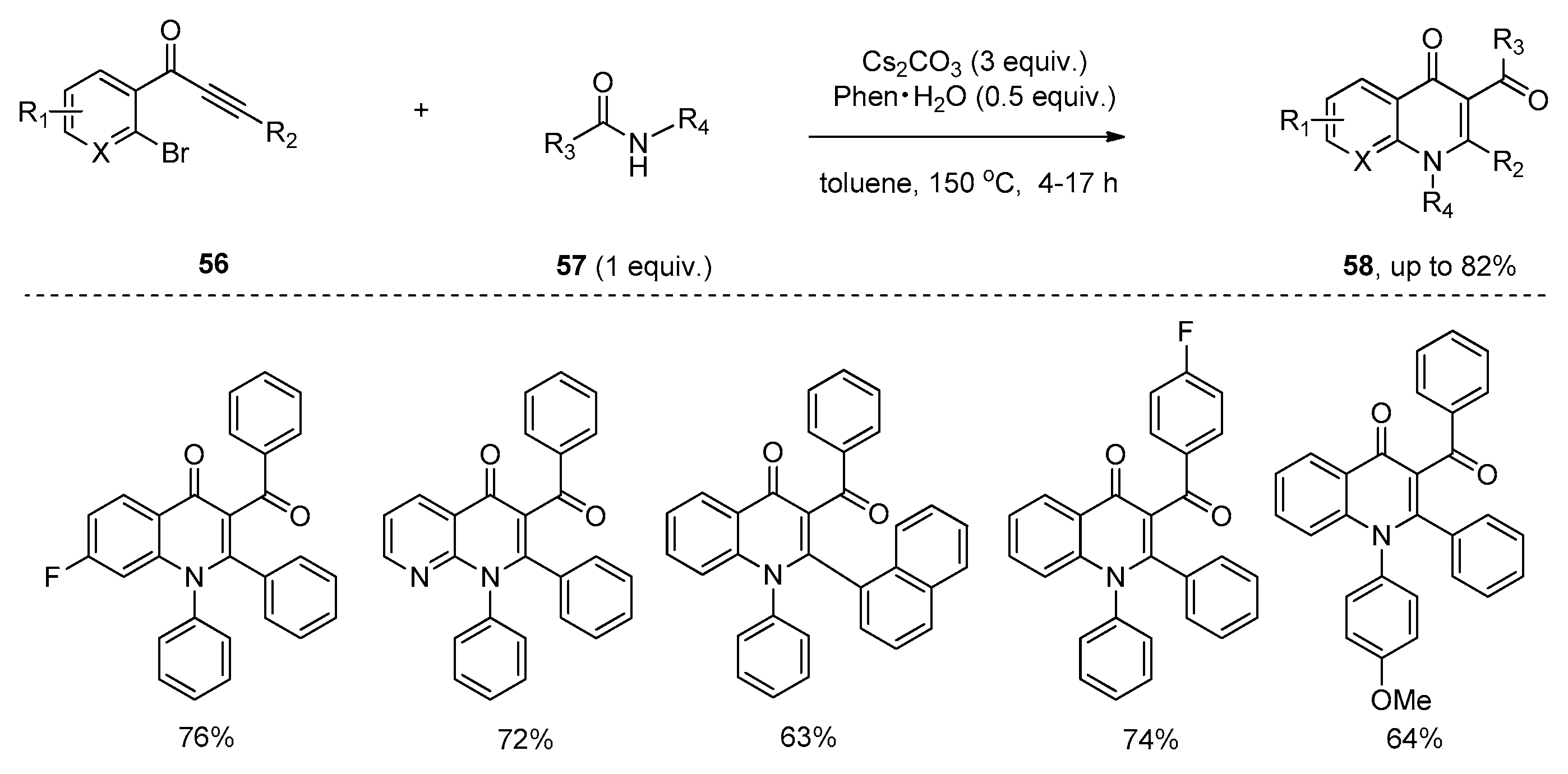{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
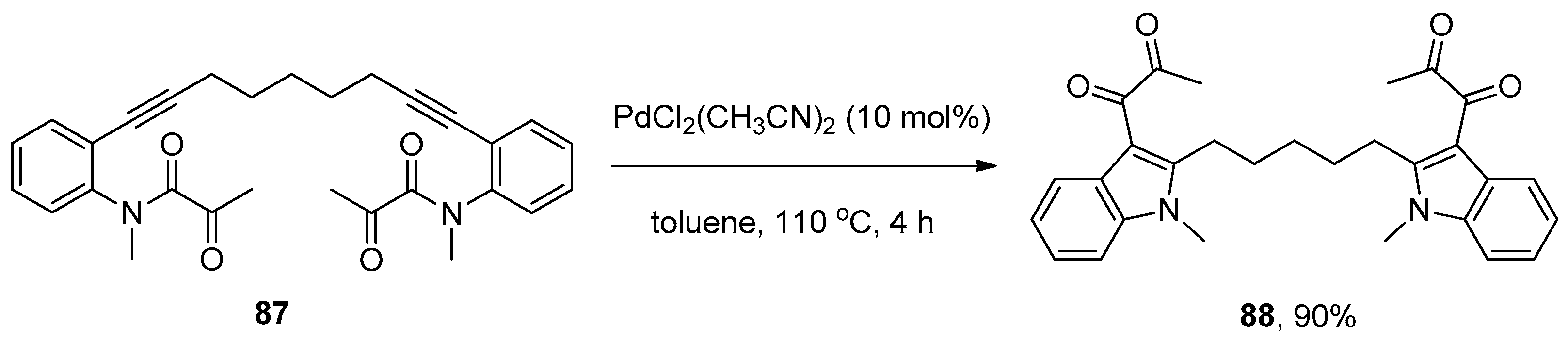{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Addition of Amide Bonds to Alkynes
2.1. Direct Addition of Amide Bonds to Alkynes without Catalysts and Additives
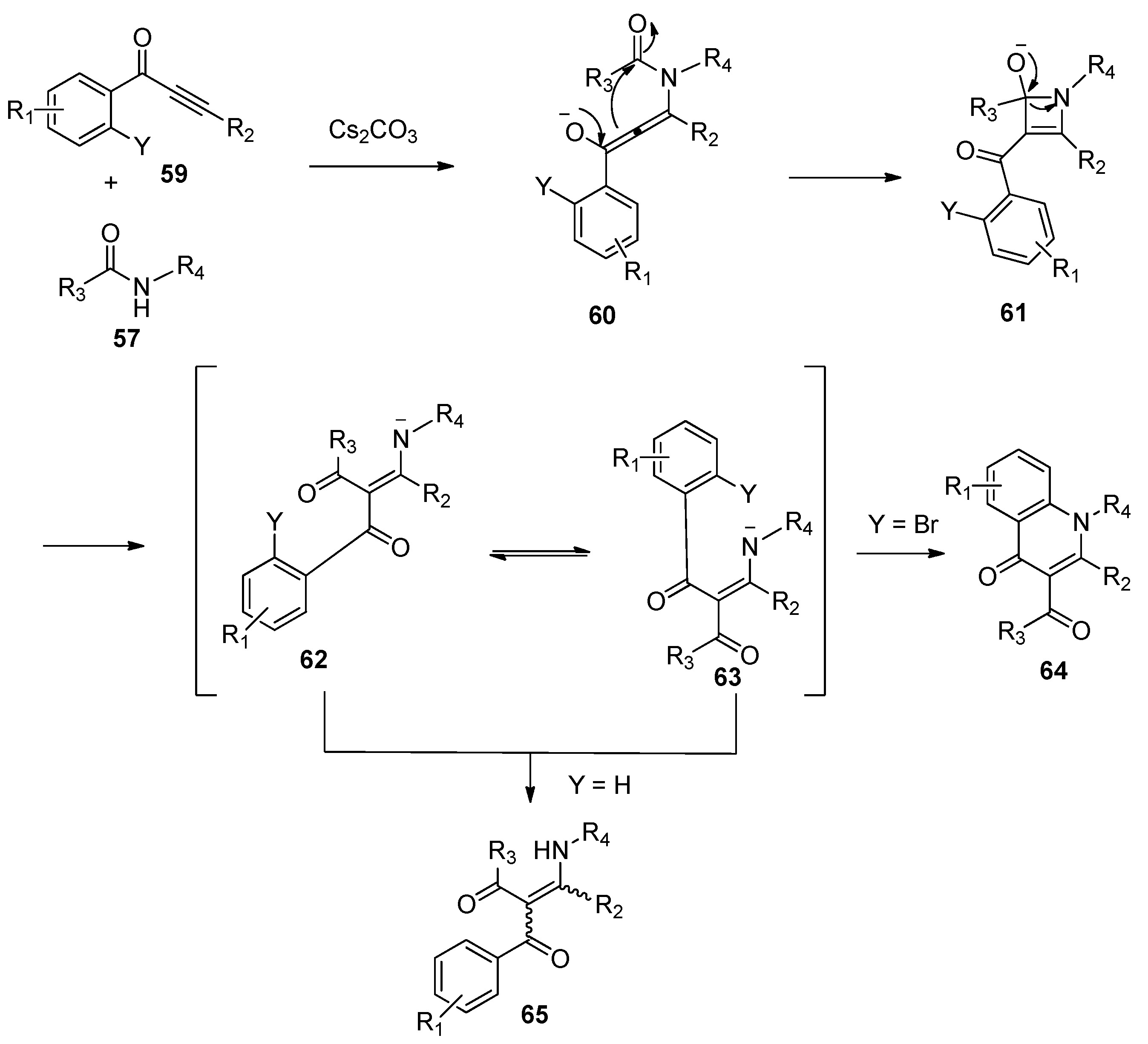
2.2. Base-Promoted Addition of Amide Bonds to Alkynes
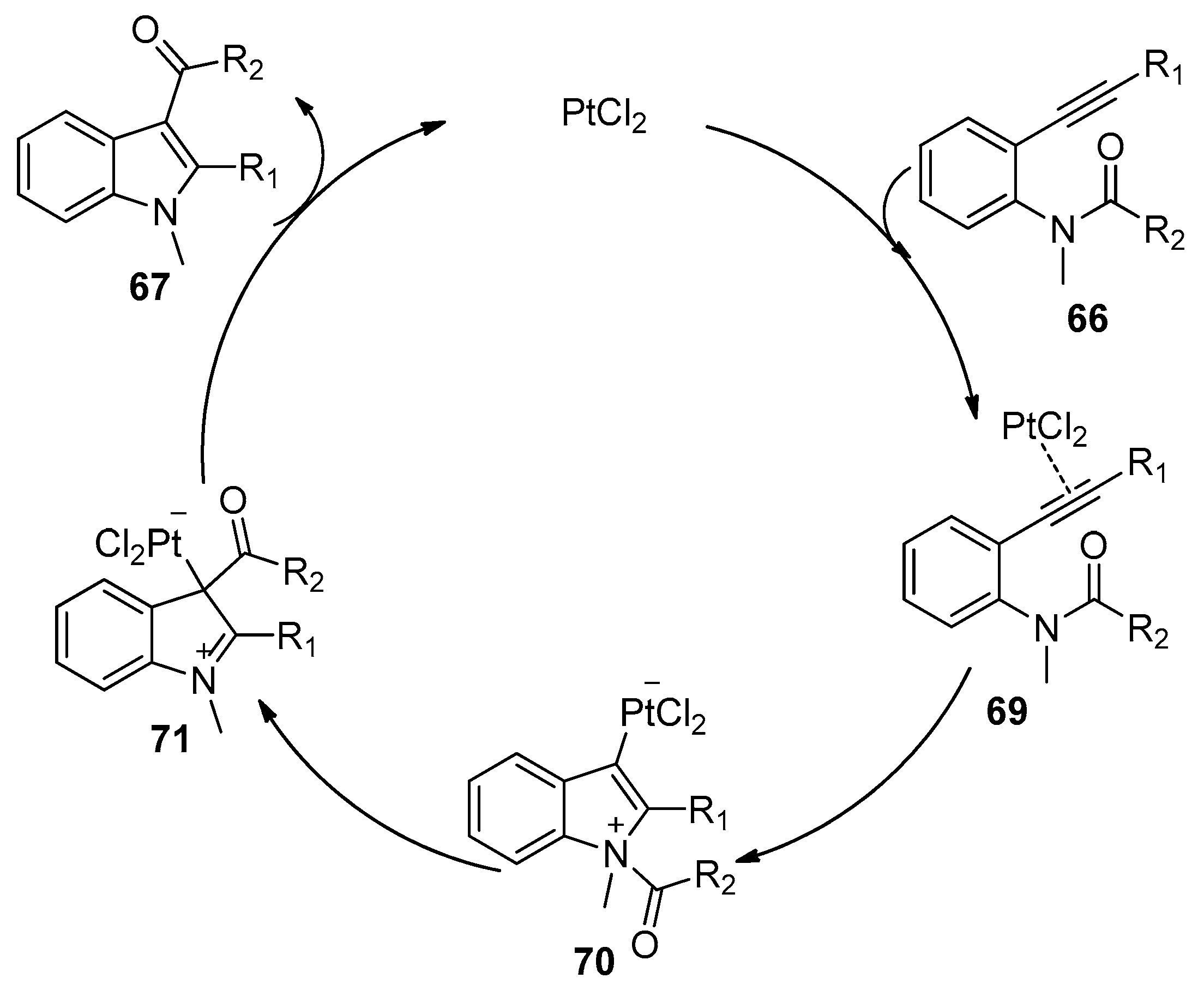
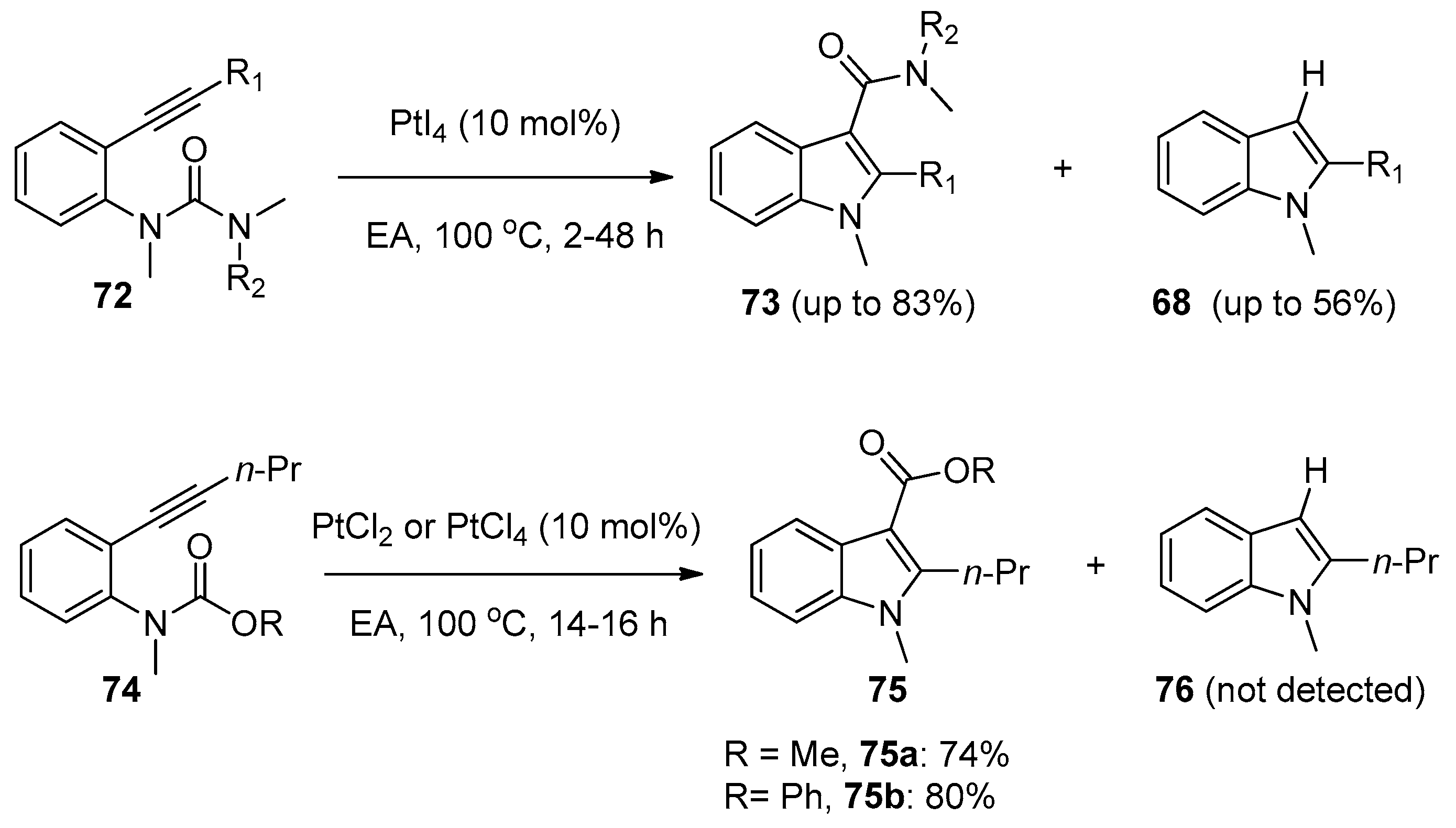
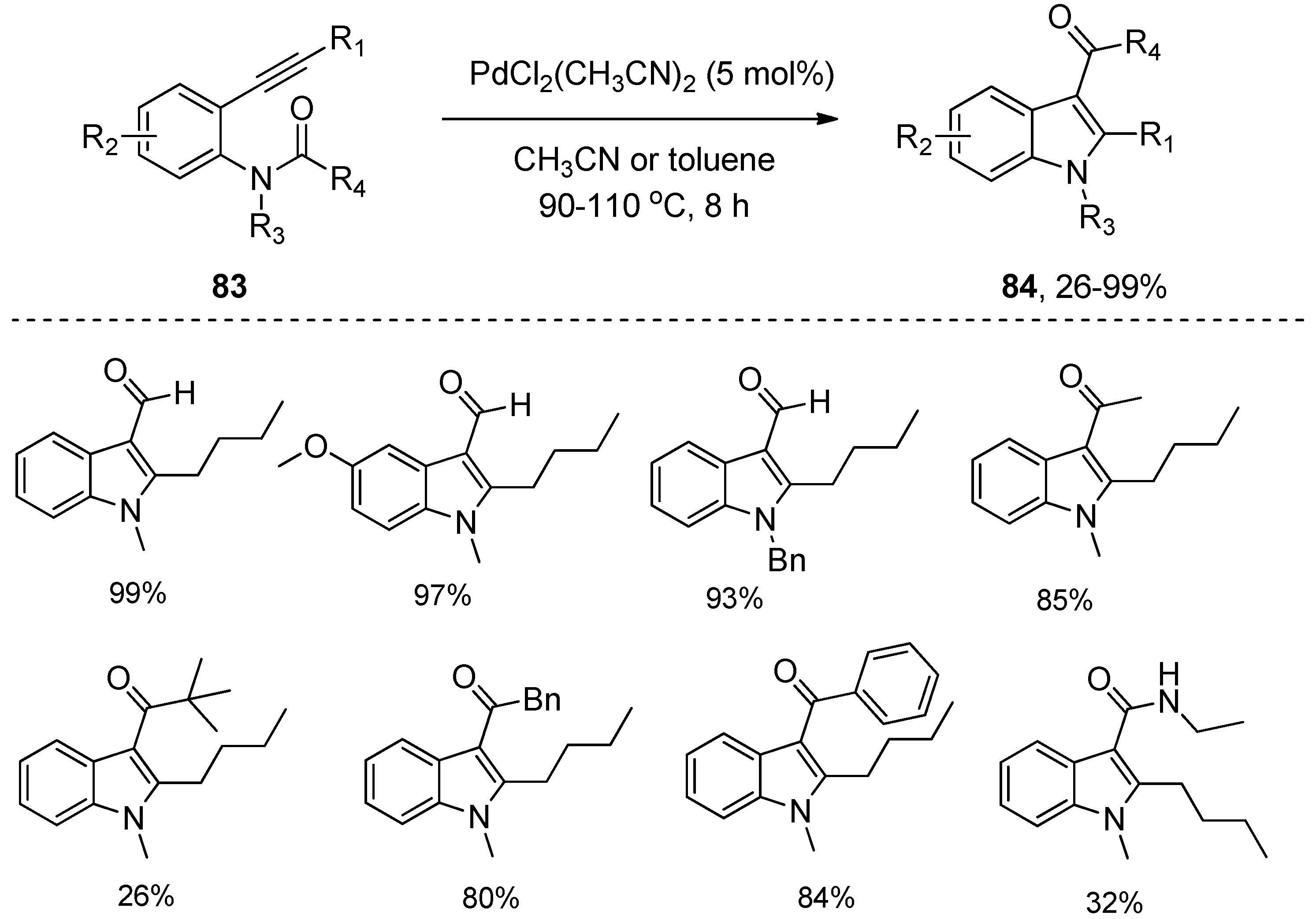
2.3. Transition-Metal-Catalyzed Addition of Amide Bonds to Alkynes
2.4. Addition of Amide Bonds to Alkynes through Organocatalysis
3. Transition-Metal-Catalyzed Addition of Sulfonamide Bonds to Alkynes
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Irvine, G.J.; Lesley, M.J.G.; Marder, T.B.; Norman, N.C.; Rice, C.R.; Robins, E.G.; Roper, W.R.; Whittell, G.R.; Wright, L.J. Transition metal-boryl compounds: Synthesis, reactivity, and structure. Chem. Rev. 1998, 98, 2685–2722. [Google Scholar] [CrossRef] [PubMed]
- Marder, T.B.; Norman, N.C. Transition metal catalysed diboration. Top. Catal. 1998, 5, 63–73. [Google Scholar] [CrossRef]
- Han, L.B.; Tanaka, M. Transition metal-catalysed addition reactions of H-heteroatom and inter-heteroatom bonds to carbon-carbon unsaturated linkages via oxidative additions. Chem. Commun. 1999, 395–402. [Google Scholar] [CrossRef]
- Beletskaya, I. Element-element addition to alkynes catalyzed by the group 10 metals. Chem. Rev. 1999, 99, 3435–3461. [Google Scholar] [CrossRef] [PubMed]
