Convenient Synthesis of Functionalized Cyclopropa[c]coumarin-1a-carboxylates

 , ,
, , 
Abstract
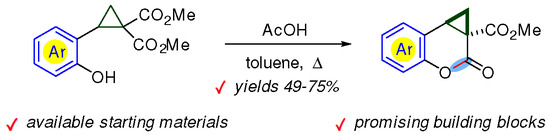
:
1. Introduction
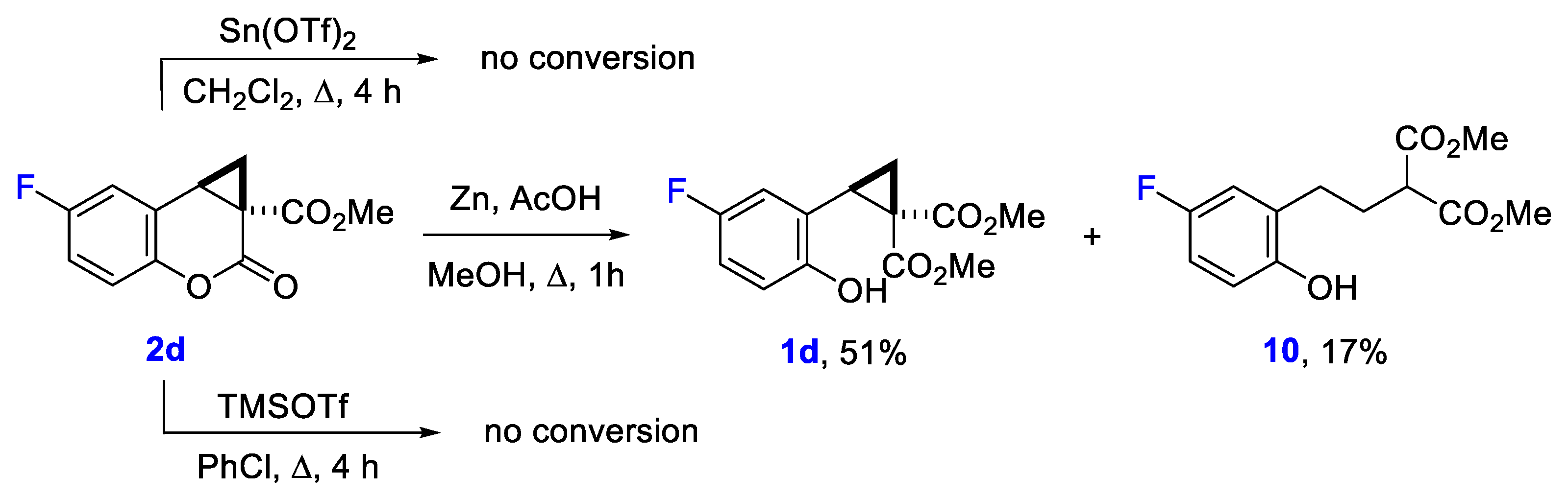
2. Results and Discussion
3. Experimental
3.1. General Information
3.2. General Procedure for the Synthesis of Cyclopropa[c]coumarins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef]
- Pereira, T.M.; Franco, D.P.; Vitorio, F.; Kummerle, A.E. Coumarin Compounds in Medicinal Chemistry: Some Important Examples from the Last Years. Curr. Top. Med. Chem. 2018, 18, 124–148. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, D.; Godugu, C.; Dubey, P.K. A Review of Pharmacological Properties of Coumarins. Mini-Rev. Med. Chem. 2018, 18, 113–141. [Google Scholar] [CrossRef] [PubMed]
- Detsi, A.; Kontogiorgis, C.; Hajipavlou-Litina, D. Coumarin derivatives: An updated patent review (2015–2016). Exp. Opin. Ther. Patents 2017, 27, 1201–1226. [Google Scholar] [CrossRef] [PubMed]
- Tejada, S.; Martorell, M.; Capo, X.; Tur, J.A.; Pons, A.; Sureda, A. Coumarin and Derivates as Lipid Lowering Agents. Curr. Top. Med. Chem. 2017, 17, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-Q.; Xu, Z.; Zhang, S.; Wu, X.; Ding, J.-W.; Lv, Z.-S.; Feng, L.-S. Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 136, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Dandriyal, J.; Singla, R.; Kumar, M.; Jaitak, V. Recent developments of C-4 substituted coumarin derivatives as anticancer agents. Eur. J. Med. Chem. 2016, 119, 141–168. [Google Scholar] [CrossRef]
- Penta, S. (Ed.) Advances in Structure and Activity Relationship of Coumarin Derivatives; Academic Press-Elsevier: Amsterdam, The Netherlands, 2015; ISBN 978-0-12-803797-3. [Google Scholar]
- Medina, F.G.; Marrero, J.G.; Macias-Alonso, M.; Gonzalez, M.C.; Cordova-Guerrero, I.; Teissier Garcia, A.G.; Osegueda-Robles, S. Coumarin heterocyclic derivatives: Chemical synthesis and biological activity. Nat. Prod. Rep. 2015, 32, 1472–1507. [Google Scholar] [CrossRef]
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur. J. Med. Chem. 2015, 102, 611–630. [Google Scholar] [CrossRef]
- Keri, R.S.; Sasidhar, B.S.; Nagaraja, B.M.; Santos, M.A. Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents. Eur. J. Med. Chem. 2015, 100, 257–269. [Google Scholar] [CrossRef]
- Hoerr, R.; Noeldner, M. Ensaculin (KA-672.HCl): A Multitransmitter Approach to Dementia Treatment. CNS Drug Rev. 2002, 8, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Steglich, W.; Fugmann, B.; Lang-Fugmann, S. RÖMPP Encyclopedia Natural Products, 1st ed.; Georg Thieme Verlag: Stuttgart, Germany, 2000; pp. 187–188. ISBN 3-13-117711-X. [Google Scholar]
- Enejoh, O.S.; Suleiman, M.M. Anthelmintics and Their Application in Veterinary Medicine. Res. Med. Eng. Sci. 2017, 2. [Google Scholar] [CrossRef]
