
Computational Screening and Analysis of Lung Cancer Related Non-Synonymous Single Nucleotide Polymorphisms on the Human Kirsten Rat Sarcoma Gene




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Prediction of Disease Related SNPs
2.2.1. Prediction of Functional Consequences of nsSNPs
2.2.2. Estimation of Evolutionary Conservation of nsSNPs
2.2.3. Prediction of Protein Change Stability of nsSNPs
2.2.4. Identification of Somatic Mutations that can Cause Cancer
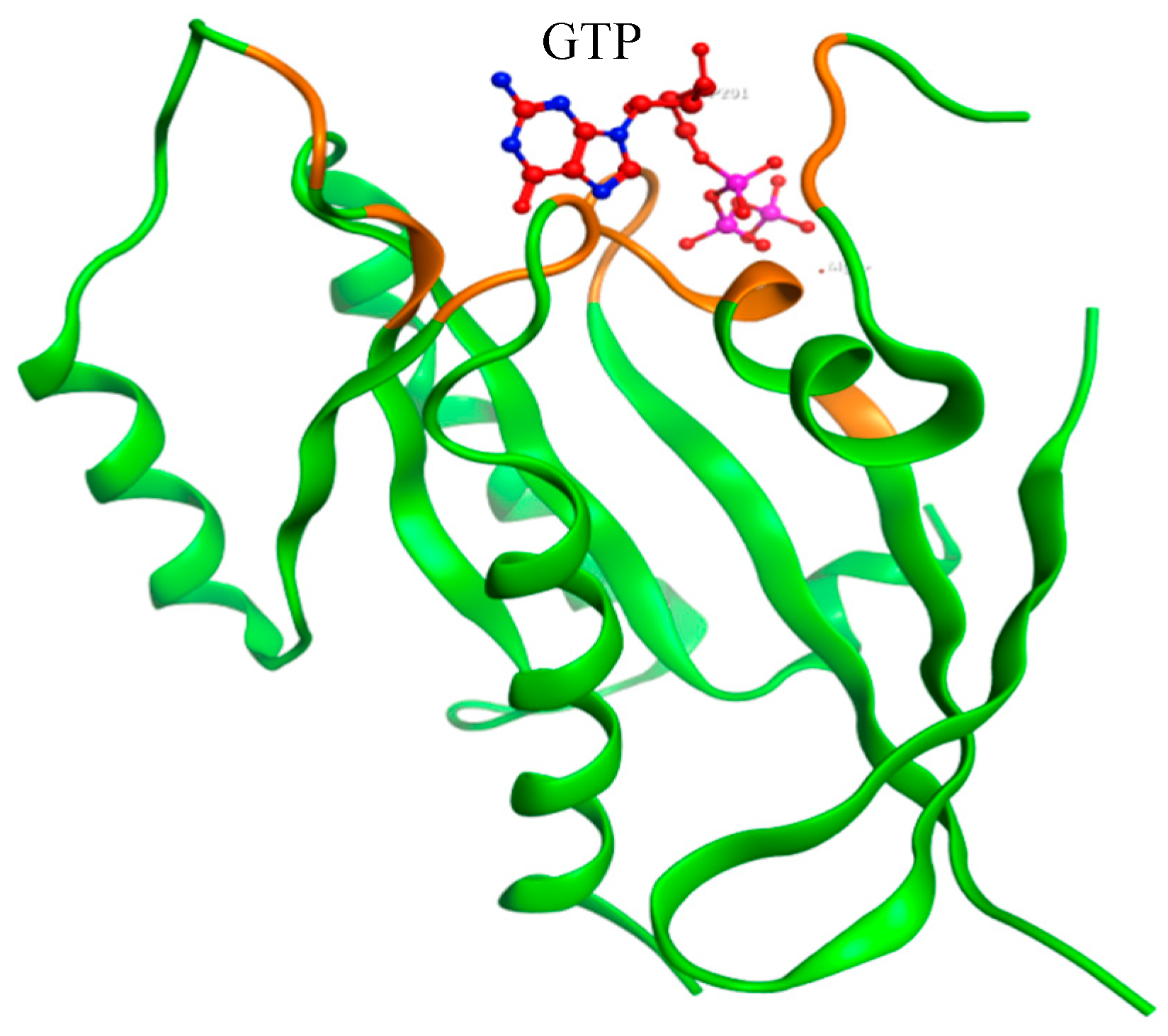
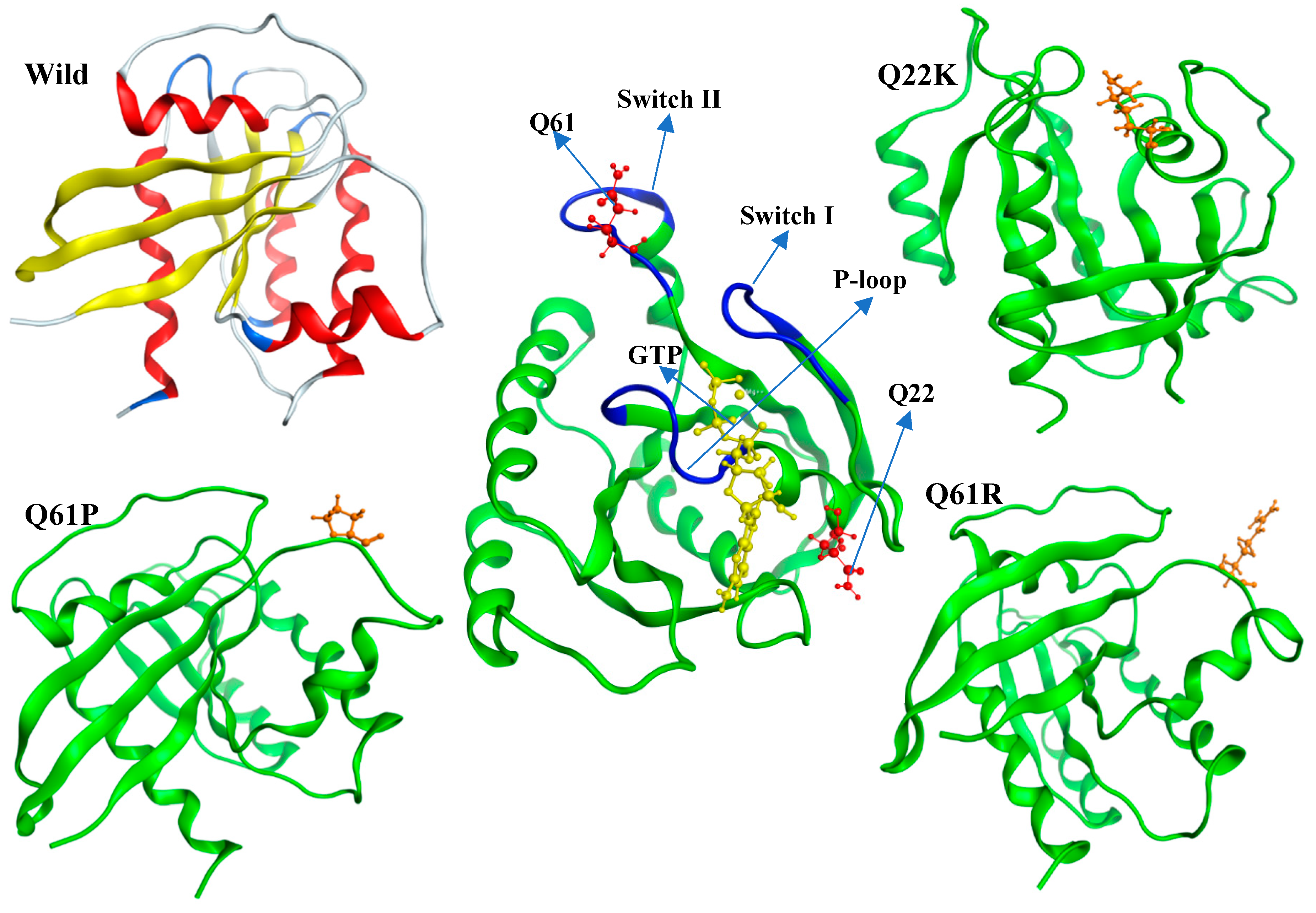
2.3. Modeling of Native and Mutant KRAS Proteins
2.4. Molecular Dynamics Simulation and Trajectories Analysis
2.5. Principal Component Analysis
3. Results and Discussion
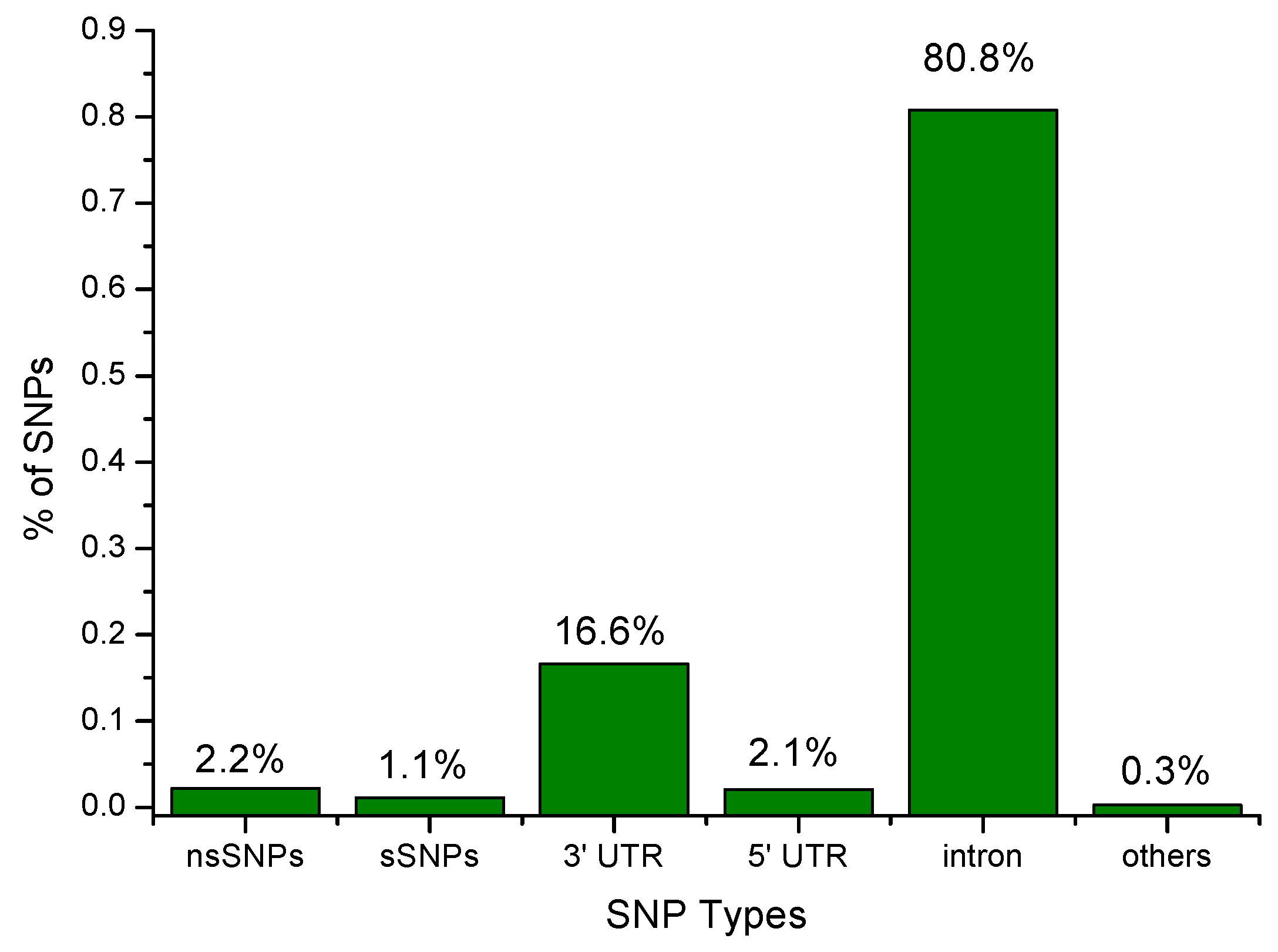
3.1. SNP Data Set from dbSNP
3.1.1. Screening of Missense SNPs Based on Functional Analysis
3.1.2. Analysis of Deleterious nsSNPs Based on the Residue Evolutionary Conservation
3.1.3. Screening of Deleterious nsSNPs Based on the Stability Analysis
3.1.4. Lung Cancer Related Mutations by COSMIC Database
3.2. Molecular Dynamics Simulation
3.2.1. Structural Stability Analysis
3.2.2. Structural Flexibility Analysis
3.2.3. Structural Compactness Analysis
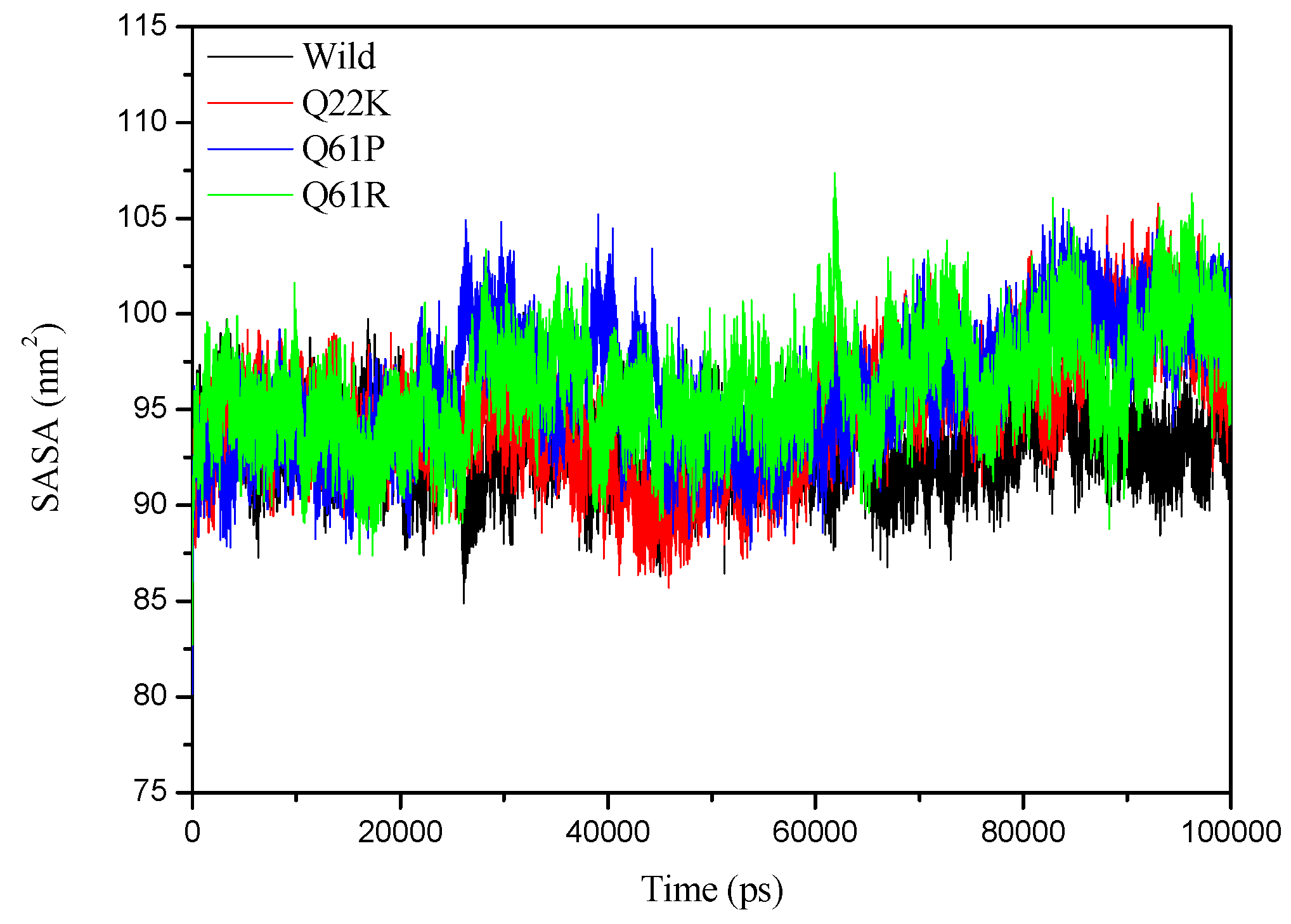
3.2.4. SASA Analysis
3.2.5. Principal Component Analysis
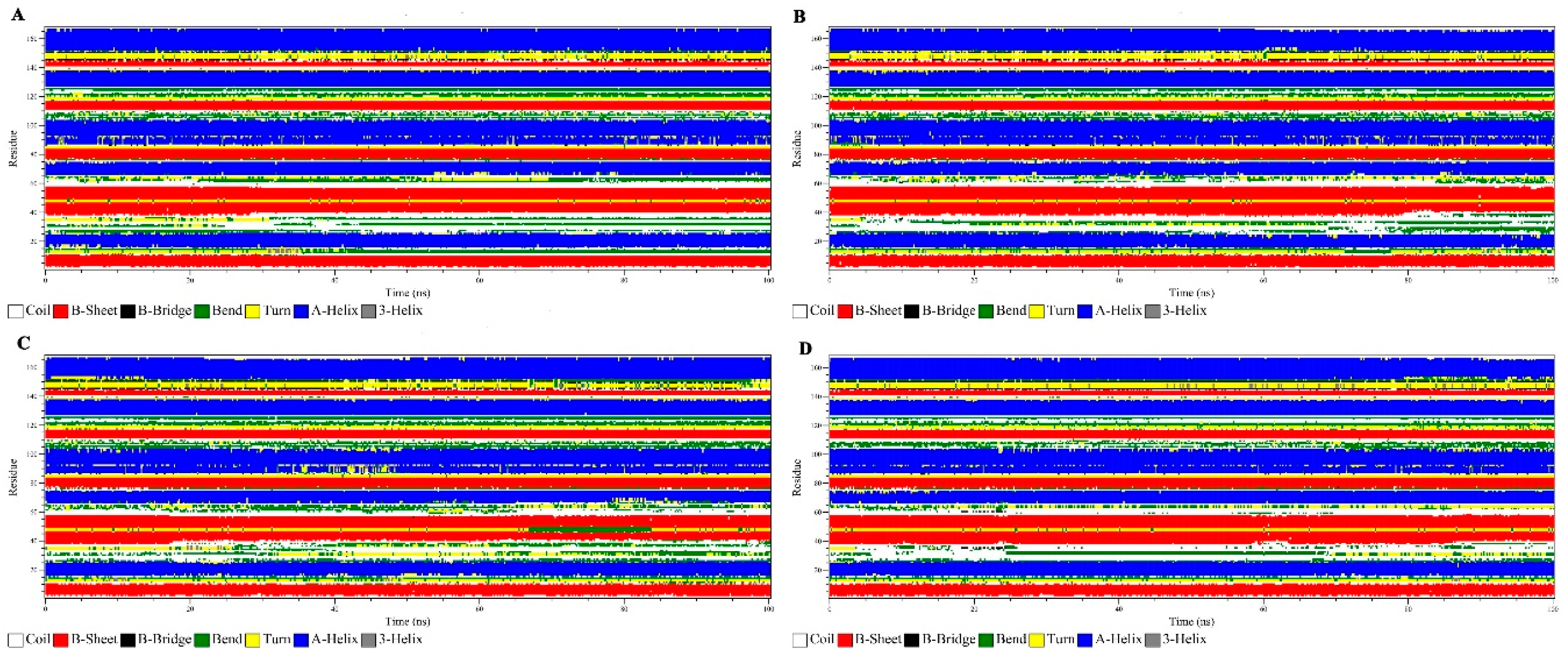
3.2.6. Secondary Structure Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl Lung Cancer Res. 2015, 4, 36. [Google Scholar] [CrossRef]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596. [Google Scholar] [CrossRef]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Leighl, N.B.; Tsao, M.S.; Shepherd, F.A. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J. Thorac Oncol 2013, 8, 530. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007, 35, 3823. [Google Scholar] [CrossRef]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16, S1. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745. [Google Scholar] [CrossRef]
- Wadood, A.; Mehmood, A.; Khan, H.; Ilyas, M.; Ahmad, A.; Alarjah, M.; Abu-Izneid, T. Epitopes based drug design for dengue virus envelope protein: A computational approach. Comput. Biol. Chem. 2017, 71, 152. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006, 62, 1125. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306. [Google Scholar] [CrossRef]
- Quan, L.; Lv, Q.; Zhang, Y. STRUM: Structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics 2016, 32, 2936. [Google Scholar] [CrossRef]