- Suginome, M.; Ito, Y. Transition-metal-catalyzed additions of silicon-silicon and silicon-heteroatom bonds to unsaturated organic molecules. Chem. Rev. 2000, 100, 3221–3256. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, T.; Miyaura, N. Chemistry of group 13 element-transition metal linkage—The platinum- and palladium-catalyzed reactions of (alkoxo)diborons. J. Organomet. Chem. 2000, 611, 392–402. [Google Scholar] [CrossRef]
- Suginome, M.; Ito, Y. Stereoselective accesses to enantioenriched allyl-, allenyl-, and propargyl-silanes via Si-Si bond activation by palladium-isocyanide catalysts. J. Organomet. Chem. 2003, 685, 218–229. [Google Scholar] [CrossRef]
- Dembitsky, V.M.; Ali, H.A.; Srebnik, M. Recent developments in bisdiborane chemistry: B–C–B, B–C–C–B, B–C=C–B and B–C≡C–B compounds. Appl. Organomet. Chem. 2003, 17, 327–345. [Google Scholar] [CrossRef]
- Ishiyama, T.; Miyaura, N. Metal-catalyzed reactions of diborons for synthesis of organoboron compounds. Chem. Rec. 2004, 3, 271–280. [Google Scholar] [CrossRef]
- Beletskaya, I. Element-element additions to unsaturated carbon-carbon bonds catalyzed by transition metal complexes. Chem. Rev. 2006, 106, 2320–2354. [Google Scholar] [CrossRef]
- Shimizu, M.; Hiyama, T. Polyborylated reagents for modern organic synthesis. Proc. Jpn. Acad. Ser. B 2008, 84, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Miyaura, N. Metal-catalyzed reactions of organoboronic acids and esters. Bull. Chem. Soc. Jpn. 2008, 81, 1535–1553. [Google Scholar] [CrossRef]
- Takaya, J.; Iwasawa, N. Catalytic, direct synthesis of bis(boronate) compounds. ACS Catal. 2012, 2, 1993–2006. [Google Scholar] [CrossRef]