- Ravina, E. The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs; Wiley-VCH: Weinheim, Germany, 2011; p. 148. ISBN 978-3-527-32669-3. [Google Scholar]
- Lee, M.M.; Peterson, B.R. Quantification of Small Molecule–Protein Interactions Using FRET between Tryptophan and the Pacific Blue Fluorophore. ACS Omega 2016, 1, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Widman, O. Über eine neue Gruppe von Cyclopropan-Derivaten. I: Die Einwirkung von Phenyl-acylhalogeniden auf 3-Acidyl-cumarine bei Gegenwart von Natrium-alkoholat. Ber. Dtsch. Chem. Ges. 1918, 51, 533–541. [Google Scholar] [CrossRef]
- Widman, O. Über eine neue Gruppe von Cyclopropan-Derivaten. II: Die Einwirkung einiger Analoga der Phenacylhalogenide auf 3-Acidyl-cumarine. Ber. Dtsch. Chem. Ges. 1918, 51, 907–911. [Google Scholar] [CrossRef]
- Bojilova, A.; Trendafilova, A.; Ivanov, C.; Rodios, N.A. Cyclopropanation reaction of 3-acyl-2H-1-benzopyran-2-ones with phenacylbromide in phase transfer systems. Tetrahedron 1993, 49, 2275–2286. [Google Scholar] [CrossRef]
- Bojilova, A.; Videnova, I.; Ivanov, C.; Rodios, N.A.; Terzis, A.; Raptopoulou, C.P. Regio- and stereo-Selective 1,3-dipolar cycloaddition reactions of ethyl diazoacetate to 3-substituted 2H-1-benzopyran-2-ones. Tetrahedron 1994, 50, 13023–13036. [Google Scholar] [CrossRef]
- Wawzonek, S.; Morreal, C.E. The Action of Alkali on 3,4-Phenacylidene-3-acetylcoumarin. J. Am. Chem. Soc. 1960, 82, 439–441. [Google Scholar] [CrossRef]
- Abdallah, H.; Gree, R.; Carrie, R. Reactions du dimethylacetal du diazoacetaldehyde avec des coumarines at une chromone electrophiles. Bull. Soc. Chim. Fr. 1984, 2, 338–344. [Google Scholar]
- Shchepin, V.V.; Silaichev, P.S.; Kodess, M.I. Reaction of Zinc Enolates Prepared from 2,2-Dibromoindan-1-one or 2,2-Dibromo-1-tetralone and Zinc with 2-Oxochromen-3-carboxylic Acid Derivatives. Russ. J. Org. Chem. 2007, 43, 1441–1445. [Google Scholar] [CrossRef]
- Shchepin, V.V.; Silaichev, P.S.; Stepanyan, Y.G.; Kalyuzhnyi, M.M.; Russkikh, N.Y.; Kodess, M.I. Cyclopropanation of N-Substituted 3-Aryl-2-cyanoprop-2-enamides and Derivatives of 5,5-Dimethyl-2-oxo-2,5-dihydrofuran-3-carboxylic Acid and 2-Oxochromene-3-carboxylic Acid with Bromine-Containing Zinc Enolates. Russ. J. Org. Chem. 2006, 42, 973–980. [Google Scholar] [CrossRef]
- Shchepin, V.V.; Silaichev, P.S.; Vakhrin, M.I.; Russkikh, N.Y. Cyclopropanation of N-Substituted 2-Oxochromene-3-carboxamides and 3-Oxobenzo[f]chromene-2-carboxamides with Bromine-containing Zinc Enolate Prepared from α,α-Dibromopinacolin and Zinc. Russ. J. Org. Chem. 2005, 41, 1219–1221. [Google Scholar] [CrossRef]
- Bojilova, A. Interaction of Trichloroacetic Acid with Some 3-Substituted 2H-1-Benzopyran-2-ones. Synth. Commun. 1990, 20, 1967–1976. [Google Scholar] [CrossRef]