- Jia, M.; Yang, B.; Li, Z.; Shen, H.; Song, X.; Gu, W. Computational analysis of functional single nucleotide polymorphisms associated with the CYP11B2 gene. PLoS One 2014, 9, e104311. [Google Scholar] [CrossRef]
- Kumar, R.; Bansal, A.; Shukla, R.; Singh, T.R.; Ramteke, P.W.; Singh, S.; Gautam, B. In silico screening of deleterious single nucleotide polymorphisms (SNPs) and molecular dynamics simulation of disease associated mutations in gene responsible for Oculocutaneous Albinism type 6 (OCA 6) disorder. J. Biomol. Struct. Dyn. 2018. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235. [Google Scholar] [CrossRef]
- Xu, S.; Long, B.N.; Boris, G.H.; Chen, A.; Ni, S.; Kennedy, M.A. Structural insight into the rearrangement of the switch I region in GTP-bound G12A K-Ras. Acta Crystallogr. D Struct. Biol. 2017, 73, 970. [Google Scholar] [CrossRef]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163. [Google Scholar] [CrossRef]
- Berezin, C.; Glaser, F.; Rosenberg, J.; Paz, I.; Pupko, T.; Fariselli, P.; Casadio, R.; Ben-Tal, N. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics 2004, 20, 1322. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatics 2016, 54, 5.6.1. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Chikan, N.A.; Bukhari, S.; Shabir, N.; Amin, A.; Shafi, S.; Qadri, R.A.; Patel, T.N.C. Atomic insight into the altered O6-Methylguanine-DNA methyltransferase protein architecture in gastric cancer. PloS One 2015, 10, e0127741. [Google Scholar] [CrossRef]
- Jänne, P.A.; Shaw, A.T.; Pereira, J.R.; Jeannin, G.; Vansteenkiste, J.; Barrios, C.; Franke, F.A.; Grinsted, L.; Zazulina, V.; Smith, P. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013, 14, 38. [Google Scholar] [CrossRef]
- Kumar, M.S.; Hancock, D.C.; Molina-Arcas, M.; Steckel, M.; East, P.; Diefenbacher, M.; Armenteros-Monterroso, E.; Lassailly, F.; Matthews, N.; Nye, E. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 2012, 149, 642. [Google Scholar] [CrossRef]
- Lim, S.M.; Westover, K.D.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Edit. 2014, 53, 199. [Google Scholar] [CrossRef]
- Choi, H.S.; Min, K.T.; Cha, Y.S.; Hong, S.P. Multiplex detection of KRAS mutations by a matrix-assisted laser desorption/ionization-time of flight mass spectrometry assay. Clin. Biochem. 2014, 47, 1091. [Google Scholar] [CrossRef]
- Seo, A.; Yang, J.; Kim, H.; Jheon, S.; Kim, K.; Lee, C.; Jin, Y.; Yun, S.; Chung, J.; Paik, J. Clinicopathologic and prognostic significance of c-MYC copy number gain in lung adenocarcinomas. Brit. J. Cancer 2014, 110, 2688. [Google Scholar] [CrossRef]
