Permeabilities of CO2, H2S and CH4 through Choline-Based Ionic Liquids: Atomistic-Scale Simulations



Abstract
:1. Introduction
2. Computational Method
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Betts, R.A.; Boucher, O.; Collins, M.; Cox, P.M.; Falloon, P.D.; Gedney, N.; Hemming, D.L.; Huntingford, C.; Jones, C.D.; Sexton, D.M.H.; et al. Projected increase in continental runoff due to plant responses to increasing carbon dioxide. Nature 2007, 448, 1037–1041. [Google Scholar] [CrossRef]
- Meyer, J. Crisis reading. Nature 2008, 455, 733. [Google Scholar] [CrossRef]
- Haszeldine, R.S. Carbon capture and storage: How green can black be? Science 2009, 325, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Keith, D.W. Why capture CO2 from the atmosphere? Science 2009, 325, 1654. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Ramdin, M.; de Loos, T.W.; Vlugt, T.J.H. State-of-the-art of CO2 capture with ionic liquids. Ind. Eng. Chem. Res. 2012, 51, 8149. [Google Scholar] [CrossRef]
- Yu, C.-H.; Huang, C.-H.; Tan, C.-S. A review of CO2 capture by absorption and adsorption. Aerosol Air Qual. Res. 2012, 12, 745–769. [Google Scholar] [CrossRef]
- Theo, W.L.; Lim, J.S.; Hashim, H.; Mustaffa, A.A.; Ho, W.S. Review of pre-combustion capture and ionic liquid in carbon capture and storage. Appl. Energy 2016, 183, 1633–1663. [Google Scholar] [CrossRef]
- Luo, X.; Wang, C. The development of carbon capture by functionalized ionic liquids. Curr. Opin. Green Sustain. Chem. 2016, 3, 33–38. [Google Scholar] [CrossRef]
- Torralba-Calleja, E.; Skinner, J.; Gutiérrez-Tauste, D. CO2 capture in ionic liquids: A review of solubilities and experimental methods. J. Chem. 2013, 2013, 473584. [Google Scholar] [CrossRef]
- Chaban, V.V.; Prezhdo, O.V. Ionic and molecular liquids: Working together for robust engineering. J. Phys. Chem. Lett. 2013, 4, 1423–1431. [Google Scholar] [CrossRef]
- Banat, F.; Younas, O.; Didarul, I. Energy and exergical dissection of a natural gas sweetening plant using methyldiethanol amine (MDEA) solution. J. Nat. Gas Sci. Eng. 2014, 16, 1–7. [Google Scholar] [CrossRef]
- Rochelle, G.; Chen, E.; Freeman, S.; Van Wagener, D.; Xu, Q.; Voice, A. Aqueous piperazine as the new standard for CO2 capture technology. Chem. Eng. J. 2011, 171, 725–733. [Google Scholar] [CrossRef]
- Abotaleb, A.; El-Naas, M.H.; Amhamed, A. Enhancing gas loading and reducing energy consumption in acid gas removal systems: A simulation study based on real NGL plant data. J. Nat. Gas Sci. Eng. 2017, 55, 565–574. [Google Scholar] [CrossRef]
- Amhamed, A.; Abotaleb, A. Novel integrated acid gas removal and forward osmosis draw solution regeneration system for saving energy and water treatment. J. Oil Gas Petrochem. Sci. 2018, 1, 91–99. [Google Scholar] [CrossRef]
- Yadav, A.; Pandey, S. Densities and viscosities of (choline chloride+Urea) deep eutectic solvent and its aqueous mixtures in the temperature range 293.15 K to 363.15 K. J. Chem. Eng. Data 2014, 59, 2221. [Google Scholar] [CrossRef]
- Gan, Q.; Rooney, D.; Zou, Y. Supported ionic liquid membranes in nanopore structure for gas separation and transport studies. Desalination 2006, 199, 535–537. [Google Scholar] [CrossRef]
- Holbrey, J.D. Industrial applications of ionic liquids. Chimica Oggi 2004, 22, 35–37. [Google Scholar] [CrossRef]
- Wells, A.S.; Coombe, V.T. On the freshwater ecotoxicity and biodegradation properties of some common ionic liquids. Org. Process Res. Dev. 2006, 10, 794–798. [Google Scholar] [CrossRef]
- Rima, J. Isaifan and Abdukarem Amhamed, Review on Carbon Dioxide Absorption by Choline Chloride/Urea Deep Eutectic Solvents. J. Adv. Chem. 2018, 2018, 1–6. [Google Scholar]