- Zhao, F.; Jia, X.; Wang, D.; Fei, C.; Wu, C.; Wang, J.; Liu, H. Research progress in metal-catalyzed addition of carbon-hetero bonds to alkynes. Chin. J. Org. Chem. 2017, 37, 284–300. [Google Scholar] [CrossRef]
- Zhao, F.; Jia, X.; Li, P.; Zhao, J.; Zhou, Y.; Wang, J.; Liu, H. Catalytic and catalyst-free diboration of alkynes. Org. Chem. Front. 2017, 4, 2235–2255. [Google Scholar] [CrossRef]
- Miyabe, H. Transition-metal-free activation of amide bond by arynes. Molecules 2018, 23, 2145. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Eğe, S.N.; Carter, M.L.C.; Spencer, R.L.; Nordman, C.E.; Friedman, H.Z. Formation and acid-catalyzed rearrangement of 1,2-diazepin-5-ones. J. Chem. Soc. Perkin Trans. 1976, 1, 868–876. [Google Scholar] [CrossRef]
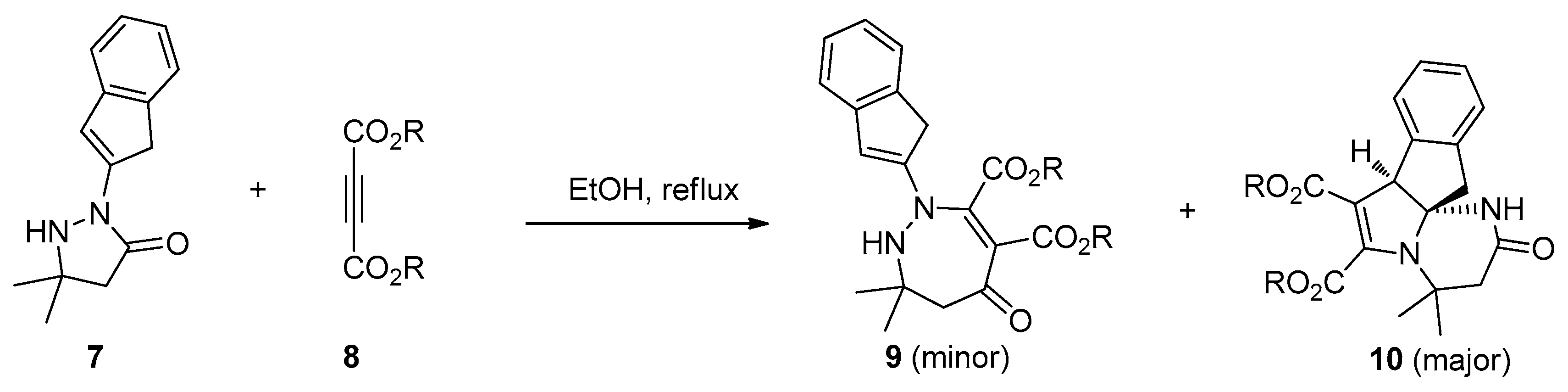
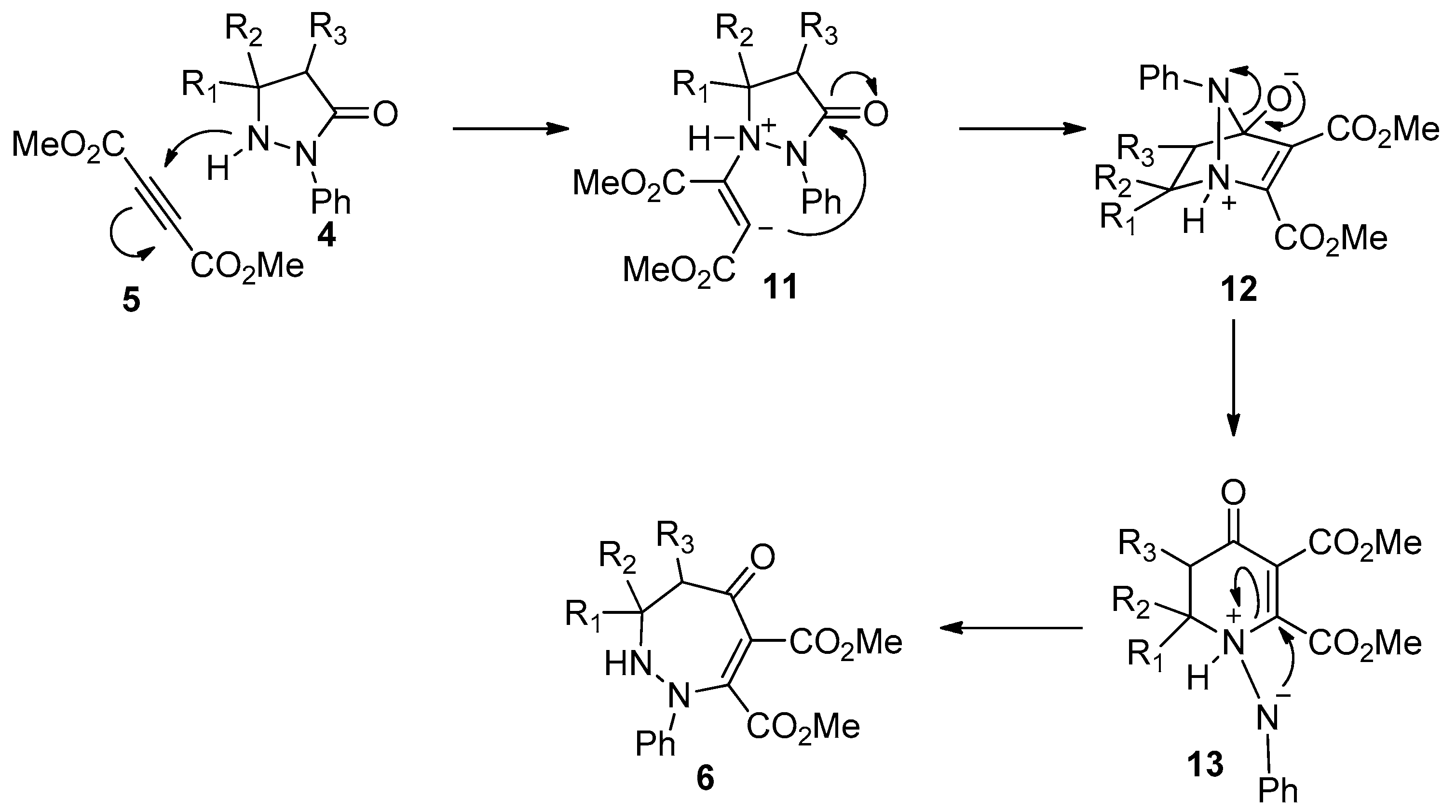
- Turk, C.; Svete, J.; Stanovnik, B.; Golič, L.; Golobič, A.; Selič, L. Unusual reactions of 5,5-dimethyl-2-(indenyl-2)-3-pyrazolidinone with acetylenedicarboxylates. Org. Lett. 2000, 2, 423–424. [Google Scholar] [CrossRef] [PubMed]
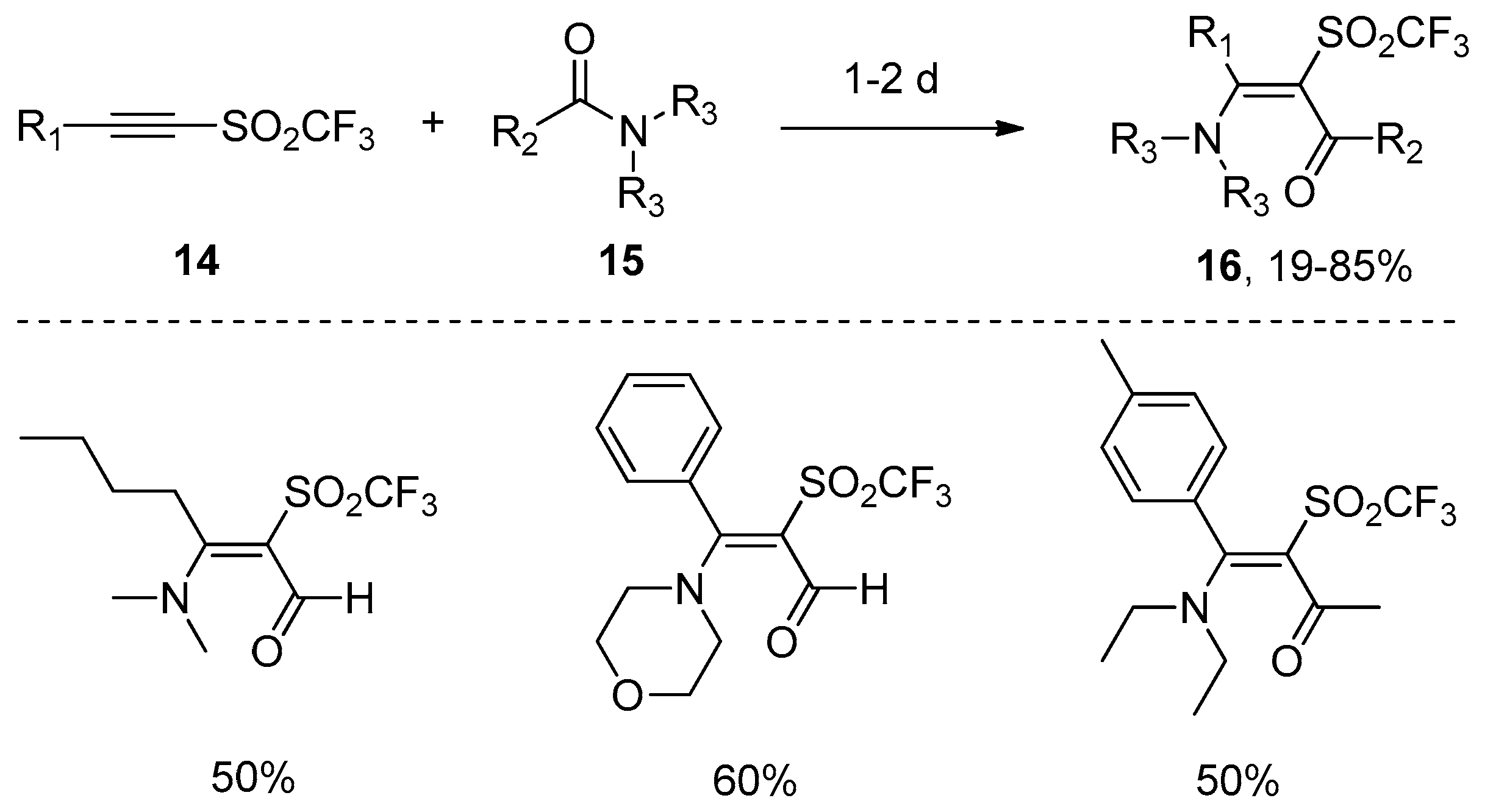
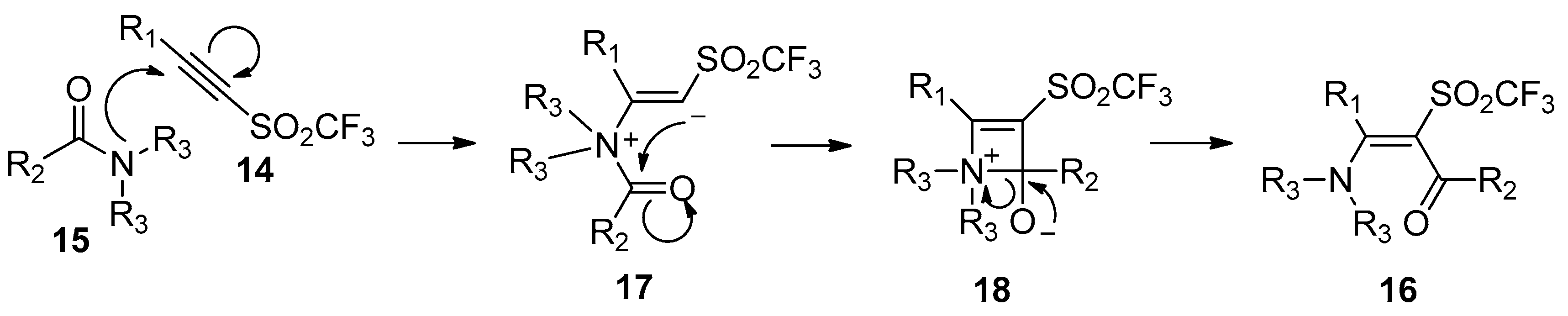
- Hanack, M.; Wilhelrn, B. Addition of carboxamides to alkynyl trifluoromethyl sulfones. Angew. Chem. Int. Ed. Engl. 1989, 28, 1057–1059. [Google Scholar] [CrossRef]
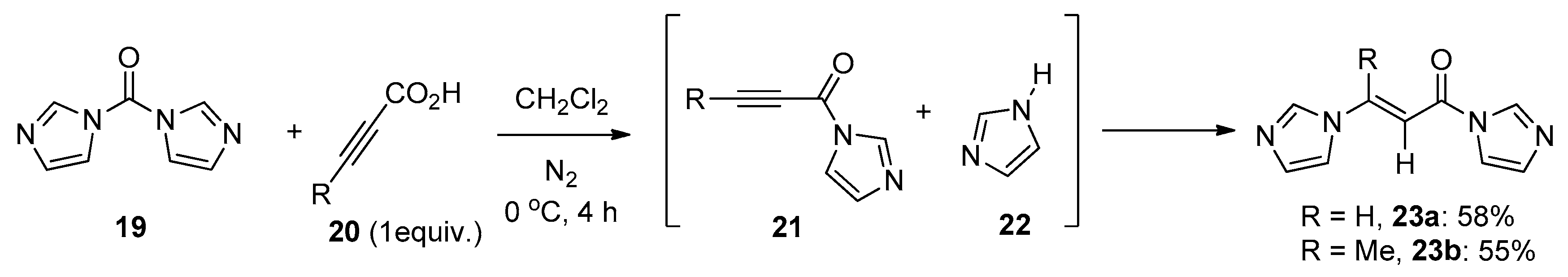