- Nagy, Á.; Pongor, L.S.; Szabó, A.; Santarpia, M.; Győrffy, B. KRAS driven expression signature has prognostic power superior to mutation status in non-small cell lung cancer. Int. J. Cancer 2017, 140, 930. [Google Scholar] [CrossRef]
Sample Availability: Not available. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rs ID | Variant | SIFT | SNAP2 | PROVEAN | |||
|---|---|---|---|---|---|---|---|
| Prediction | Score | Prediction | Score | Prediction | Score | ||
| rs17851045 | Q61H | intolerant | 0 | effect | 94 | Deleterious | −4.556 |
| rs104886028 | M72I | tolerated | 0.25 | effect | 7 | Deleterious | −3.376 |
| rs104886029 | A59V | intolerant | 0 | effect | 38 | Deleterious | −3.839 |
| rs104894359 | G60S | intolerant | 0 | effect | 73 | Deleterious | −5.817 |
| rs104894359 | G60R | intolerant | 0 | effect | 99 | Deleterious | −7.758 |
| rs104894361 | K5N | intolerant | 0 | effect | 90 | Deleterious | −3.836 |
| rs104894362 | F156L | intolerant | 0 | effect | 65 | Deleterious | −5.310 |
| rs104894364 | T58I | intolerant | 0 | effect | 97 | Deleterious | −5.823 |
| rs104894365 | V14I | intolerant | 0 | effect | 96 | Neutral | −0.819 |
| rs104894366 | P34R | intolerant | 0 | effect | 96 | Deleterious | −7.598 |
| rs104894367 | V152G | intolerant | 0 | effect | 75 | Deleterious | −5.873 |
| rs112445441 | G13D | intolerant | 0 | effect | 98 | Deleterious | −5.403 |
| rs121913236 | Q22K | intolerant | 0 | effect | 55 | Deleterious | −3.313 |
| rs121913238 | Q61K | intolerant | 0.01 | effect | 69 | Deleterious | −3.588 |
| rs121913239 | Q61E | intolerant | 0.01 | effect | 58 | Deleterious | −2.772 |
| rs121913240 | Q61P | intolerant | 0.01 | effect | 63 | Deleterious | −5.602 |
| rs121913241 | Q61R | intolerant | 0.01 | effect | 63 | Deleterious | −3.455 |
| rs121913242 | Q61L | intolerant | 0.01 | effect | 93 | Deleterious | −6.507 |
| rs121913527 | A146P | intolerant | 0 | effect | 82 | Deleterious | −4.513 |
| rs121913528 | A59T | intolerant | 0.01 | effect | 40 | Deleterious | −3.727 |
| rs121913528 | A59S | intolerant | 0 | effect | 35 | Deleterious | −2.701 |
| rs121913529 | G12D | intolerant | 0 | effect | 99 | Deleterious | −5.373 |
| rs121913531 | G12A | intolerant | 0 | effect | 96 | Deleterious | −4.621 |
| rs121913534 | G12V | intolerant | 0 | effect | 98 | Deleterious | −7.113 |
| rs121913530 | G12S | intolerant | 0 | effect | 97 | Deleterious | −4.443 |
| rs121913532 | G12R | intolerant | 0.04 | effect | 99 | Deleterious | −6.183 |