- Guo, J.; Liu, Y.; Li, X.; Liu, X.; Lin, L.; Feng, X. Nickel(II)-catalyzed enantioselective cyclopropanation of 3-alkenyl-oxindoles with phenyliodonium ylide via free carbene. Chem. Sci. 2016, 7, 2717–2721. [Google Scholar] [CrossRef]
- Huang, X.; Klimczyk, S.; Veiros, L.F.; Maulide, N. Stereoselective intramolecular cyclopropanation through catalytic olefin activation. Chem. Sci. 2013, 4, 1105–1110. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Gololobov, Y.G.; Nesmeyanov, A.N.; Lysenko, V.P.; Bordeskul, I.E. Twenty-five years of dimethylsulfoxonium methylide (Corey’s reagent). Tetrahedron 1987, 43, 2609–2651. [Google Scholar] [CrossRef]
- Yamashita, M.; Okuyama, K.; Kawajiri, T.; Takada, A.; Inagaki, Y.; Nakano, H.; Tomiyama, M.; Ohnaka, A.; Terayama, I.; Kawasaki, I.; et al. A novel tandem reaction of 3-substituted coumarins with two equivalents of dimethylsulfoxonium ylide to 2-substituted cyclopenta[b]benzofuran-3-ol derivatives. Tetrahedron 2002, 58, 1497–1505. [Google Scholar] [CrossRef]
- Arimitsu, K.; Nomura, S.; Iwasaki, H.; Ozeki, M.; Yamashita, M. First total synthesis of (±)-adunctin B. Tetrahedron Lett. 2011, 52, 7046–7048. [Google Scholar] [CrossRef]
- Yamashita, M.; Okuyama, K.; Kawasaki, I.; Ohta, S. One-Step Synthesis of 2-Substituted Cyclopenta[b]benzofuran-3-ol Derivatives from 3-Substituted Coumarins. Tetrahedron Lett. 1995, 36, 5603–5605. [Google Scholar] [CrossRef]
- Lee, J.; Ko, K.M.; Kim, S.-G. Ni(ClO4)2-Catalyzed Friedel-Crafts Reaction of Coumarin-Fused Donor-Acceptor Cyclopropanes with Indoles: Stereoselective Synthesis of trans-3,4-Disubstituted-3,4-dihydrocoumarins. Eur. J. Org. Chem. 2018, 4166–4170. [Google Scholar] [CrossRef]
- Choi, S.; Kim, S.-G. (±)-Methyl 1,1a,2,7b-Tetrahydro-2-oxocyclopropa[c]chromene-1a-carboxylate. Molbank 2017, 2017, M966. [Google Scholar] [CrossRef]
- Ivanova, O.A.; Andronov, V.A.; Vasin, V.S.; Shumsky, A.N.; Rybakov, V.B.; Voskressensky, L.G.; Trushkov, I.V. Expanding the Reactivity of Donor-Acceptor Cyclopropanes: Synthesis of Benzannulated Five-Membered Heterocycles via Intramolecular Attack of a Pendant Nucleophilic Group. Org. Lett. 2018, 20, 7947–7952. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Bezzubov, S.I.; Melnikov, M.Y.; Bludynina, E.M. Donor-acceptor cyclopropanes as ortho-quinone methide equivalents in formal (4 + 2)-cycloaddition to alkenes. Org. Biomol. Chem. 2018, 16, 3897–3909. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Trushkov, I.V.; Melnikov, M.Y. Ring Opening of Donor-Acceptor Cyclopropanes with the Azide Ion: A Tool for Construction of N-Heterocycles. Chem. Eur. J. 2015, 21, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Borpatra, P.J.; Deka, B.; Rajbongshi, B.K.; Deb, M.L.; Baruah, P.K. One-pot sequential multi-component reaction: Synthesis of 3-substituted indoles. Synth. Commun. 2018, 48, 2074–2082. [Google Scholar] [CrossRef]
