
Discovery of 2-(1-(3-(4-Chloroxyphenyl)-3-oxo- propyl)pyrrolidine-3-yl)-1H-benzo[d]imidazole-4-carboxamide: A Potent Poly(ADP-ribose) Polymerase (PARP) Inhibitor for Treatment of Cancer

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. PARP Inhibition Assay
2.3. Cell Proliferation Assay
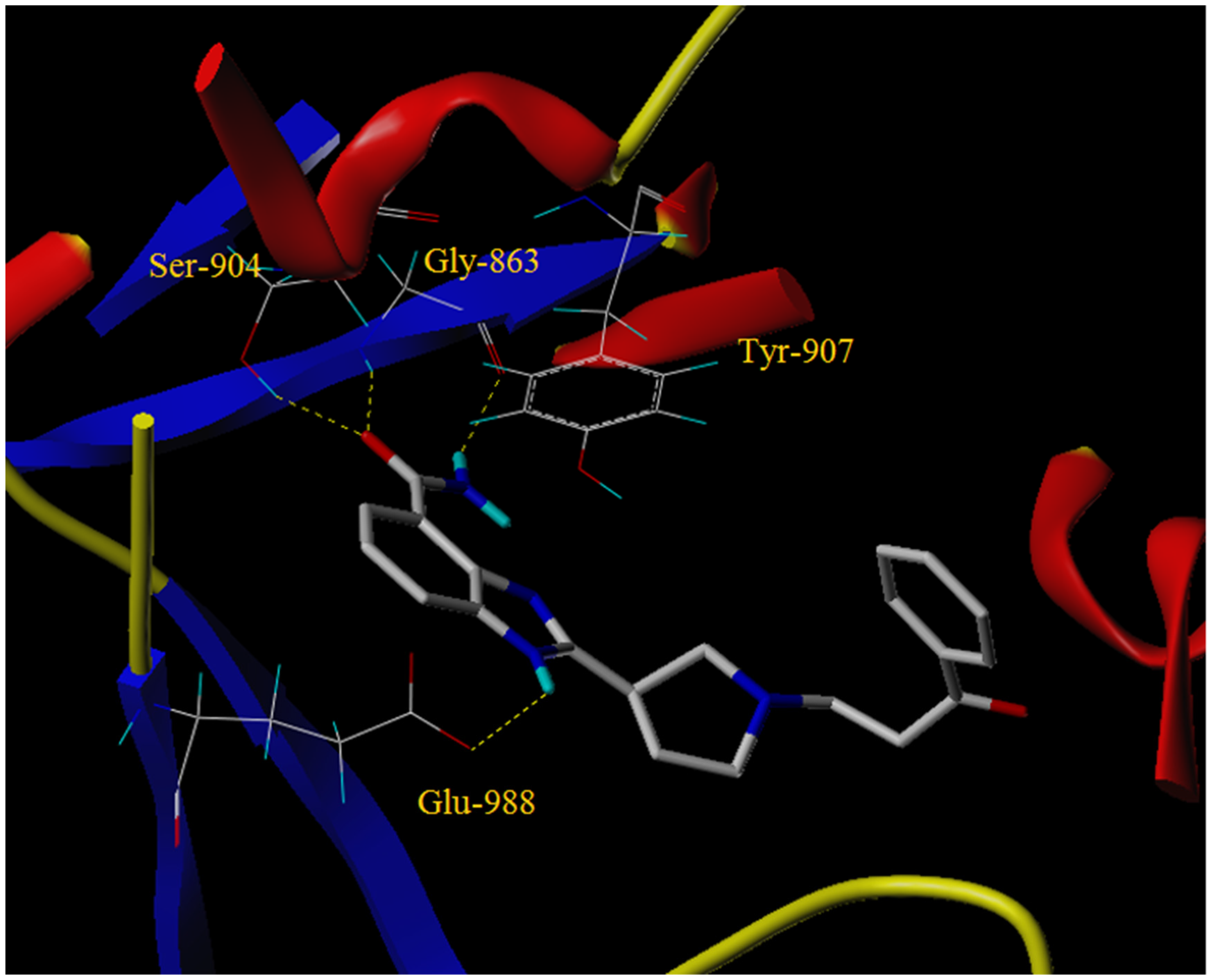
2.4. Molecular Docking
3. Materials and Methods
3.1. Genereral Informations
3.2. Chemistry
3.2.1. Procedure A: Synthesis of 2-(Pyrrolidin-3-yl)-1H-benzo[d]imidazole-4-carboxamide (N3)
3.2.2. Procedure B: Synthesis of 5ca, 5cb, 5cc, 5cd, 5ce, 5ch, 5ci, 5cj, 5ck and 5cp
3.2.3. Procedure C: Synthesis of 5cf and 5cg
3.2.4. Procedure D: Synthesis of 5cl and 5cm
3.2.5. Procedure E: Synthesis of 5cn and 5co
3.3. PARP Inhibition Assay
3.4. Cell Proliferation Assay
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Virág, L.; Szabó, C. The therapeutic potential of poly(ADP-ribose)polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Szabó, C. Poly(ADP-Ribose)polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Ame´, J.-C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. BioEssays 2004, 26, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Burkle, A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. BioEssays 2001, 23, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Almahli, H.; Hadchity, E.; Jaballah, M.Y.; Daher, R.; Ghabbour, H.A.; Kabil, M.M.; Al-shakliah, N.S.; Eldehna, W.M. Development of novel synthesized phthalazinone-based PARP-1 inhibitors with apoptosis inducing mechanism in lung cancer. Bioorg. Chem. 2018, 77, 443–456. [Google Scholar] [CrossRef]
- Malyuchenko, N.V.; Kotova, E.Y.; Kulaeva, O.I.; Kirpichnikov, M.P.; Studitskiy, V.M. PARP1 inhibitors: Antitumor drug design. Acta Nat. 2015, 7, 27–37. [Google Scholar]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phtha-lazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Loh, V.M.; Cockcroft, X.L.; Dillon, K.J.; Dixon, L.; Drzewiecki, J.; Eversley, P.J.; Gomez, S.; Hoare, J.; Kerrigan, F.; Matthews, I.T.W.; et al. Phthalazinones. Part 1: The design and synthesis of a novel series of potent inhibitors of poly(ADP ribose) polymerase. Bioorg. Med. Chem. Lett. 2005, 15, 2235–2238. [Google Scholar] [CrossRef]
- Scott, L.J. Niraparib: First global approval. Drugs 2017, 77, 1029–1034. [Google Scholar] [CrossRef]
- Kanjanapan, Y.; Lheureux, S.; Oza, A.M. Niraparib for the treatment of ovarian cancer. Expert Opin. Pharmaco. 2017, 18, 631–640. [Google Scholar] [CrossRef]