| rs121913533 | G12C | intolerant | 0 | effect | 94 | Deleterious | −7.161 |
| rs121913535 | G13C | intolerant | 0 | effect | 92 | Deleterious | −7.619 |
| rs121913538 | L19F | intolerant | 0 | effect | 54 | Deleterious | −3.373 |
| rs193929331 | K5E | intolerant | 0 | effect | 83 | Deleterious | −3.154 |
| rs200970347 | G179S | tolerated | 0.06 | neutral | -36 | Neutral | −1.374 |
| rs201170656 | M189L | tolerated | 1 | neutral | -67 | Neutral | −0.162 |
| rs202247812 | N116S | intolerant | 0 | effect | 81 | Deleterious | −4.564 |
| rs372793780 | R164Q | intolerant | 0.02 | effect | 53 | Deleterious | −2.610 |
| rs387907205 | Y71H | intolerant | 0 | effect | 75 | Deleterious | −4.454 |
| rs387907205 | Y71D | intolerant | 0 | effect | 86 | Deleterious | −9.374 |
| rs387907206 | K147E | intolerant | 0 | effect | 83 | Deleterious | −3.624 |
| rs397517041 | V152F | intolerant | 0 | effect | 65 | Deleterious | −4.424 |
| rs397517042 | F156I | intolerant | 0 | effect | 62 | Deleterious | −5.312 |
| rs397517042 | F156V | intolerant | 0 | effect | 64 | Deleterious | −6.196 |
| rs397517476 | Y166N | tolerated | 0.28 | effect | 18 | Deleterious | −3.387 |
| rs397517476 | Y166H | tolerated | 0.49 | effect | 4 | Neutral | −1.392 |
| rs727503106 | R97K | tolerated | 0.29 | effect | 53 | Neutral | −2.130 |
| rs727503108 | G60V | intolerant | 0 | effect | 77 | Deleterious | −8.727 |
| rs727503110 | Q22R | intolerant | 0 | effect | 20 | Deleterious | −3.324 |
| rs727503110 | Q22L | intolerant | 0 | effect | 49 | Deleterious | −5.834 |
| rs727504662 | M72L | intolerant | 0.01 | effect | 46 | Deleterious | −2.549 |
| rs730880470 | T50S | tolerated | 0.51 | neutral | -44 | Deleterious | −3.144 |
| rs730880471 | D119N | intolerant | 0 | effect | 83 | Deleterious | −4.566 |
| rs730880472 | L23R | intolerant | 0 | effect | 77 | Deleterious | −4.925 |
| rs730880473 | A130V | intolerant | 0.03 | effect | 17 | Neutral | −2.446 |
| rs770248150 | K117N | intolerant | 0 | effect | 86 | Deleterious | −4.558 |
| rs794727277 | N26Y | intolerant | 0.01 | effect | 51 | Deleterious | −5.455 |
| rs794727720 | Y157C | intolerant | 0.02 | effect | 3 | Deleterious | −6.787 |
| rs1057517885 | I171T | tolerated | 0.59 | neutral | -20 | Neutral | 0.121 |
| rs1057519725 | A146V | intolerant | 0 | effect | 53 | Deleterious | −3.625 |
| rs1135401776 | K147R | intolerant | 0.01 | effect | 18 | Deleterious | −2.718 |
| rs138669124 | F141L | intolerant | 0.01 | effect | 65 | Deleterious | −4.534 |
| rs368557003 | Q165R | tolerated | 0.85 | neutral | -56 | Neutral | −0.013 |