- Knölker, H.J.; El-Ahl, A.A. Imidazole derivatives, part VIII. Stereoselective formation of 1-[(E) 3-(1-imidazolyl)-2-alkenoyl]imidazoles. Heterocycles 1993, 36, 1381–1385. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohkuma, T.; Tsuchihashi, G. Preparation of enaminones by two-carbon homologation of amides with lithium (triphenylsilyl)acetylide. J. Org. Chem. 1987, 52, 2930–2932. [Google Scholar] [CrossRef]
- Jeong, I.H.; Jeon, S.L.; Min, Y.K.; Kim, B.T. A novel approach to β-trifluoromethyl enaminones. Tetrahedron Lett. 2002, 43, 7171–7174. [Google Scholar] [CrossRef]
- Jeong, I.H.; Jeon, S.L.; Kim, M.S.; Kim, B.T. New approaches to β-trifluoromethylated enone derivatives. J. Fluorine Chem. 2004, 125, 1629–1638. [Google Scholar] [CrossRef]
- Jeon, S.L.; Kim, J.K.; Son, J.B.; Kim, B.T.; Jeong, I.H. One pot synthesis of novel α,β-dichloro-β-trifluoromethylated enones and their application to the synthesis of trifluoromethylated heterocycles. J. Fluorine Chem. 2007, 128, 153–157. [Google Scholar] [CrossRef]
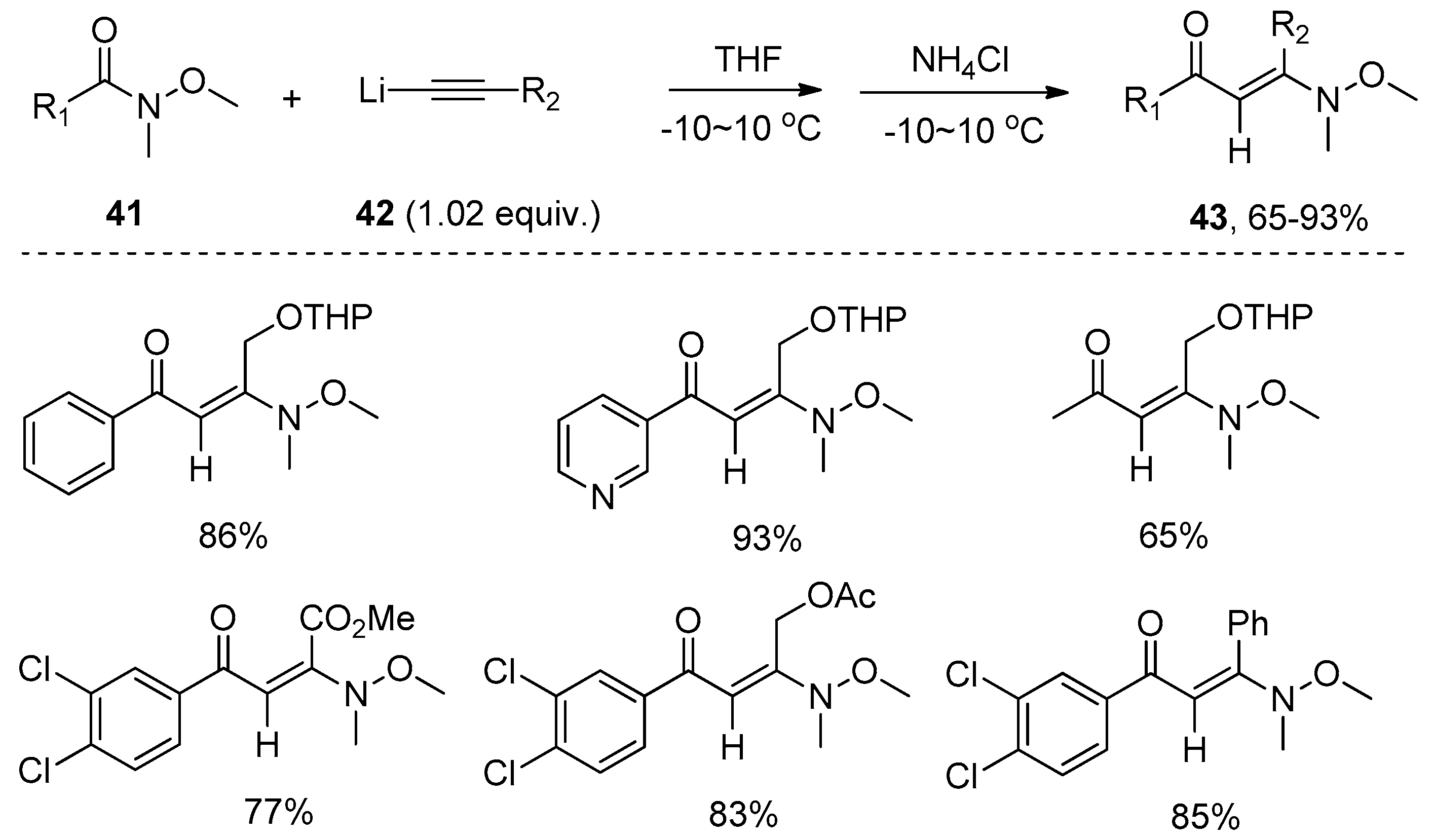
- Persson, T.; Nielsen, J. Synthesis of N-methoxy-N-methyl-β-enaminoketoesters: New synthetic precursors for the regioselective synthesis of heterocyclic compounds. Org. Lett. 2006, 8, 3129–3222. [Google Scholar] [CrossRef]
- Choudhury, A.; Breslav, M.; Grimm, J.S.; Xiao, T.; Xu, D.; Sorgi, K.L. A facile one-pot synthesis of acyclic β-enamino ketones, an important class of versatile synthetic intermediates. Tetrahedron Lett. 2007, 48, 3069–3072. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, Y.; Xu, M.; Kong, L.; Wang, M.; Li, Y. Transition-metal-free insertion reactions of alkynes into the C–N r-bonds of imides: Synthesis of substituted enamides or chromones. Chem. Commun. 2018, 54, 6192–6195. [Google Scholar] [CrossRef]
