1. Introduction
Recent computational [
1,
2,
3,
4] and experimental [
5,
6,
7,
8] studies have called attention to the multiplicity of halogen [
9,
10,
11,
12,
13], chalcogen [
14,
15], pnicogen [
16], and tetrel bonds [
17,
18,
19] (corresponding to Groups 17, 16, 15, and 14, respectively) that appear necessary to supplement the familiar hydrogen bonds of intermolecular interaction theory [
20]. Such proliferating “bond” forms naturally reignite age-old controversies concerning the relative importance of “covalent” versus “electrostatic” contributions in the general theory of intermolecular forces [
21,
22,
23,
24]. Of course, all protagonists concur that the only relevant potential terms in the non-relativistic Hamiltonian are of Coulombic form (hence, tautologically “electrostatic” in nature). However, the deeper issue is whether supramolecular complexation (like molecule formation) is primarily governed by exchange-type (exponential) interactions of the short-range quantal regime or by the exchange-free (power–law) behavior of the long-range classical limit. In short, we ask whether orbital-based (quantal) or “dipole–dipole”-type (classical) conceptions provide the more useful starting point for describing intermolecular forces. For present purposes, the former is exemplified by a natural bond orbital [
25] (NBO)-based description, and the latter by is exemplified by symmetry adapted perturbation theory (SAPT) [
26,
27], electrostatic potential models (e.g., of CHELP [
28,
29] or “sigma hole” [
30,
31,
32] types), or related classical force field [
33,
34] models.
In the case of hydrogen bonding, the primacy of short-range covalency forces is now widely recognized [
20,
35,
36,
37,
38]. In the NBO framework [
39], a general B:···H—A hydrogen bond is identified with the strength of a
nB→σ*
AH donor–acceptor interaction between the lone pair (
nB) of the Lewis base B and the valence antibond (σ*
AH) of the Lewis acid A—H. In natural resonance theory (NRT) [
40,
41,
42,
43] terms, such a
nB→σ*
AH interaction represents resonance mixing between the parent natural Lewis structure (NLS)
I and the secondary “charge–transfer” resonance structure
II (with respective NRT weightings
wI and
wII):
in which the “covalent” (—) and “hydrogen” bond (···) symbols are interchanged [
44]. As recognized by Coulson [
45], the resonance hybrid (1a) is also the resonance-theoretic equivalent of the Pimentel–Rundle 3-center, 4-electron (3c/4e) molecular obital (MO) formulation of hypervalency [
46,
47]. In the simple two-resonance limit, the formal
bAH,
bBH bond orders are necessarily related by the characteristic “bond conservation” relationship [
48],
which merely reflects the resonance-averaging condition (
wI +
wII = 1) for the composite resonance hybrid. Even in the absence of NRT bond order descriptors, the importance of NBO
nB→σ*
AH delocalization can be assessed by 2
nd-order perturbative energy (Δ
E(2)nσ*), intermolecular charge-transfer (
Qnσ*), or the deletion of this NBO interaction (Δ
E$DELnσ*), as well as the variational recalculation of structure, energetics, and vibrational frequencies [
49]. In contrast to the superficial “on-off” picture of H-bonding that is sometimes suggested by graphical software or inflexible analysis methods, the NBO/NRT descriptors all reflect the continuously variable character of resonance-type bonding, as quantified by fractional bond orders ranging over integer and sub-integer values. All such characteristic NBO/NRT signatures are found in all known H-bonded species, including the paradoxical “anti-electrostatic” H-bonds between like-charged ions [
50] that most clearly demonstrate the relatively secondary influence of “electrostatic” modulation on the authentic H-bonding phenomenon.