| rs373500216 | A134G | intolerant | 0.04 | effect | 53 | Deleterious | −3.378 |
| rs374681135 | P178S | tolerated | 0.77 | neutral | -29 | Neutral | −0.199 |
| rs529925358 | I183V | tolerated | 0.47 | effect | 5 | Neutral | 0.071 |
| rs539423712 | V160A | tolerated | 0.05 | effect | 28 | Deleterious | −3.533 |
| rs542902732 | M1I | intolerant | 0.01 | neutral | -13 | Neutral | −2.480 |
| rs575569675 | T124S | tolerated | 0.65 | neutral | -41 | Neutral | −0.801 |
| rs746609817 | K128R | tolerated | 0.27 | effect | 11 | Neutral | −0.496 |
| rs749177256 | T158I | tolerated | 0.14 | effect | 5 | Deleterious | −4.132 |
| rs754870563 | G138E | intolerant | 0.02 | effect | 41 | Deleterious | −5.915 |
| rs755177746 | A155G | tolerated | 0.08 | effect | 53 | Deleterious | −3.310 |
| rs755877953 | V160M | intolerant | 0 | effect | 20 | Deleterious | −2.648 |
| rs755967833 | I188V | intolerant | 0.5 | effect | 26 | Neutral | −0.257 |
| rs756890312 | G77A | intolerant | 0.01 | effect | 58 | Deleterious | −5.942 |
| rs757674707 | V160I | intolerant | 0.04 | effect | 19 | Neutral | −0.885 |
| rs757816355 | S136N | tolerated | 0.3 | neutral | -26 | Neutral | −1.842 |
| rs766231905 | I171M | tolerated | 0.12 | neutral | -26 | Neutral | 0.484 |
| rs770020203 | T74A | tolerated | 0.13 | effect | 21 | Deleterious | −4.434 |
| rs771629239 | E174K | tolerated | 0.58 | neutral | -47 | Neutral | −0.392 |
| rs772985440 | S172C | intolerant | 0.02 | effect | 16 | Deleterious | −2.706 |
| rs775836436 | V112I | tolerated | 0.47 | neutral | -14 | Neutral | −0.580 |
| rs778702415 | G138R | tolerated | 0.07 | effect | 20 | Deleterious | −5.963 |
| rs779951033 | I187V | tolerated | 1 | neutral | -69 | Neutral | 0.083 |
| rs780974222 | G75A | intolerant | 0.02 | effect | 34 | Deleterious | −5.938 |
| rs781634879 | T127R | tolerated | 0.63 | effect | 3 | Deleterious | −2.928 |
| rs868857258 | L79P | intolerant | 0 | effect | 76 | Deleterious | −6.881 |
| rs904755552 | I46M | intolerant | 0 | effect | 20 | Deleterious | −2.794 |
| rs953088090 | K88E | tolerated | 0.24 | effect | 47 | Deleterious | −2.537 |
| rs989151052 | D154G | intolerant | 0 | effect | 57 | Deleterious | −4.537 |
| rs1024789250 | K182E | tolerated | 0.23 | effect | 35 | Neutral | −1.118 |
| rs1191739287 | K170E | tolerated | 0.83 | effect | 2 | Neutral | −0.754 |
| rs1199162369 | R68C | intolerant | 0 | effect | 68 | Deleterious | −7.900 |
| rs1265970615 | T158P | tolerated | 0.23 | effect | 53 | Deleterious | −3.632 |
| rs1296330213 | L6I | tolerated | 0.09 | neutral | -53 | Neutral | −1.566 |