- Landry, T.D.; Brooks, K.; Poche, D.; Woolhiser, M. Acute toxicity profile of 1-Butyl-3-Methylimidazolium Chloride. Bull. Environ. Contam. Toxicol. 2005, 74, 559–565. [Google Scholar] [CrossRef]
- Earle, M.J.; Seddon, K.R. Ionic liquids. Green solvents for the future. Pure Appl. Chem. 2000, 72, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Bates, E.D.; Mayton, R.D.; Ntai, I.; Davis, J.H., Jr. CO2 Capture by a task-specific ionic liquid. J. Am. Chem. Soc. 2002, 124, 926–927. [Google Scholar] [CrossRef]
- Brennecke, J.F.; Gurkan, B.E. Ionic liquids for CO2 capture and emission reduction. J. Phys. Chem. Lett. 2010, 1, 3459–3464. [Google Scholar] [CrossRef]
- Plechkova, N.V.; Rogers, R.D.; Seddon, K.R. (Eds.) Ionic Liquids: From Knowledge to Application. ACS Symp. Ser. 2009, 1030, 1–20. [Google Scholar]
- Holbrey, J.D.; Seddon, K.R. Ionic liquids. Clean Prod. Process. 1999, 1, 223–236. [Google Scholar] [CrossRef]
- Huang, X.; Margulis, C.J.; Li, M.; Berne, B.J. Why is the partial molar volume of CO2 so small when dissolved in a room temperature ionic liquid? structure and dynamics of CO2 dissolved in [Bmim+] [PF6-]. J. Am. Chem. Soc. 2005, 127, 17842–17851. [Google Scholar] [CrossRef] [PubMed]
- Babarao, R.; Dai, S.; Jiang, D. Understanding the high solubility of CO2 in an ionic liquid with the tetracyanoborate anion. J. Phys. Chem. B 2011, 115, 9789–9794. [Google Scholar] [CrossRef]
- Maginn, E. Molecular simulation of ionic liquids: Current status and future opportunities. J. Phys. Condens. Matter 2009, 21, 373101. [Google Scholar] [CrossRef]
- Yu, Y.; Lu, X.; Zhou, Q.; Dong, K.; Yao, H.; Zhang, S. Biodegradable naphthenic acid ionic liquids: Synthesis, characterization, and quantitative structure-biodegradation relationship. Chem. Eur. J. 2008, 14, 11174–11182. [Google Scholar] [CrossRef]
- Petkovic, M.; Ferguson, J.L.; Nimal, H.Q.; Ferreira, R.; Leitao, M.C.; Seddon, K.R.; Silva, C. Novel biocompatible cholinium-based ionic liquids–toxicity and biodegradability. Green Chem. 2010, 12, 643. [Google Scholar] [CrossRef]
- Costa, A.J.L.; Soromenho, M.R.C.; Shimizu, K.; Marrucho, I.M.; Esperanca, J.M.S.S.; Canongia, J.N.; Rebelo, L.P.N. Density, thermal expansion and viscosity of Cholinium-derived ionic liquids. Chem. Phys. Chem. 2012, 13, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Atilhan, M.; Khraisheh, M.; Alcalde, R.; Fernández, J. Study on hydroxylammonium-based ionic liquids. II. Computational analysis of CO2 absorption. J. Phys. Chem. B 2011, 115, 12487–12498. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Atilhan, M. Water effect on CO2 absorption for hydroxylammonium based ionic liquids: A molecular dynamics study. Chem. Phys. 2012, 400, 118–125. [Google Scholar] [CrossRef]
- Aparicio, S.; Atilhan, M. A computational study on Choline Benzoate and Choline Salicylate ionic liquids in the pure state and after CO2 adsorption. J. Phys. Chem. B 2012, 116, 9171–9185. [Google Scholar] [CrossRef]
- García, G.; Atilhan, M.; Aparicio, S. Water effect on acid-gas capture using Choline Lactate: A DFT insight beyond moleculemolecule pair simulations. J. Phys. Chem. B 2015, 119, 5546–5557. [Google Scholar] [CrossRef]
- Hu, Y.F.; Liu, Z.C.; Xu, C.M.; Ming, X. The molecular characteristics dominating the solubility of gases in ionic liquids. Chem. Soc. Rev. 2011, 40, 3802–3823. [Google Scholar] [CrossRef]
- Bazhenov, S.; Ramdin, M.; Volkov, A.; Volkov, V.; Vlugt, T.J.H.; de Loos, T.W. CO2 solubility in biodegradable hydroxylammonium-based ionic liquids. J. Chem. Eng. Data 2014, 59, 702–708. [Google Scholar] [CrossRef]
- Klähn, M.; Seduraman, A. What determines CO2 solubility in ionic liquids? A molecular simulation study. J. Phys. Chem. B 2015, 119, 10066–10078. [Google Scholar] [CrossRef]
- ATB version 3.0. Available online: https://atb.uq.edu.au/ (accessed on 5 May 2019).