- Zheng, Z.; Tao, Q.; Ao, Y.; Xu, M.; Li, Y. Transition-metal-free aminoacylation of ynones with amides: Synthesis of 3-carbonyl-4-quinolinones or functionalized enaminones. Org. Lett. 2018, 20, 3907–3910. [Google Scholar] [CrossRef]
- Shimada, T.; Nakamura, I.; Yamamoto, Y. Intramolecular C-N bond addition of amides to alkynes using platinum catalyst. J. Am. Chem. Soc. 2004, 126, 10546–10547. [Google Scholar] [CrossRef]
- Nakamura, I.; Sato, Y.; Konta, S.; Terada, M. Platinum-catalyzed consecutive C-N bond formation-[1,3] shift of carbamoyl and ester groups. Tetrahedron Lett. 2009, 50, 2075–2077. [Google Scholar] [CrossRef]
- Nakamura, I.; Mizushima, Y.; Yamagishi, U.; Yamamoto, Y. Synthesis of 2,3-disubstituted benzofurans and indoles by π-Lewis acidic transition metal-catalyzed cyclization of ortho-alkynylphenyl O,O- and N,O-acetals. Tetrahedron 2007, 63, 8670–8676. [Google Scholar] [CrossRef]
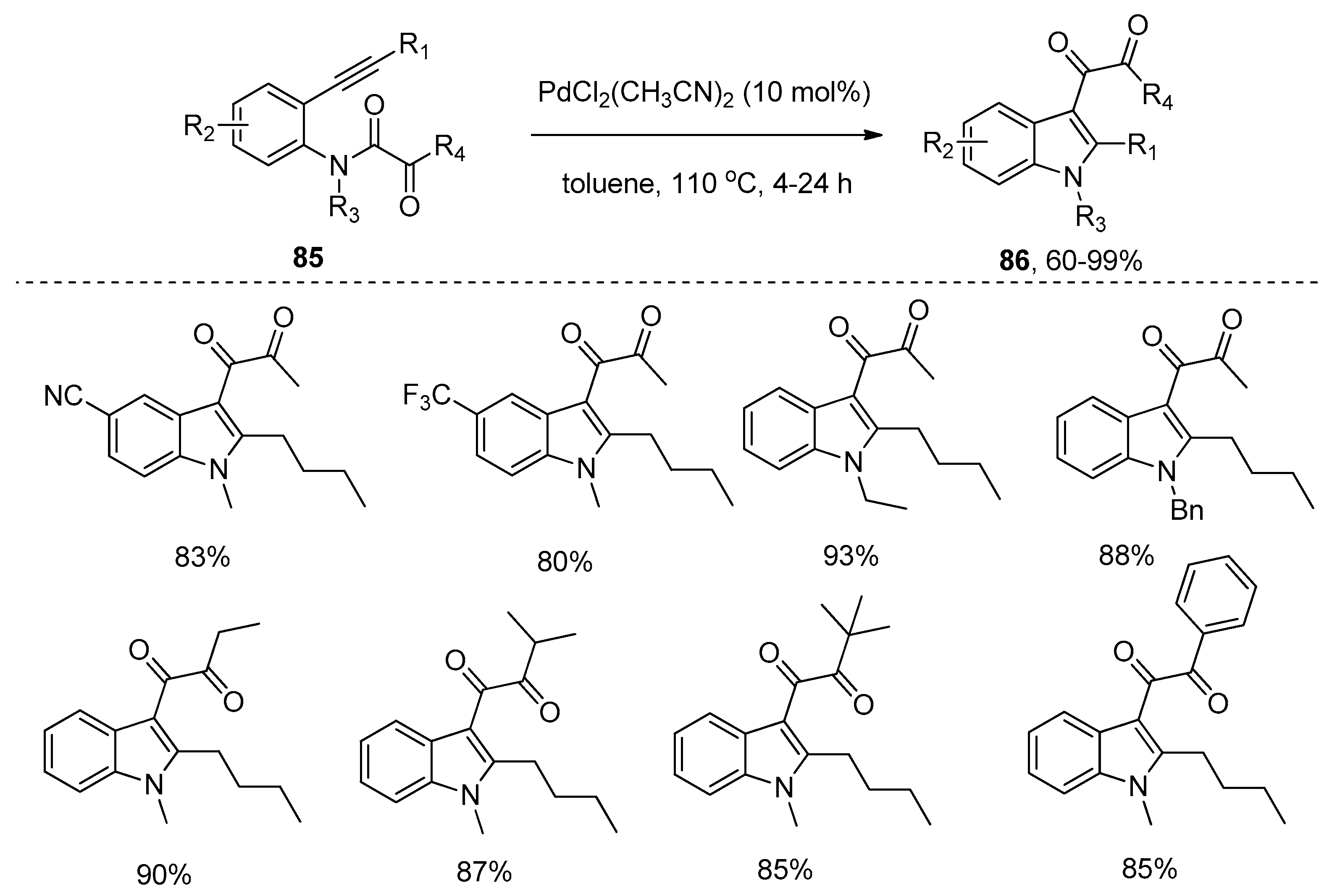
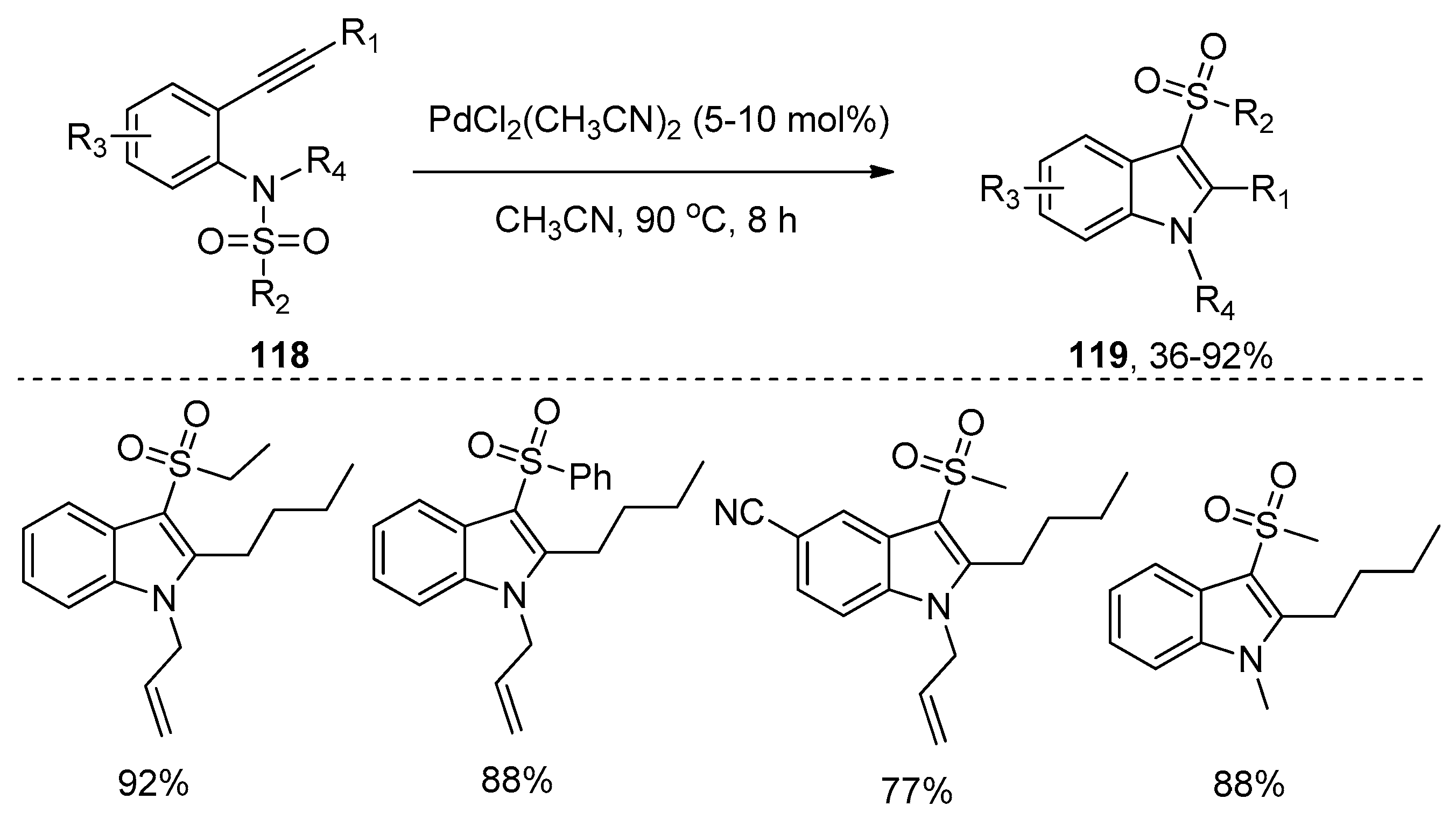
- Zhao, F.; Zhang, D.; Nian, Y.; Zhang, L.; Yang, W.; Liu, H. Palladium-catalyzed difunctionalization of alkynes via C–N and S–N cleavages: A versatile approach to highly functional indoles. Org. Lett. 2014, 16, 5124–5127. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, F.; Shu, S.; Wang, J.; Liu, H. Palladium-catalyzed intramolecular addition of C–N bond to alkynes: A novel approach to 3-diketoindoles. RSC Adv. 2015, 5, 90396–90399. [Google Scholar] [CrossRef]