| rs1296330213 | L6V | tolerated | 0.06 | effect | 3 | Neutral | −2.337 |
| rs1307793966 | R164G | intolerant | 0.01 | effect | 72 | Deleterious | −4.743 |
| rs1308177469 | M189I | tolerated | 0.24 | effect | 11 | Neutral | −0.817 |
| rs1309399018 | H95N | tolerated | 0.5 | effect | 2 | Neutral | 0.028 |
| rs1340281106 | N86H | intolerant | 0.04 | effect | 64 | Deleterious | −3.195 |
| rs1344202459 | I142T | tolerated | 0.1 | effect | 26 | Deleterious | −3.283 |
| rs1363431968 | D126H | intolerant | 0.03 | effect | 59 | Deleterious | −3.361 |
| rs1407509439 | T50I | tolerated | 0.15 | neutral | -20 | Deleterious | −3.410 |
| rs1434157586 | R123W | tolerated | 0.08 | effect | 52 | Deleterious | −6.229 |
| rs1437657227 | D92H | intolerant | 0 | effect | 37 | Deleterious | −4.092 |
| rs1463850736 | A130T | intolerant | 0 | effect | 32 | Deleterious | −2.556 |
| rs1463850736 | A130P | intolerant | 0.01 | effect | 51 | Deleterious | −3.248 |
| rs1463850736 | A130S | intolerant | 0.01 | effect | 24 | Neutral | −1.591 |
| rs1470495974 | I163V | tolerated | 0.24 | neutral | -22 | Neutral | −0.817 |
| rs ID | Variant | Conservation Score | ||
|---|---|---|---|---|
| SWISS-PROT | UniProt | UniRef90 | ||
| rs17851045 | Q61H | 9 | 8 | 9 |
| rs104886029 | A59V | 9 | 9 | 9 |
| rs104894359 | G60S | 9 | 9 | 9 |
| rs104894359 | G60R | 9 | 9 | 9 |
| rs104894361 | K5N | 8 | 8 | 8 |
| rs104894362 | F156L | 9 | 9 | 9 |
| rs104894364 | T58I | 9 | 9 | 9 |
| rs104894366 | P34R | 6 | 6 | 5 |
| rs104894367 | V152G | 8 | 9 | 8 |
| rs112445441 | G13D | 6 | 6 | 6 |
| rs121913236 | Q22K | 8 | 7 | 7 |
| rs121913238 | Q61K | 9 | 8 | 9 |
| rs121913239 | Q61E | 9 | 8 | 9 |
| rs121913240 | Q61P | 9 | 8 | 9 |
| rs121913241 | Q61R | 9 | 8 | 9 |
| rs121913242 | Q61L | 9 | 8 | 9 |
| rs121913527 | A146P | 9 | 9 | 9 |
| rs121913528 | A59T | 9 | 9 | 9 |
| rs121913528 | A59S | 9 | 9 | 9 |
| rs121913529 | G12D | 7 | 8 | 8 |
| rs121913531 | G12A | 7 | 8 | 8 |
| rs121913534 | G12V | 7 | 8 | 8 |
| rs121913530 | G12S | 7 | 8 | 8 |
| rs121913532 | G12R | 7 | 8 | 8 |
| rs121913533 | G12C | 7 | 8 | 8 |
| rs121913535 | G13C | 6 | 6 | 6 |
| rs121913538 | L19F | 6 | 7 | 7 |
| rs193929331 | K5E | 8 | 8 | 8 |
| rs202247812 | N116S | 9 | 9 | 9 |
| rs387907205 | Y71H | 8 | 7 | 7 |
| rs387907205 | Y71D | 8 | 7 | 7 |
| rs387907206 | K147E | 8 | 7 | 7 |
| rs397517041 | V152F | 8 | 9 | 8 |
| rs397517042 | F156I | 9 | 9 | 9 |
| rs397517042 | F156V | 9 | 9 | 9 |
| rs727503108 | G60V | 9 | 8 | 9 |
| rs727503110 | Q22R | 8 | 7 | 7 |