- Jones, P.; Altamura, S.; Boueres, J.; Ferrigno, F.; Fonsi, M.; Giomini, C.; Lamartina, S.; Monteagudo, E.; Ontoria, J.M.; Orsale, M.V.; Palumbi, M.C.; et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A novel oral poly (ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J. Med. Chem. 2009, 52, 7170–7185. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP-1 and PARP-2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Canan Koch, S.S.; Thoresen, L.H.; Tikhe, J.G.; Maegley, K.A.; Almassy, R.J.; Li, J.; Yu, X.-H.; Zook, S.E.; Kumpf, R.A.; et al. Novel tricyclic poly(ADP-ribose) polymerase-1 inhibitors with potent anticancer chemopotentiating activity: Design, synthesis, and X-ray cocrystal structure. J. Med. Chem. 2002, 45, 4961–4974. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Kaye, S.; Yap, T. PARP inhibitors: The race is on. Brit. J. Cancer 2016, 114, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.D.; Zhu, G.-D.; Gandhi, V.B.; Gong, J.; Liu, X.; Shi, Y.; Klinghofer, V.; Johnson, E.F.; Donawho, C.K.; Frost, D.J.; et al. Discovery of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the Treatment of Cancer. J. Med. Chem. 2009, 52, 514–523. [Google Scholar] [CrossRef]
- Penning, T.D.; Zhu, G.-D.; Gandhi, V.B.; Gong, J.; Thomas, S.; Lubisch, W.; Grandel, R.; Wernet, W.; Park, C.H.; Fry, E.H.; et al. Discovery and SAR of 2-(1-propylpiperidin-4-yl)-1H-benzimidazole-4-carboxamide: A potent inhibitor of poly (ADP-ribose) polymerase (PARP) for the treatment of cancer. Bioorg. Med. Chem. 2008, 16, 6965–6975. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.; Ficco, R.P.; Dain, D.; Ginski, M.; Lautar, S.; Lee Wisdom, K.; Linag, S.; Lin, Q.; Lu, M.X.-C.; Morgan, L.; et al. Design and synthesis of poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors. Part 4: Biological evaluation of imidazobenzodiazepines as potent PARP-1 inhibitors for treatment of ischemic injuries. Bioorg. Med. Chem. 2003, 11, 3695–3707. [Google Scholar] [CrossRef]
- Costatino, G.; Macchiarulo, A.; Camaioni, E.; Pellicciari, R. Modeling of poly(ADP- ribose)polymerase (PARP) inhibitors. Docking of ligands and quantitative structure- activity relationship analysis. J. Med. Chem. 2001, 44, 3786–3794. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Promier, Y. Stereospecific PARP trapping by BMN-673 and comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Pommier, Y.; O’ Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Barkalow, J.H.; Breting, J.; Gaede, B.J.; Haight, A.R.; Henry, R.; Kotecki, B.; Mei, J.; Pearl, K.B.; Tedrow, J.S.; Viswanath, S.K. Process development for ABT-472, a benzimidazole PARP inhibitor. Org. Process Res. Dev. 2007, 11, 693–698. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5ca–5cp are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | PARP-1 Inhibition % (10 nM) | PARP-2 Inhibition % (10 nM) | PARP-1 IC50 (nM) | PARP-2 IC50 (nM) |