- Misztal, S.; Mokrosz, J.L.; Bielecka, Z. Oxidation of 2,3-Bis(hydroxymethyl)indole. J. Prakt. Chem. 1989, 331, 751–756. [Google Scholar] [CrossRef]
- Fusco, R.; Sannicolo, F. Rearrangement of Arylhydrazones of α,β-Unsaturated Carbonyl Compounds in Polyphosphoric Acid. 6. J. Org. Chem. 1984, 49, 4374–4378. [Google Scholar] [CrossRef]
- Ivanov, K.L.; Villemson, E.V.; Latyshev, G.V.; Bezzubov, S.I.; Majouga, A.G.; Melnikov, M.Y.; Bludynina, E.M. Regioselective Hydrogenolysis of Donor-Acceptor Cyclopropanes with Zn-AcOH Reductive System. J. Org. Chem. 2017, 82, 9537–9549. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Ivanova, O.A.; Rakhmankulov, E.R.; Budynina, E.M.; Trushkov, I.V.; Melnikov, M.Y. Lewis Acid-Catalyzed Isomerization of 2-Arylcyclopropane-1,1-dicarboxylates: A New Efficient Route to 2-Styrylmalonates. Adv. Synth. Catal. 2010, 352, 3179–3184. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Initiator (mol %) | R | Solvent | Temp (°C) | t (h) | Yield, % 1 |
|---|---|---|---|---|---|---|
| 1 | K2CO3 (130) 2 | H | DMSO | 20 | 3 | 30 |
| 2 | K2CO3 (130) 2 | H | DMSO | 20 | 25 | 30 |
| 3 | DIPEA (120) 3 | H | PhCl | 100 | 5 | 45 |
| 4 | TFA (110) 3 | H | PhCl | reflux | 7 | - 4 |
| 5 | TsOH (5) 3 | H | CHCl3 | reflux | 5 | - 4 |
| 6 | AcOH (200) 3 | H | PhMe | 110 5 | 8 | 30 |
| 7 | AcOH (200) 3 | H | PhMe | reflux | 6 | 46 |
| 8 | AcOH (200) 3 | H | PhMe | reflux | 9 | 61 |
| 9 | AcOH (100) 3 | H | PhCl | reflux | 6.5 | 55 6 |
| 10 | AcOH (200) 3 | H | PhMe | reflux | 16 | 72 |
| 11 | AcOH (200) 3 | MOM | PhMe | reflux | 16 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, O.A.; Andronov, V.A.; Levina, I.I.; Chagarovskiy, A.O.; Voskressensky, L.G.; Trushkov, I.V. Convenient Synthesis of Functionalized Cyclopropa[c]coumarin-1a-carboxylates. Molecules 2019, 24, 57. https://doi.org/10.3390/molecules24010057
Ivanova OA, Andronov VA, Levina II, Chagarovskiy AO, Voskressensky LG, Trushkov IV. Convenient Synthesis of Functionalized Cyclopropa[c]coumarin-1a-carboxylates. Molecules. 2019; 24(1):57. https://doi.org/10.3390/molecules24010057
Chicago/Turabian StyleIvanova, Olga A., Vladimir A. Andronov, Irina I. Levina, Alexey O. Chagarovskiy, Leonid G. Voskressensky, and Igor V. Trushkov. 2019. "Convenient Synthesis of Functionalized Cyclopropa[c]coumarin-1a-carboxylates" Molecules 24, no. 1: 57. https://doi.org/10.3390/molecules24010057
APA StyleIvanova, O. A., Andronov, V. A., Levina, I. I., Chagarovskiy, A. O., Voskressensky, L. G., & Trushkov, I. V. (2019). Convenient Synthesis of Functionalized Cyclopropa[c]coumarin-1a-carboxylates. Molecules, 24(1), 57. https://doi.org/10.3390/molecules24010057