- Okauchi, T.; Itonaga, M.; Minami, T.; Owa, T.; Kitoh, K.; Yoshino, H. A general method for acylation of indoles at the 3-position with acyl chlorides in the presence of dialkylaluminum chloride. Org. Lett. 2000, 2, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Merkul, E.; Dohe, J.; Gers, C.; Rominger, F.; Müller, T.J.J. Three-component synthesis of ynediones by a Glyoxylation/Stephens-Castro coupling sequence. Angew. Chem. Int. Ed. 2011, 50, 2966–2969. [Google Scholar] [CrossRef]
- Wu, J.C.; Song, R.J.; Wang, Z.Q.; Huang, X.C.; Xie, Y.X.; Li, J.H. Copper-catalyzed C-H oxidation/cross-coupling of α-amino carbonyl compounds. Angew. Chem. Int. Ed. 2012, 51, 3453–3457. [Google Scholar] [CrossRef]
- Tang, R.Y.; Guo, X.K.; Xiang, J.N.; Li, J.H. Palladium-catalyzed synthesis of 3-acylated indoles involving oxidative cross-coupling of indoles with α-amino carbonyl compounds. J. Org. Chem. 2013, 78, 11163–11171. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, J.; Wu, X.; Liu, S.; Wu, A. Direct regioselective oxidative cross-coupling of indoles with methyl ketones: A novel route to C3-dicarbonylation of indoles. Org. Lett. 2015, 17, 134–137. [Google Scholar] [CrossRef] [PubMed]
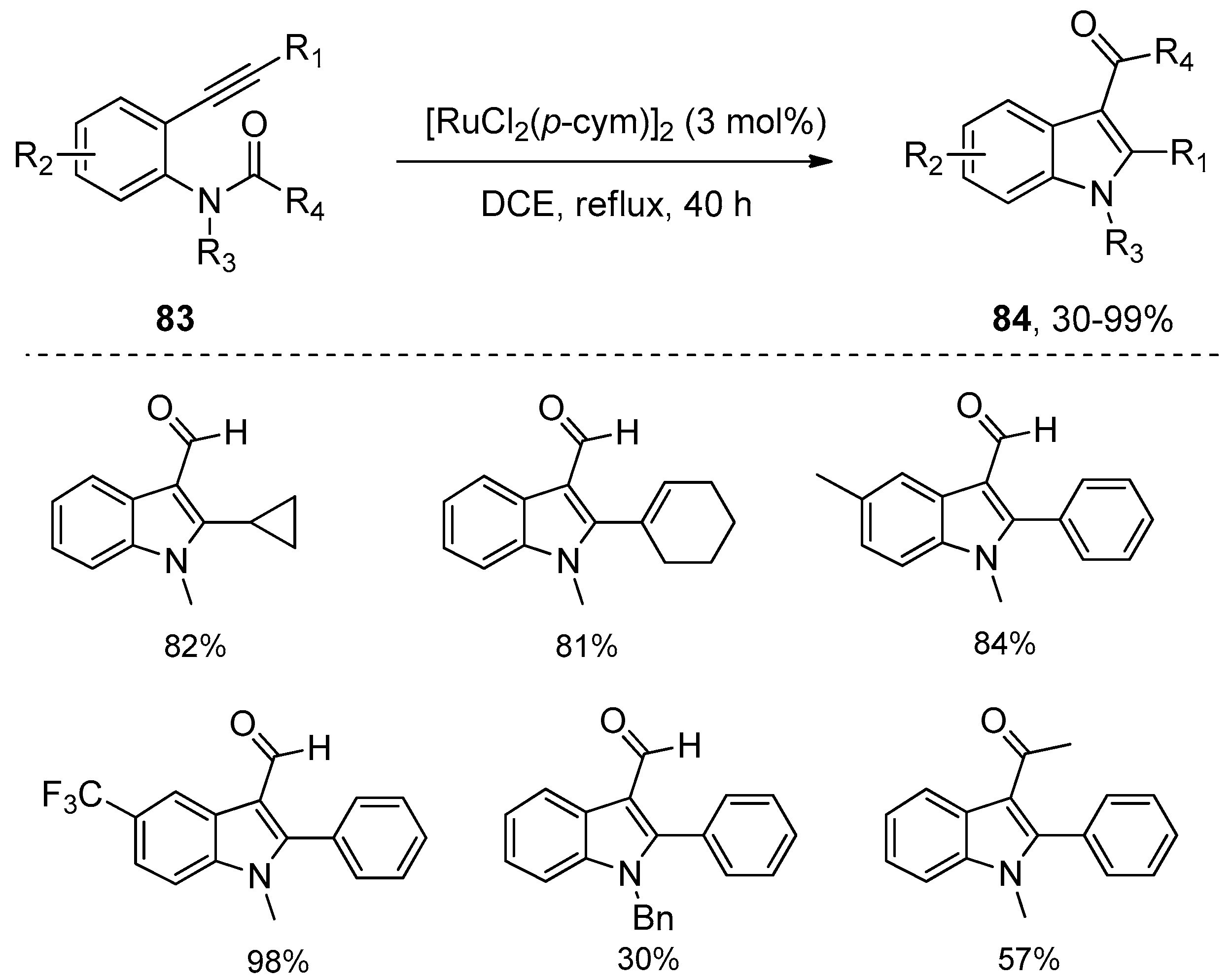
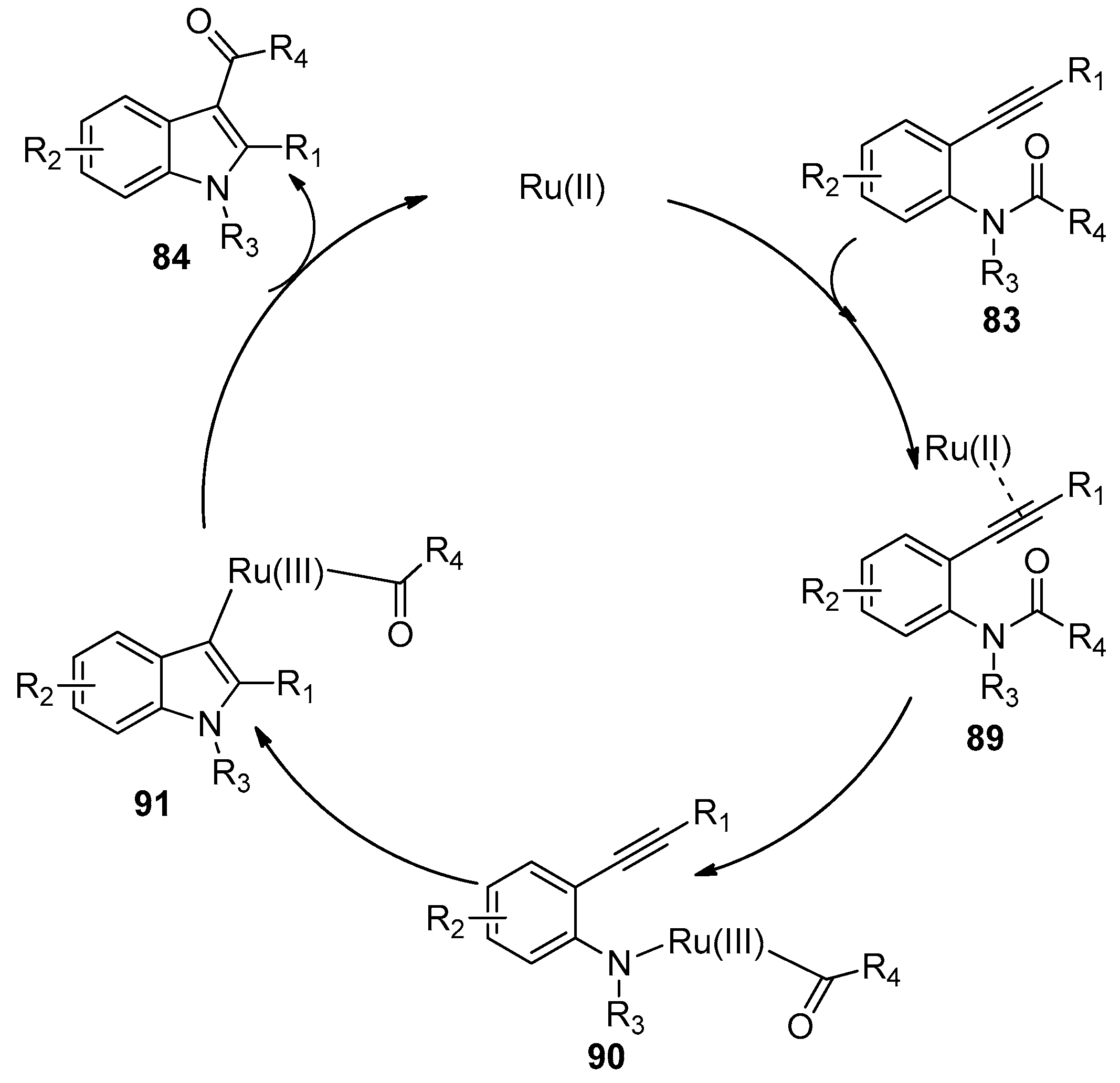
- Wu, C.Y.; Hu, M.; Liu, Y.; Song, R.J; Lei, Y.; Tang, B.X.; Li, R.J.; Li, J.H. Ruthenium-catalyzed annulation of alkynes with amides via formyl translocation. Chem. Commun. 2012, 48, 3197–3199. [Google Scholar] [CrossRef] [PubMed]
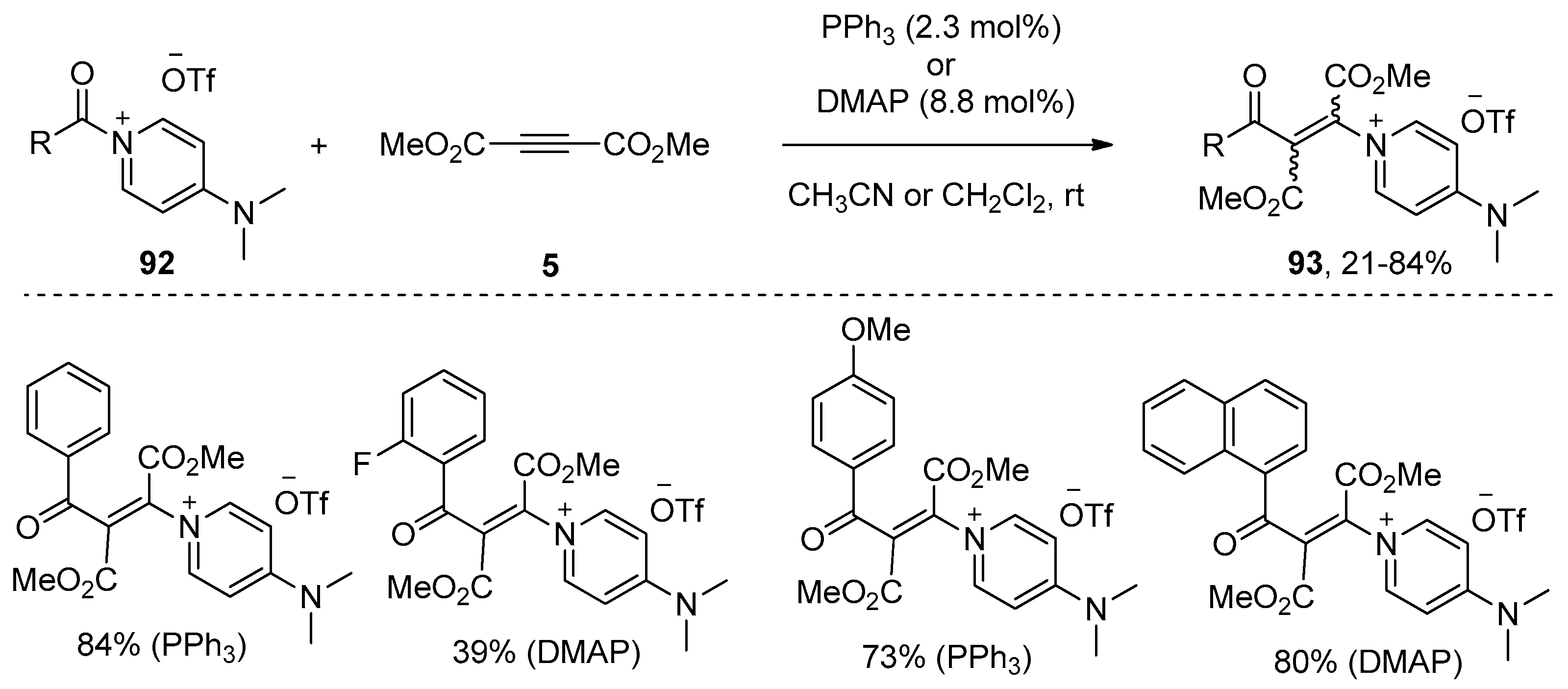
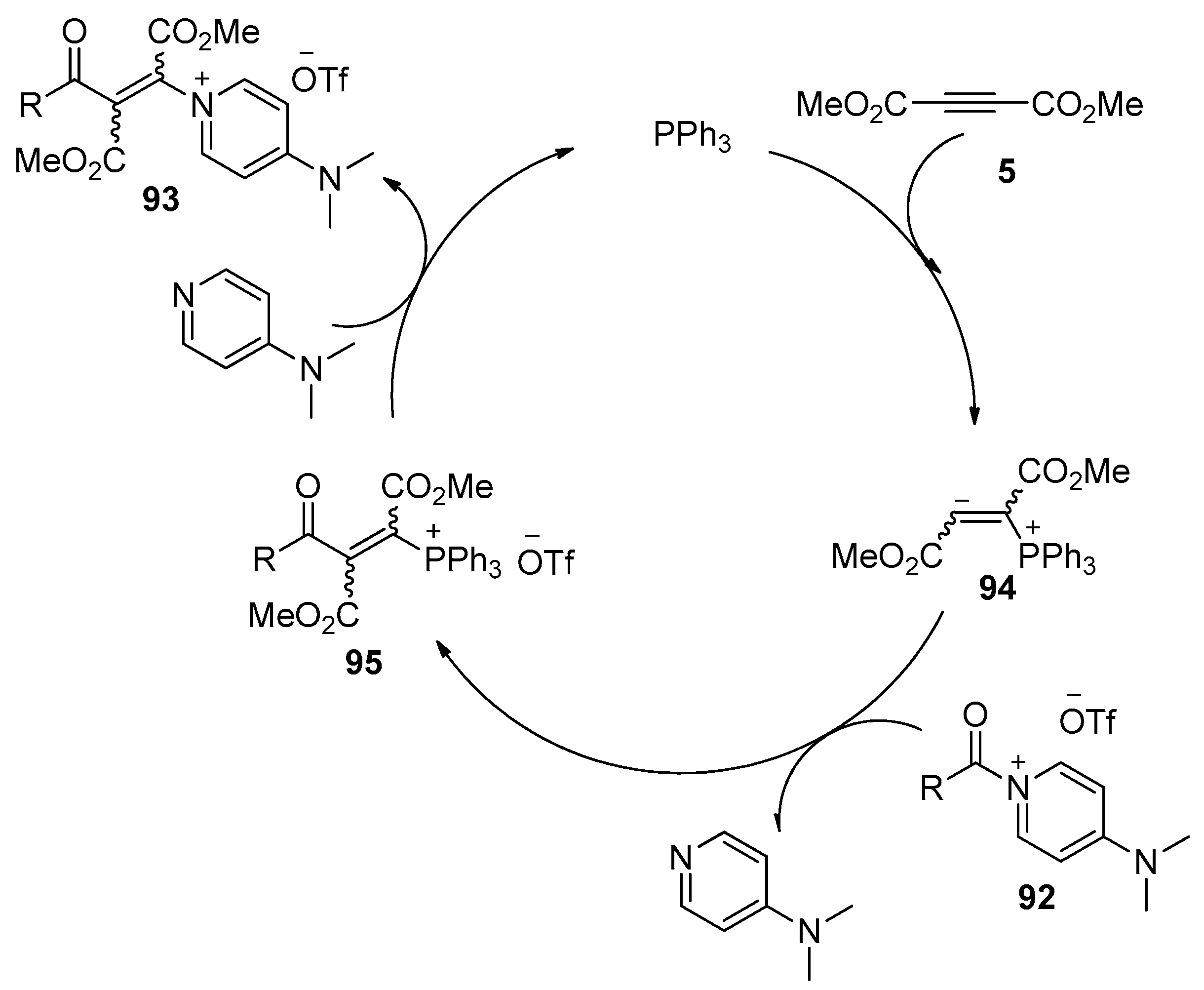