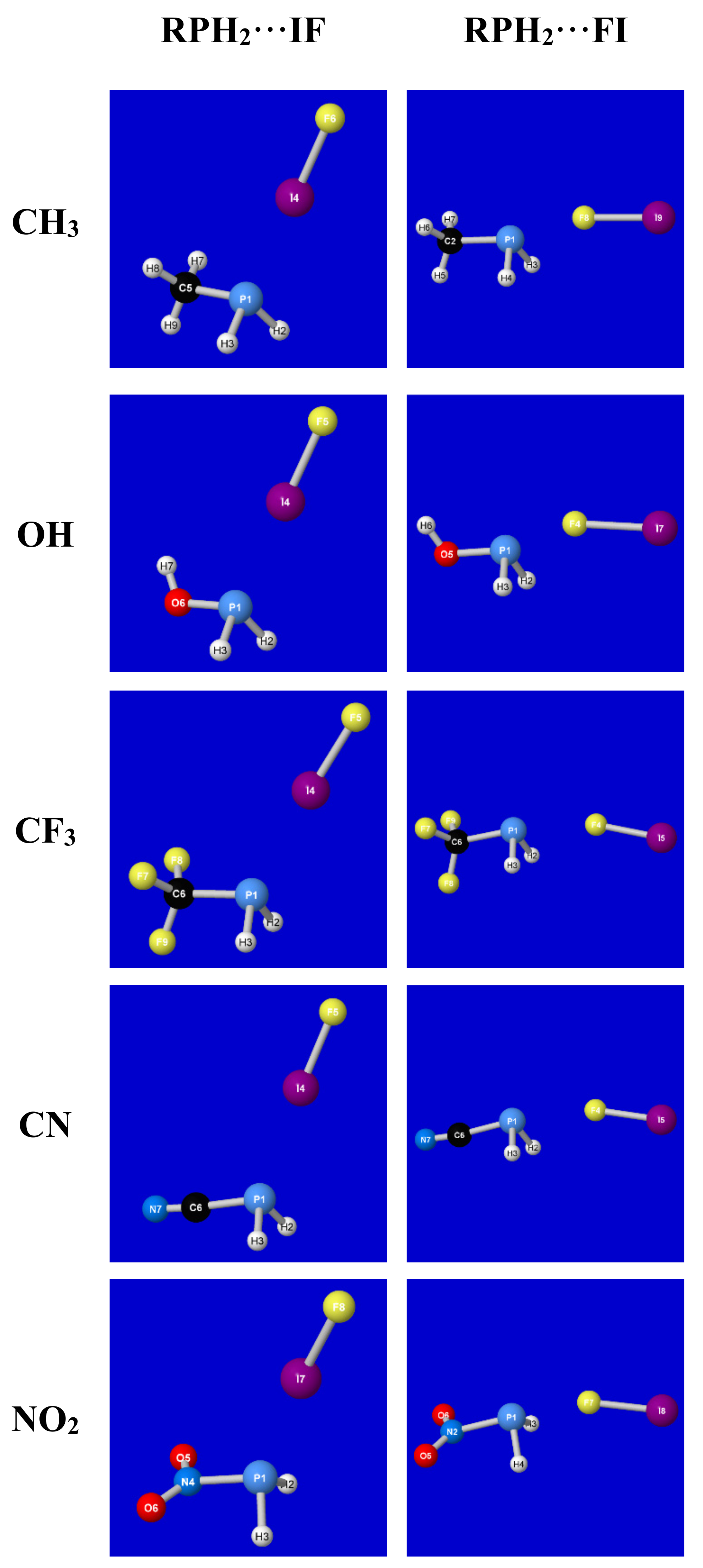
In the present work, we take up the analogous covalency versus electrostatics questions for title complexes RPH
2···IF/FI (R = CH
3, OH, CF
3, CN, NO
2) that exhibit a wide range of binding strengths. A smaller subset of such species was previously examined [
51] with an NBO/NRT analysis of MP2/aug-cc-pVTZ level computations. The present study extends the choice of species and the variety of NBO/NRT analysis tools to specifically address the broader questions of covalency versus electrostatics in halogen and pnicogen bonding, as well as the general relationships to H-bonding and other types of “X-ogen bonding” that signal a grand unified picture of all such donor-acceptor phenomena. In each case, we exhibit the mutually consistent correlations between NBO/NRT-based descriptors and experimentally measurable properties that illustrate the interpretative and predictive advantages of the unified NBO/NRT conceptual framework compared to alternative analysis methodologies [
25].
Note that we ignore the weaker effects of the London dispersion interaction [
52], a pure correlation effect that (unlike the
n-σ* interaction) is strictly absent in uncorrelated Hartree–Fock theory. Though an empirical dispersion correction [
53] is often found to improve the performance of density functional theory (DFT) calculations, such correction is only an additive term in the total energy expression, with no direct effect on Kohn–Sham orbitals, electron density, or other properties. Effects of this correction are therefore “invisible” to NBO/NRT analysis at any specific geometry, but, in principle, they can lead to shifts on the potential energy surface at which an analysis is performed. We evaluated the B3LYP-D3 properties for CH
3PH
2···IF (see
Supplementary Materials for full details) and found that the changes are relatively negligible in magnitude (smaller, e.g., than variations among alternative basis levels discussed below) as well as opposite in sign to that expected for the authentic London dispersion attraction (acting instead to reduce Δ
Ebind). Such “corrections” are not considered further in the present work.