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An automated force field topology builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Hummer, G. Position-dependent diffusion coefficients and free energies from Bayesian analysis of equilibrium and replica molecular dynamics simulations. New J. Phys. 2005, 7, 34. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Rowley, C.N. Molecular simulation of nonfacilitated membrane permeation. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 1672–1687. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Chernick, M.R.; Gonzlez-Manteiga, W.; Crujeiras, R.M.; Barrios, E.B. Bootstrap methods. In International Encyclopedia of Statistical Science; Springer: Berlin/Heidelberg, Germany, 2011; p. 169. [Google Scholar]
- Youngs, T.G.A.; Hardacre, C. Application of Static Charge Transfer within an Ionic-Liquid Force Field and Its Effect on Structure and Dynamics. Chem. Phys. Chem. 2008, 9, 1548. [Google Scholar] [CrossRef]
- Zhang, Y.; Maginn, E.J. A Simple AIMD Approach to Derive Atomic Charges for Condensed Phase Simulation of Ionic Liquids. J. Phys. Chem. B 2012, 116, 10036–10048. [Google Scholar] [CrossRef] [PubMed]
- Morrow, T.I.; Maginn, E.J. Molecular Dynamics Study of the Ionic Liquid 1-n-Butyl-3-methylimidazolium Hexafluorophosphate. J. Phys. Chem. B 2002, 106, 12807. [Google Scholar] [CrossRef]
- Porter, A.R.; Liem, S.Y.; Popelier, P.L.A. Room temperature ionic liquids containing low water concentrations—A molecular dynamics study. Phys. Chem. Chem. Phys. 2008, 10, 4240. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.N.; Lancaster, J.R., Jr.; Denicola, A. The interaction of reactive oxygen and nitrogen species with membranes. Curr. Top. Membr. 2008, 61, 23–42. [Google Scholar]
- Subczynski, W.K.; Hyde, J.S.; Kusumi, A. Oxygen permeability of phosphatidylcholine-cholesterol membranes. Proc. Natl. Acad. Sci. USA 1989, 86, 4474–4478. [Google Scholar] [CrossRef] [PubMed]
- Missner, A.; Kügler, P.; Saparov, S.M.; Sommer, K.; Mathai, J.C.; Zeidel, M.L.; Pohl, P.J. Carbon dioxide transport through membranes. Biol. Chem. 2008, 283, 25340. [Google Scholar] [CrossRef]