- Weiss, R.; Bess, M.; Huber, S.M.; Heinemann, F.W. Nucleophilic β-Oniovinylation: Concept, mechanism, scope, and applications. J. Am. Chem. Soc. 2008, 130, 4610–4617. [Google Scholar] [CrossRef] [PubMed]
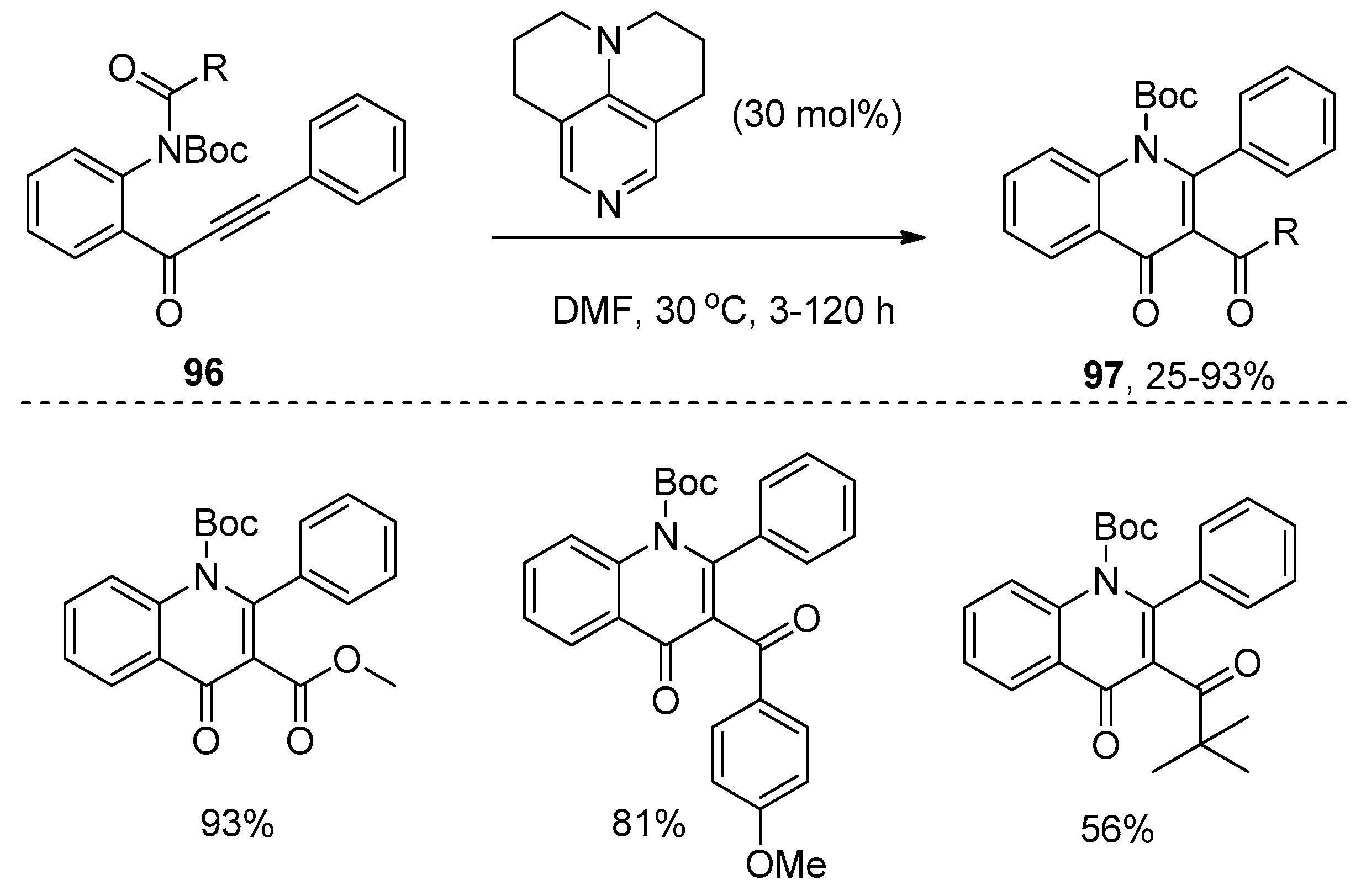
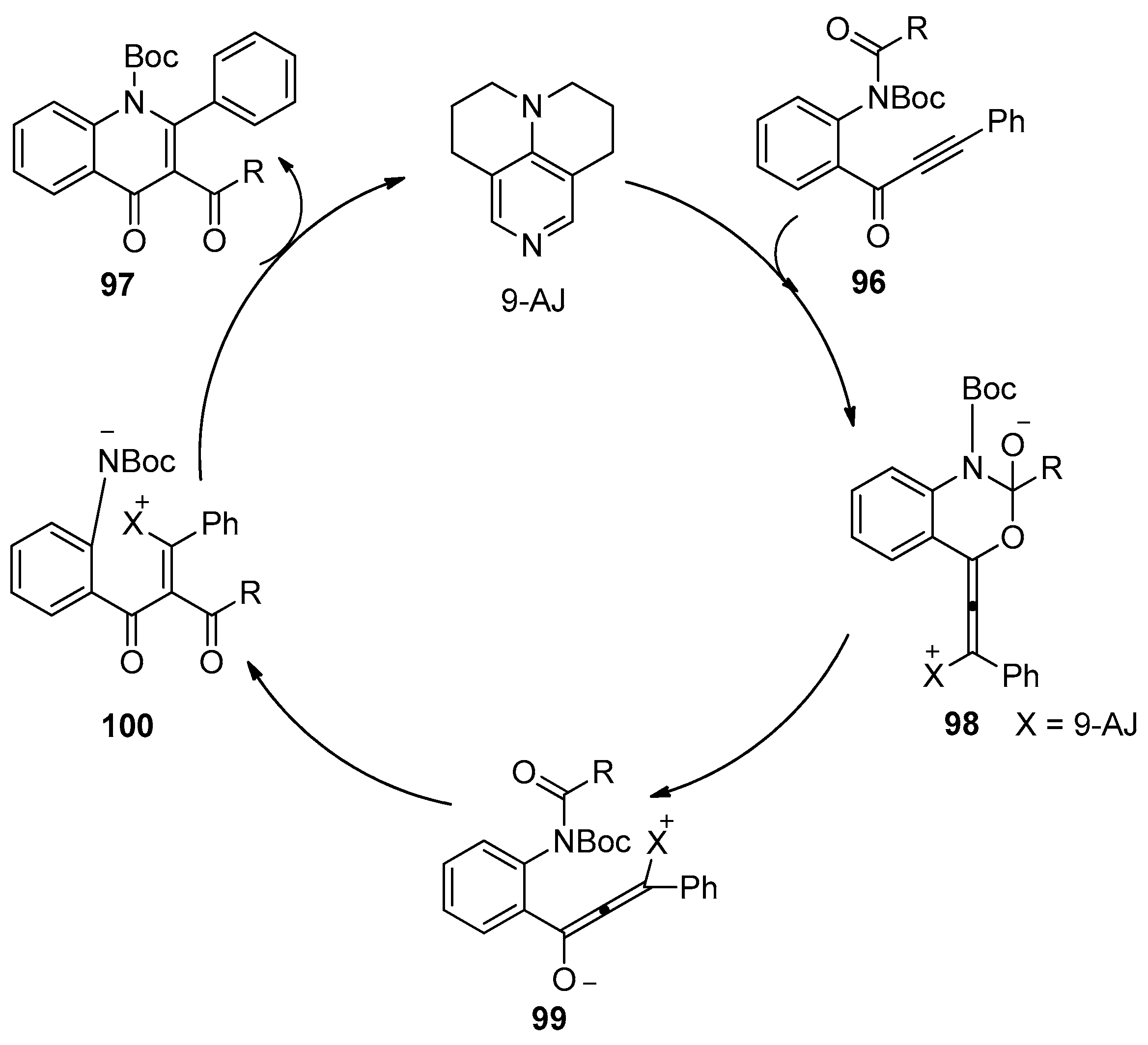
- Saito, K.; Yoshida, M.; Uekusa, H.; Doi, T. Facile synthesis of pyrrolyl 4-quinolinone alkaloid quinolactacide by 9-AJ-catalyzed tandem acyl transfer-cyclization of o-alkynoylaniline derivatives. ACS Omega. 2017, 2, 4370–4381. [Google Scholar] [CrossRef]
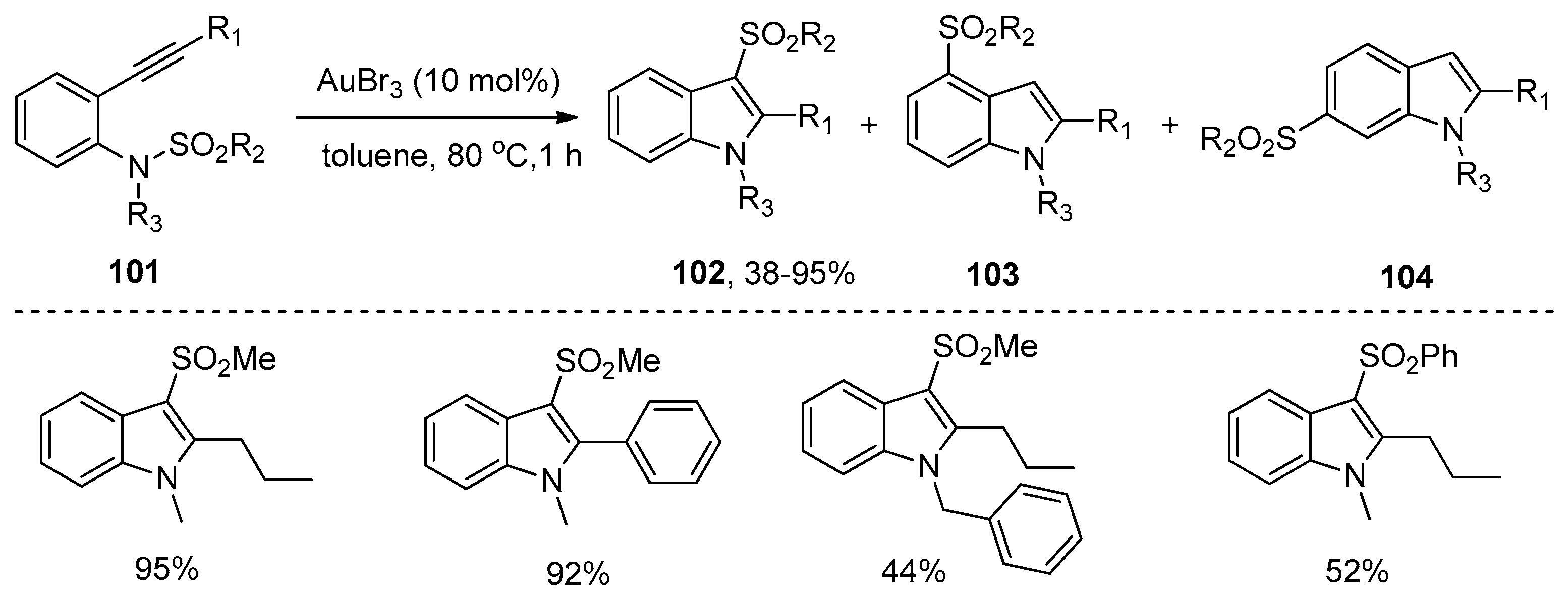
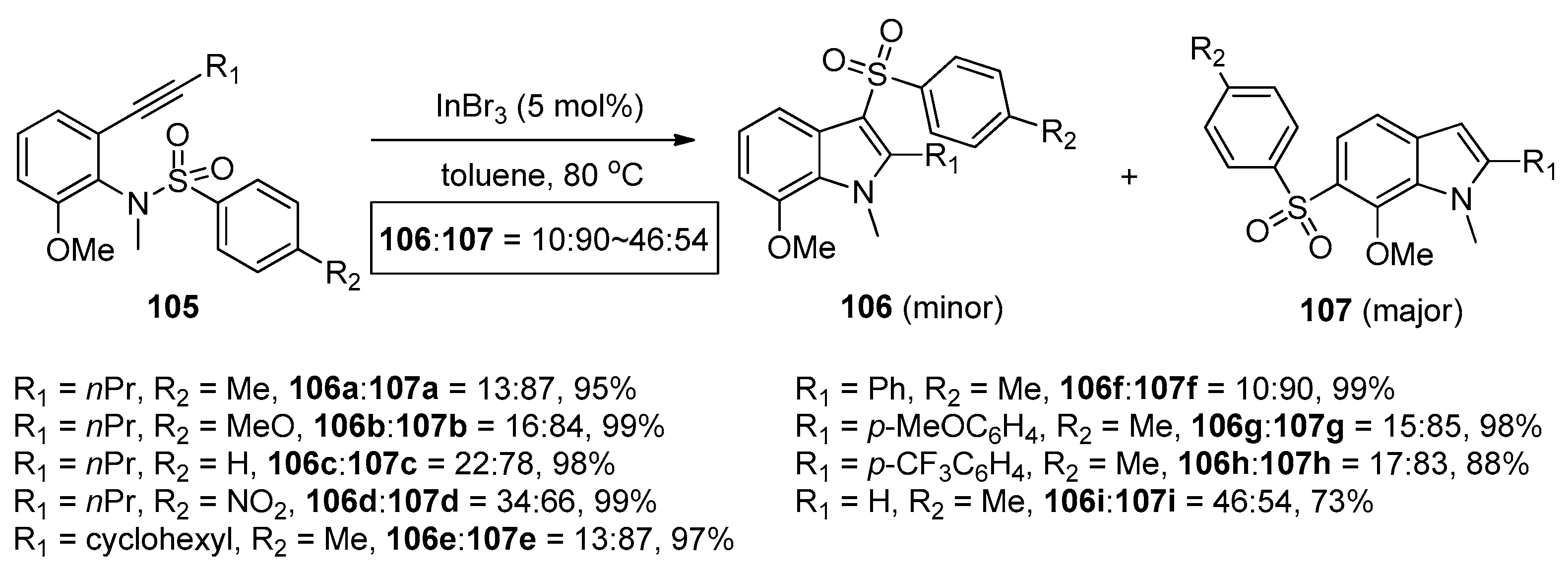
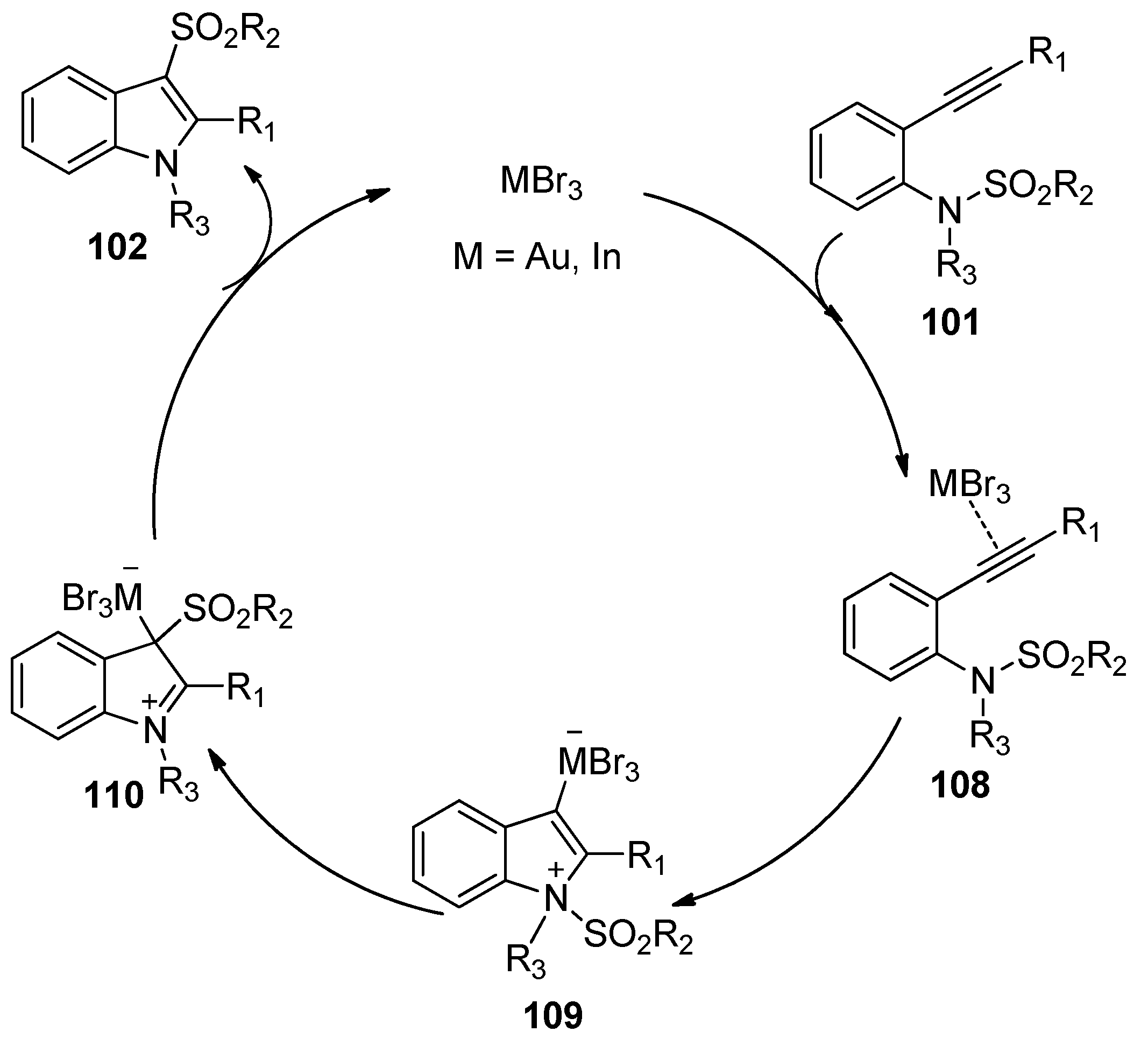
- Nakamura, I.; Yamagishi, U.; Song, D.; Konta, S.; Yamamoto, Y. Gold- and indium-catalyzed synthesis of 3- and 6-sulfonylindoles from ortho-alkynyl-N-sulfonylanilines. Angew. Chem. Int. Ed. 2007, 46, 2284–2287. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Yamagishi, U.; Song, D.; Konta, S.; Yamamoto, Y. Synthesis of 3- and 6-sulfonylindoles from ortho-alkynyl-N-sulfonylanilines by the use of Lewis acidic transition-metal catalysts. Chem. Asian J. 2008, 3, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Ciccarone, T.M.; MacTough, S.C.; Rooney, C.S.; Balani, S.K.; Condra, J.H.; Emini, E.A.; Goldman, M.E.; Greenlee, W.J.; Kauffman, L.R.; et al. 5-Chloro-3-(phenylsulfonyl)indole-2-carboxamide: A novel, non-nucleoside inhibitor of HIV-1 reverse transcriptase. J. Med. Chem. 1993, 36, 1291–1294. [Google Scholar] [CrossRef]
- Silvestri, R.; Martino, G.D.; Regina, G.L.; Artico, M.; Massa, S.; Vargiu, L.; Mura, M.; Loi, A.G.; Marceddu, T.; Colla, P.L. Novel Indolyl Aryl Sulfones Active against HIV-1 Carrying NNRTI Resistance Mutations: Synthesis and SAR Studies. J. Med. Chem. 2003, 46, 2482–2493. [Google Scholar] [CrossRef]