| rs727503110 | Q22L | 8 | 7 | 7 |
| rs727504662 | M72L | 8 | 7 | 7 |
| rs730880471 | D119N | 9 | 9 | 9 |
| rs730880472 | L23R | 7 | 7 | 7 |
| rs770248150 | K117N | 9 | 9 | 9 |
| rs794727277 | N26Y | 7 | 4 | 5 |
| rs794727720 | Y157C | 1 | 1 | 1 |
| rs1057519725 | A146V | 9 | 9 | 9 |
| rs1135401776 | K147R | 8 | 7 | 7 |
| rs138669124 | F141L | 7 | 6 | 7 |
| rs373500216 | A134G | 8 | 8 | 8 |
| rs754870563 | G138E | 1 | 1 | 1 |
| rs755877953 | V160M | 6 | 7 | 7 |
| rs756890312 | G77A | 7 | 7 | 7 |
| rs372793780 | R164Q | 4 | 6 | 5 |
| rs772985440 | S172C | 1 | 1 | 1 |
| rs780974222 | G75A | 7 | 7 | 7 |
| rs868857258 | L79P | 6 | 5 | 5 |
| rs904755552 | I46M | 5 | 5 | 5 |
| rs989151052 | D154G | 2 | 1 | 3 |
| rs1199162369 | R68C | 8 | 8 | 7 |
| rs1307793966 | R164G | 4 | 6 | 5 |
| rs1340281106 | N86H | 5 | 5 | 5 |
| rs1363431968 | D126H | 3 | 3 | 4 |
| rs1437657227 | D92H | 6 | 5 | 5 |
| rs1463850736 | A130T | 5 | 6 | 6 |
| rs1463850736 | A130P | 5 | 6 | 6 |
| rs ID | Variant | I-Mutant2.0 | Mupro | ||
|---|---|---|---|---|---|
| DDG | Stability | DDG | Stability | ||
| rs17851045 | Q61H | −0.96 | Decrease | −0.48 | Decrease |
| rs104886029 | A59V | −0.06 | Decrease | −0.35 | Decrease |
| rs104894359 | G60S | −1.39 | Decrease | −0.78 | Decrease |
| rs104894359 | G60R | −1.36 | Decrease | −0.63 | Decrease |
| rs104894361 | K5N | −0.25 | Decrease | −0.70 | Decrease |
| rs104894364 | T58I | 0.28 | Increase | −0.003 | Decrease |
| rs121913236 | Q22K | −0.55 | Decrease | −1.27 | Decrease |
| rs121913238 | Q61K | −0.16 | Decrease | −0.56 | Decrease |
| rs121913239 | Q61E | 0.22 | Increase | −0.13 | Decrease |
| rs121913240 | Q61P | −1.35 | Decrease | −0.69 | Decrease |
| rs121913241 | Q61R | −0.55 | Decrease | −0.18 | Decrease |
| rs121913242 | Q61L | 0.47 | Increase | 0.54 | Increase |
| rs121913527 | A146P | −1.58 | Decrease | −1.32 | Decrease |
| rs121913528 | A59T | −1.36 | Decrease | −1.34 | Decrease |
| rs121913528 | A59S | −0.82 | Decrease | −1.03 | Decrease |
| rs121913529 | G12D | −0.83 | Decrease | −0.75 | Decrease |
| rs121913531 | G12A | −0.53 | Decrease | −1.19 | Decrease |
| rs121913534 | G12V | −0.36 | Decrease | −0.66 | Decrease |
| rs121913530 | G12S | −1.49 | Decrease | −1.08 | Decrease |
| rs121913532 | G12R | −1.47 | Decrease | −0.93 | Decrease |
| rs121913533 | G12C | −1.34 | Decrease | −0.58 | Decrease |
| rs193929331 | K5E | −0.23 | Decrease | −0.43 | Decrease |
| rs202247812 | N116S | −0.81 | Decrease | −1.84 | Decrease |
| rs387907206 | K147E | −0.73 | Decrease | −0.38 | Decrease |