- Riahi, S.; Rowley, C.N. Why can hydrogen sulfide permeate cell membranes? J. Am. Chem. Soc. 2014, 136, 15111–15113. [Google Scholar] [CrossRef] [PubMed]
- Karkhanechi, H.; Salmani, S.; Asghari, M. A review on gas separation applications of supported ionic liquid membranes. ChemBioEng Rev. 2015, 2, 290–302. [Google Scholar] [CrossRef]
- Uchytil, P.; Schauer, J.; Petrychkovych, R. Ionic liquid membranes for carbon dioxide methane separation. J. Membr. Sci. 2011, 383, 262–271. [Google Scholar] [CrossRef]




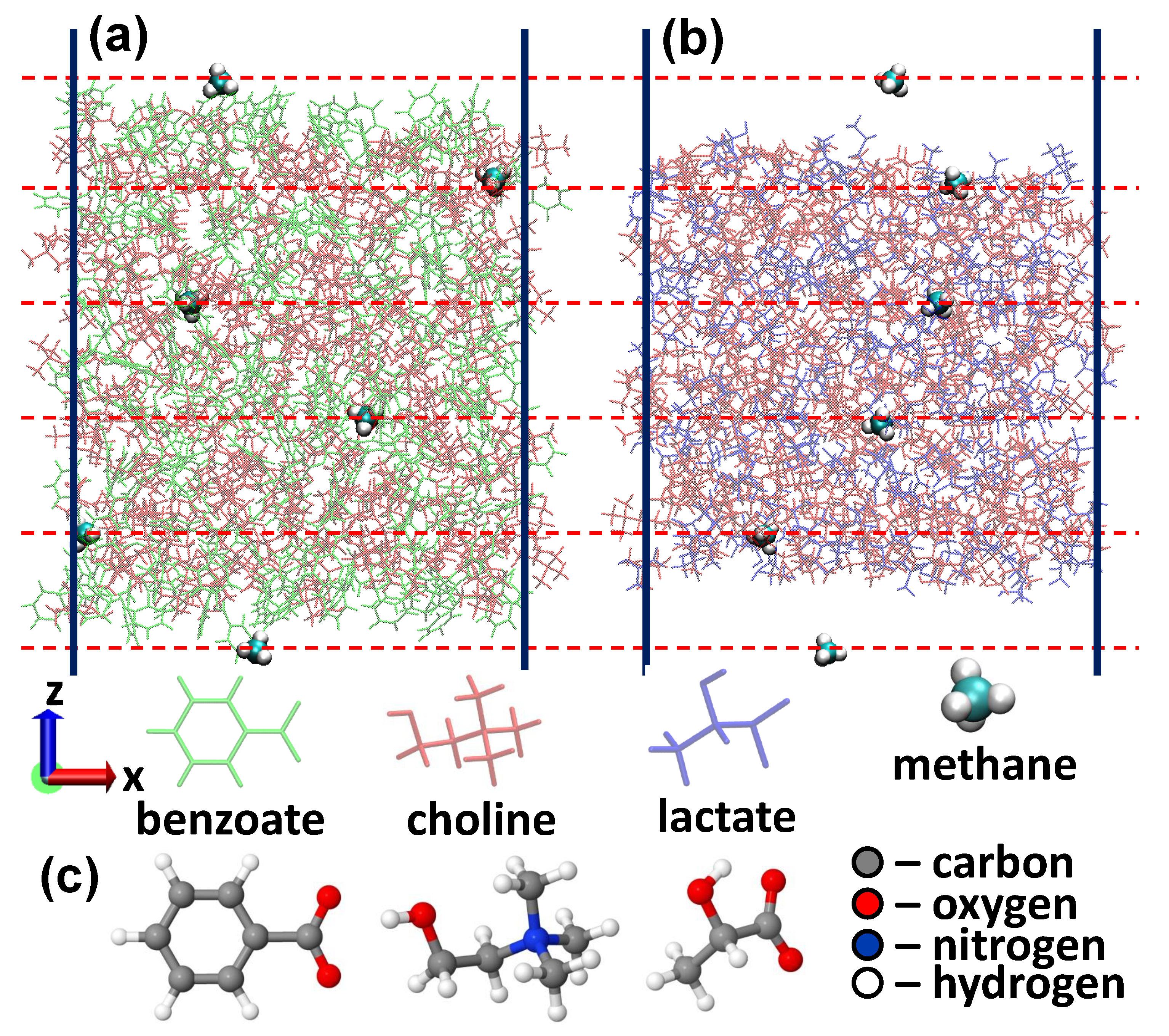{kind=link}
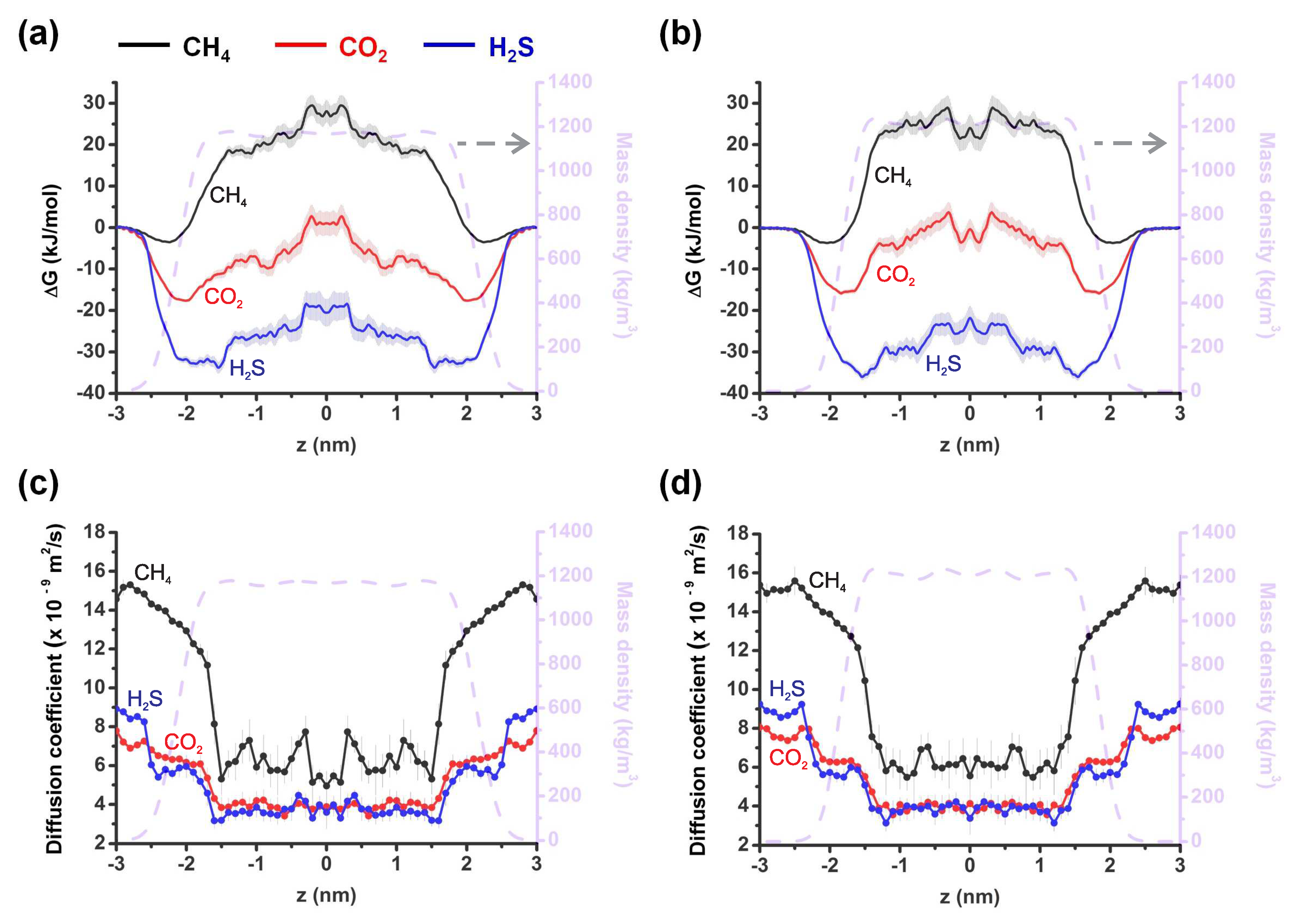{kind=link}
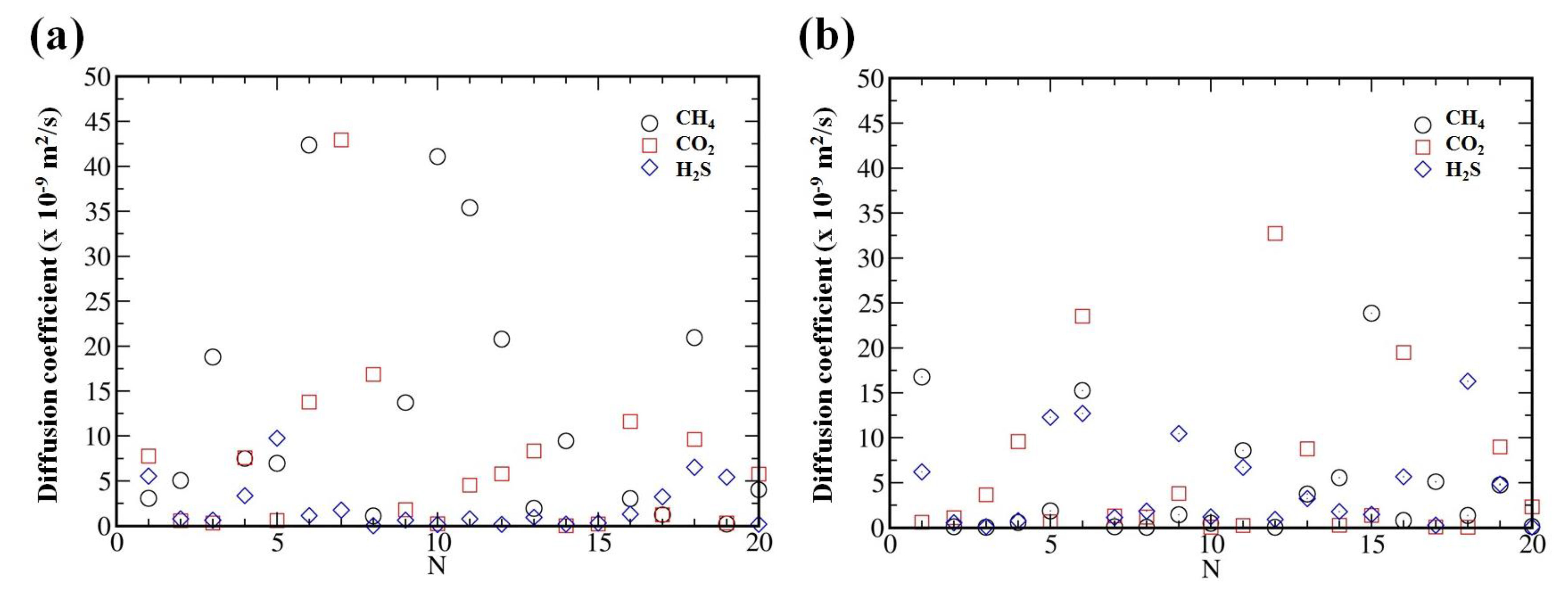{kind=link}
| P (m/s) | P (m/s) | P (m/s) | CO/CH Selectivity | HS/CH Selectivity | HS/CO Selectivity | |
|---|---|---|---|---|---|---|
| BENZ-CHOL | 8.3 × 10 | 2.25 | 13.21 | 2.71 × 10 | 1.59 × 10 | 5.87 |
| LACT-CHOL | 7.6 × 10 | 1.22 | 8.27 | 1.61 × 10 | 1.09 × 10 | 6.78 |
| P (m/s) | P (m/s) | P (m/s) | CO/CH Selectivity | HS/CH Selectivity | HS/CO Selectivity | |
|---|---|---|---|---|---|---|