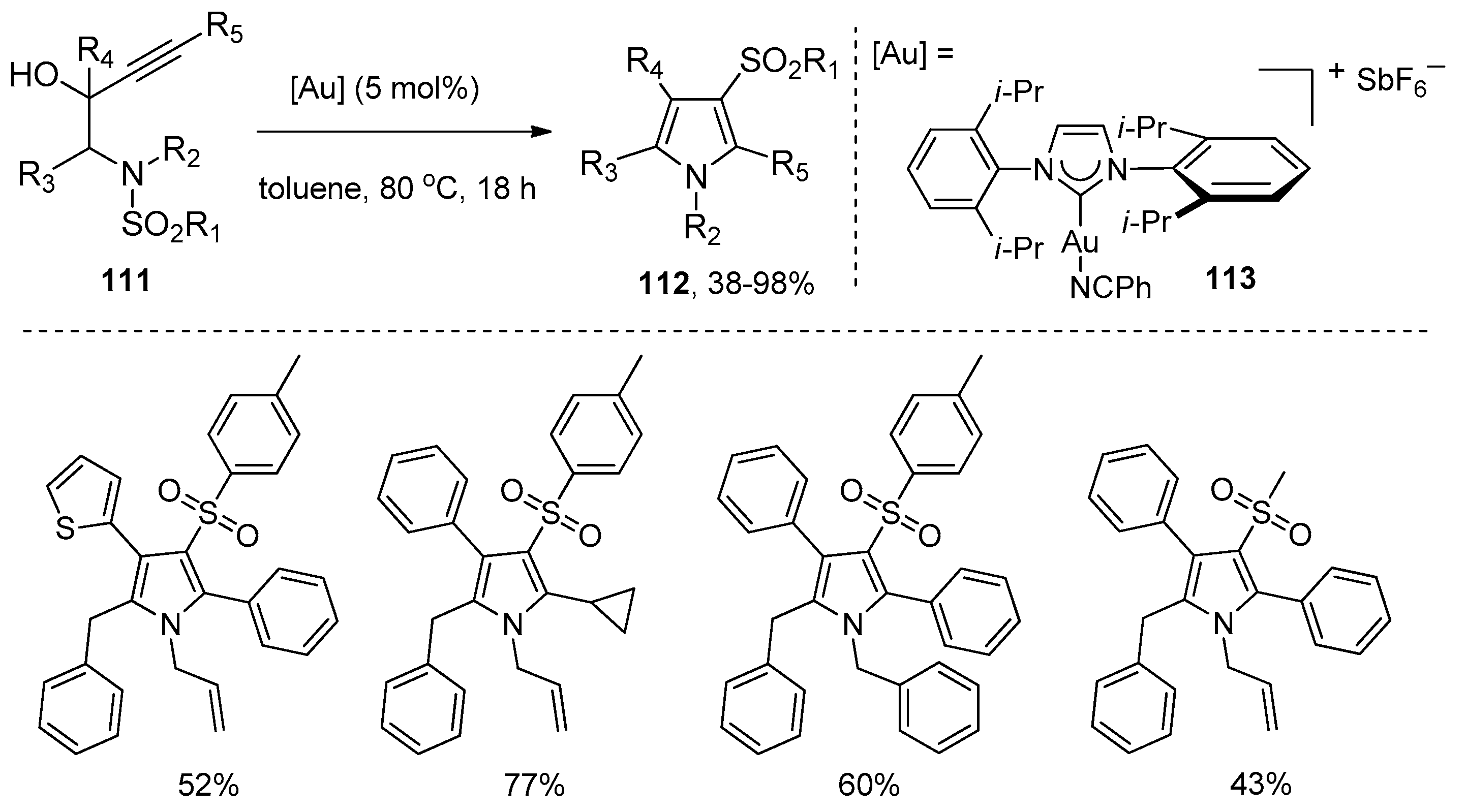
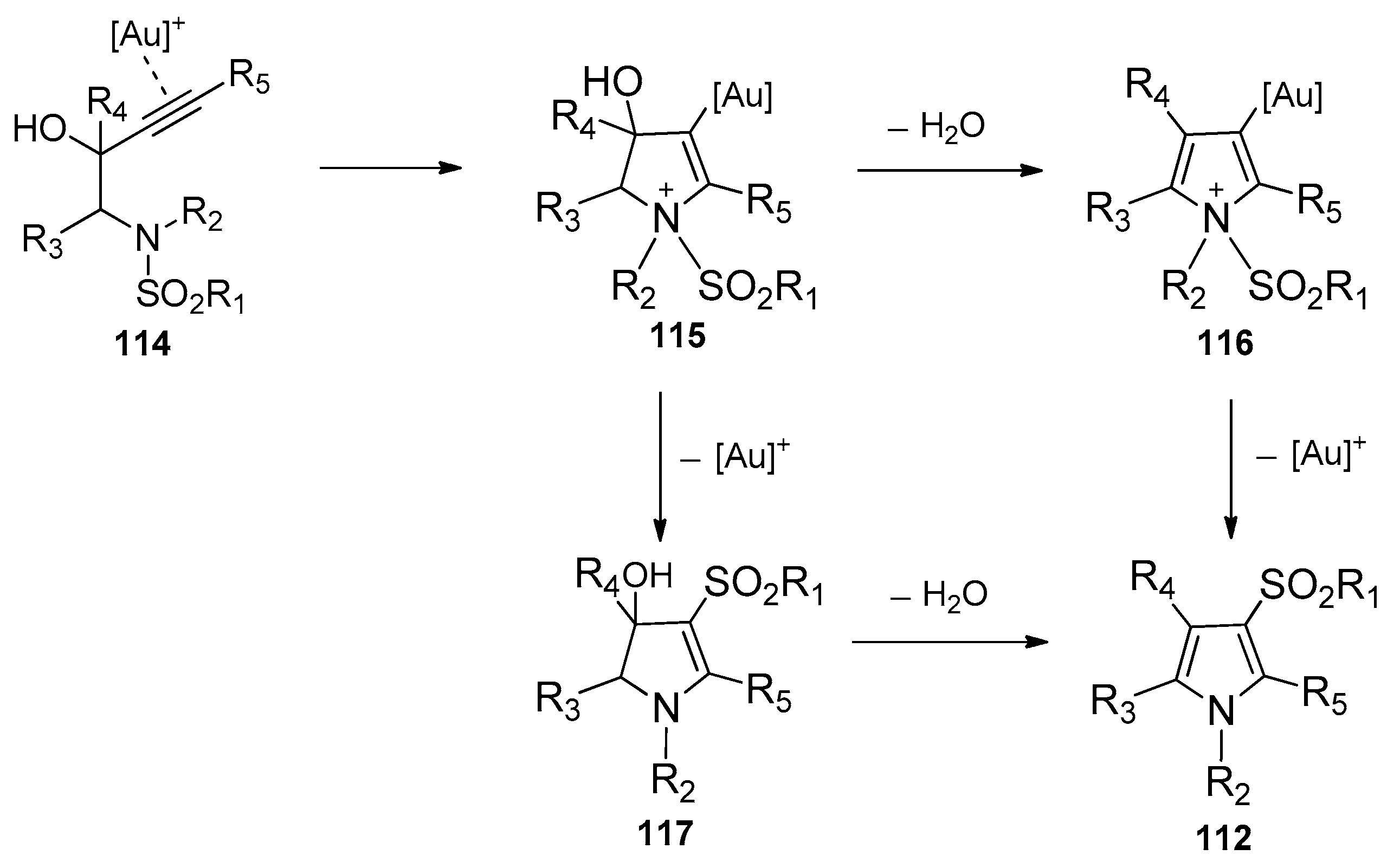
- Teo, W.T.; Rao, W.; Koh, M.J.; Chan, P.W.H. Gold-catalyzed domino aminocyclization/1,3-sulfonyl migration of N-substituted N-sulfonyl-aminobut-3-yn-2-ols to 1-substituted 3-sulfonyl-1H-pyrroles. J. Org. Chem. 2013, 78, 7508–7517. [Google Scholar] [CrossRef]
- Wu, C.; Zhao, F.; Du, Y.; Zhao, L.; Chen, L.; Wang, J.; Liu, H. Highly selective intramolecular addition of C–N and S–N bonds to alkynes catalyzed by palladium: A practical access to two distinct functional indoles. RSC Adv. 2016, 6, 70682–70690. [Google Scholar] [CrossRef]





































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Li, P.; Liu, X.; Jia, X.; Wang, J.; Liu, H. Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes. Molecules 2019, 24, 164. https://doi.org/10.3390/molecules24010164
Zhao F, Li P, Liu X, Jia X, Wang J, Liu H. Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes. Molecules. 2019; 24(1):164. https://doi.org/10.3390/molecules24010164
Chicago/Turabian StyleZhao, Fei, Pinyi Li, Xiaoyan Liu, Xiuwen Jia, Jiang Wang, and Hong Liu. 2019. "Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes" Molecules 24, no. 1: 164. https://doi.org/10.3390/molecules24010164
APA StyleZhao, F., Li, P., Liu, X., Jia, X., Wang, J., & Liu, H. (2019). Recent Advances in the Addition of Amide/Sulfonamide Bonds to Alkynes. Molecules, 24(1), 164. https://doi.org/10.3390/molecules24010164