| rs727503108 | G60V | −1.22 | Decrease | −0.38 | Decrease |
| rs727503110 | Q22R | −1.31 | Decrease | −0.84 | Decrease |
| rs727503110 | Q22L | −0.08 | Decrease | −0.10 | Decrease |
| rs730880471 | D119N | −1.43 | Decrease | −0.78 | Decrease |
| rs770248150 | K117N | −0.68 | Decrease | −0.17 | Decrease |
| rs1057519725 | A146V | −0.34 | Decrease | −0.97 | Decrease |
| rs1135401776 | K147R | −0.63 | Decrease | −0.41 | Decrease |
| rs1199162369 | R68C | 0.33 | Increase | −1.33 | Decrease |
| WT | Q22K | Q61P | Q61R | |
|---|---|---|---|---|
| Backbone rmsd (nm) | 0.1853(0.0187) | 0.2108(0.0771) | 0.2504(0.0652) | 0.2240(0.0741) |
| Cα-rmsd (nm) | 0.1933(0.0195) | 0.2173(0.0766) | 0.2562(0.0654) | 0.2294(0.0743) |
| Cα-rmsf (nm) | 0.0939(0.0666) | 0.1251(0.1068) | 0.1183(0.0985) | 0.1289(0.0845) |
| Rg-Cα (nm) | 1.4960(0.0086) | 1.5072(0.0169) | 1.5130(0.0181) | 1.5128(0.0148) |
| Rg-protein (nm) | 1.5495(0.0087) | 1.5629(0.0193) | 1.5686(0.0209) | 1.5658(0.0145) |
| SASA (nm2) | 93.008(1.9027) | 94.806(3.0942) | 96.109(3.2164) | 96.159(2.7846) |
| Sample | % | ||||
|---|---|---|---|---|---|
| α-Helices | β-Sheets | Coils | Bend | Turn | |
| Wild | 35.61 | 23.53 | 20.04 | 10.07 | 9.14 |
| Q22K | 34.94 | 24.63 | 19.60 | 9.26 | 9.89 |
| Q61P | 34.65 | 23.19 | 19.12 | 11.24 | 10.00 |
| Q61R | 35.27 | 23.23 | 20.41 | 10.12 | 9.37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Mehmood, A.; Wang, H.; Xu, Q.; Xiong, Y.; Wei, D.-Q. Computational Screening and Analysis of Lung Cancer Related Non-Synonymous Single Nucleotide Polymorphisms on the Human Kirsten Rat Sarcoma Gene. Molecules 2019, 24, 1951. https://doi.org/10.3390/molecules24101951
Wang Q, Mehmood A, Wang H, Xu Q, Xiong Y, Wei D-Q. Computational Screening and Analysis of Lung Cancer Related Non-Synonymous Single Nucleotide Polymorphisms on the Human Kirsten Rat Sarcoma Gene. Molecules. 2019; 24(10):1951. https://doi.org/10.3390/molecules24101951
Chicago/Turabian StyleWang, Qiankun, Aamir Mehmood, Heng Wang, Qin Xu, Yi Xiong, and Dong-Qing Wei. 2019. "Computational Screening and Analysis of Lung Cancer Related Non-Synonymous Single Nucleotide Polymorphisms on the Human Kirsten Rat Sarcoma Gene" Molecules 24, no. 10: 1951. https://doi.org/10.3390/molecules24101951
APA StyleWang, Q., Mehmood, A., Wang, H., Xu, Q., Xiong, Y., & Wei, D. -Q. (2019). Computational Screening and Analysis of Lung Cancer Related Non-Synonymous Single Nucleotide Polymorphisms on the Human Kirsten Rat Sarcoma Gene. Molecules, 24(10), 1951. https://doi.org/10.3390/molecules24101951