| BENZ-CHOL | 6.8 × 10 | 50.0 | 53.0 | 7.31 × 10 | 7.75 × 10 | 1.06 |
| LACT-CHOL | 1.7 × 10 | 28.4 | 33.1 | 1.62 × 10 | 1.89 × 10 | 1.17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amhamed, A.; Atilhan, M.; Berdiyorov, G. Permeabilities of CO2, H2S and CH4 through Choline-Based Ionic Liquids: Atomistic-Scale Simulations. Molecules 2019, 24, 2014. https://doi.org/10.3390/molecules24102014
Amhamed A, Atilhan M, Berdiyorov G. Permeabilities of CO2, H2S and CH4 through Choline-Based Ionic Liquids: Atomistic-Scale Simulations. Molecules. 2019; 24(10):2014. https://doi.org/10.3390/molecules24102014
Chicago/Turabian StyleAmhamed, Abdukarem, Mert Atilhan, and Golibjon Berdiyorov. 2019. "Permeabilities of CO2, H2S and CH4 through Choline-Based Ionic Liquids: Atomistic-Scale Simulations" Molecules 24, no. 10: 2014. https://doi.org/10.3390/molecules24102014
APA StyleAmhamed, A., Atilhan, M., & Berdiyorov, G. (2019). Permeabilities of CO2, H2S and CH4 through Choline-Based Ionic Liquids: Atomistic-Scale Simulations. Molecules, 24(10), 2014. https://doi.org/10.3390/molecules24102014