|---|---|---|---|---|---|
| 5ca |  | 17.6 | 34.5 | / | / |
| 5cb |  | 19.9 | 41.2 | / | / |
| 5cc |  | 42.8 | 62.3 | 12.2 | 5.8 |
| 5cd |  | −1.1 | 7.2 | / | / |
| 5ce |  | 22.0 | 74.5 | / | / |
| 5cf |  | 5.0 | 9.0 | / | / |
| 5cg |  | −2.1 | 6.5 | / | / |
| 5ch |  | 47.5 | 66.2 | 7.1 | 3.3 |
| 5ci |  | 59.2 | 62.5 | 5.9 | 4.5 |
| 5cj |  | 65.7 | 65.6 | 3.9 | 4.2 |
| 5ck |  | 6.0 | 37.1 | / | / |
| 5cl |  | 26.8 | 46.4 | / | / |
| 5cm |  | 24.8 | 32.8 | / | / |
| 5cn |  | 19.4 | 39.1 | / | / |
| 5co |  | 38.4 | 66.2 | 11.1 | 5.7 |
| 5cp |  | 68.0 | 76.7 | 3.6 | 3.2 |
| pc1 | Veliparib | 63.7 | 78.3 | 5.3 | 1.6 |

| Compound ID | R | MDA-MB-436 IC50 (μM) | CAPAN-1 IC50 (μM) |
|---|---|---|---|
| 5ca |  | 22.9 | >100 |
| 5cb |  | 90.4 | >100 |
| 5cc |  | 31.9 | 20.7 |
| 5cd |  | 55.0 | 82.5 |
| 5ce |  | >100 | >100 |
| 5cf |  | 61.0 | >100 |
| 5cg |  | 74.1 | >100 |
| 5ch |  | >100 | >100 |
| 5ci |  | 38.6 | 48.1 |
| 5cj |  | 17.4 | 11.4 |
| 5ck |  | >100 | >100 |
| 5cl |  | >100 | >100 |
| 5cm |  | >100 | >100 |
| 5cn |  | >100 | >100 |
| 5co |  | >100 | >100 |
| 5cp |  | 19.8 | 15.5 |
| pc1 | Veliparib | >100 | >100 |
| pc2 | Olaparib | 30.2 | >100 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, R.; Wu, W.; Wang, M.; Tang, L.; Chen, D.; Zhao, H.; Zhang, C.; Jiang, Y. Discovery of 2-(1-(3-(4-Chloroxyphenyl)-3-oxo- propyl)pyrrolidine-3-yl)-1H-benzo[d]imidazole-4-carboxamide: A Potent Poly(ADP-ribose) Polymerase (PARP) Inhibitor for Treatment of Cancer. Molecules 2019, 24, 1901. https://doi.org/10.3390/molecules24101901
Min R, Wu W, Wang M, Tang L, Chen D, Zhao H, Zhang C, Jiang Y. Discovery of 2-(1-(3-(4-Chloroxyphenyl)-3-oxo- propyl)pyrrolidine-3-yl)-1H-benzo[d]imidazole-4-carboxamide: A Potent Poly(ADP-ribose) Polymerase (PARP) Inhibitor for Treatment of Cancer. Molecules. 2019; 24(10):1901. https://doi.org/10.3390/molecules24101901
Chicago/Turabian StyleMin, Rui, Weibin Wu, Mingzhong Wang, Lin Tang, Dawei Chen, Huan Zhao, Cunlong Zhang, and Yuyang Jiang. 2019. "Discovery of 2-(1-(3-(4-Chloroxyphenyl)-3-oxo- propyl)pyrrolidine-3-yl)-1H-benzo[d]imidazole-4-carboxamide: A Potent Poly(ADP-ribose) Polymerase (PARP) Inhibitor for Treatment of Cancer" Molecules 24, no. 10: 1901. https://doi.org/10.3390/molecules24101901
APA StyleMin, R., Wu, W., Wang, M., Tang, L., Chen, D., Zhao, H., Zhang, C., & Jiang, Y. (2019). Discovery of 2-(1-(3-(4-Chloroxyphenyl)-3-oxo- propyl)pyrrolidine-3-yl)-1H-benzo[d]imidazole-4-carboxamide: A Potent Poly(ADP-ribose) Polymerase (PARP) Inhibitor for Treatment of Cancer. Molecules, 24(10), 1901. https://doi.org/10.3390/molecules24101901