In the following, we first describe the species, computational levels, and NBO/NRT methods to be employed. The latter include NBO Δ
E(2)nσ*, charge–transfer
Qnσ*, and
$DEL-deletion Δ
E$DELnσ* measures of donor–acceptor attraction [
49], the corresponding pairwise steric-exchange opposition (Δ
E(pw)nσ) of filled orbitals [
54] that completes the 3c/4e description of overall bonding energetics, and the various NRT-based regularities and correlations with measurable properties that are considered diagnostic of H-bonding [
22]. We then document the systematic application of these NBO/NRT methods to the title species to exhibit their mutual coherence and consistency with well-established features of H-bonding. The concluding summary emphasizes how a balanced NBO/NRT description of leading donor–acceptor interactions can lead to a unified conceptual picture of supramolecular bonding that encompasses the entire range of chemically significant (“non-innocent”) complexation and ligation phenomena.
2. Computational Methods and Results
Our studies began with a larger data set of dihalogen complexes RPH
2···XY that included all possible (X, Y = F, Cl, Br, I) dihalogen species of the first four periodic rows. However, from a qualitative conceptual perspective, it soon became apparent that the properties of such complexes vary in a smooth and chemically reasonable fashion with dihalogen polarity, as maximized in the iodine–fluorine (IF) species. Accordingly, we focus here on the “polar extremes” of RPH
2···IF versus RPH
2···FI complexation, corresponding to opposed signs of any envisioned “dipole–dipole” contributions to intermolecular binding. A direct comparison of these two extremes thereby allows one to recognize the important modulating influence of classical dipole–dipole forces (i.e., in strengthening or weakening the underlying quantal interactions), while also verifying that resonance-type covalency forces yield robust supramolecular bonding even when the presumed classical electrostatic prerequisites for such complexation are profoundly violated. While the computational results presented here focus only on the extremal RPH
2···IF versus RPH
2···FI limits of this broader picture, the
Supplementary Information (SI) includes results for all remaining dihalogen species to illustrate the generality of the resonance-type 3c/4e interaction picture to be sketched below.
Though the conceptual picture we describe is insensitive to many details of the chosen computational method and basis set [
55], the inclusion of iodine (for which common Pople-type 6-311++G** basis sets are unavailable) mandates the adoption of a relativistically corrected basis set (e.g., of the pseudopotential LANL2DZ type). For present qualitative purposes, we therefore adopt an unconventional but simple “mixed-PP” basis set of 6-311++G** for all RPH
2 monomers and LANL2DZ for dihalogen monomers of each complex. We employ simple B3LYP density functional methodology throughout this work, but alternative MP2 evaluations at different basis levels were also performed for comparison (see SI). All structures were optimized, and frequencies were computed to confirm equilibrium geometries corresponding to minimum energy points. The binding energy, Δ
Ebind =
E(AB) −
E(A) −
E(B), was calculated as the direct energy difference between optimized dimer and relaxed monomers. All electronic structure calculations were performed with the Gaussian 16 program package [
56]. NBO/NRT analyses were performed with
NBO 7.0 software [
57], and both NBO/NRT analyses and orbital imagery were obtained with the
NBOPro@Jmol program [
58].
Table 1 summarizes the key structural and binding energy parameters of RPH
2···IF and RPH
2···FI species as obtained at the adopted “B3LYP/mixed-PP” level, showing the wide range of tuning by various R-substituents. Despite the fact that all complexes are appreciably bound (by circa 3–15 kcal/mol, in defiance of superficial “dipole–dipole” or Coulombic point–charge expectations), interesting structural contrasts are immediately evident between these two classes of species. Focusing first on the orientation angles (Θ) in
Table 1, one can see that the RPH
2···IF species all maintain nearly linear P···IF alignment (Θ
PIF ≈ 180°) and a circa 120° bend angle between RP and IF axes (Θ
RPI ≈ 120°). In contrast, the RPH
2···FI species are noticeably tilted away from P···FI linearity (Θ
PFI ≈ 160°), instead adopting increasingly near-linear alignment with respect to RP and FI axes (Θ
RPF ≈ 170°). Such structural tendencies suggest that the principal underlying “linearizing” (H-bond-like) interaction differs in the two cases and that other RPH
2···XY dihalogen species of intermediate polarity will likewise exhibit intermediate orientational preferences between these two competing tendencies. Indeed, this is the broader picture obtained from the full set (X, Y = F, Cl, Br, I) of optimized dihalogen structures, with the homopolar RPH
2···X
2 species forming the approximate “dividing line” between RPH
2···IF-like versus RPH
2···FI-like structural propensity. These structural features of RPH
2···IF-like (“P···IF”) versus RPH
2···FI-like (“RP···F”) complexes are also consistently exhibited at other theory levels, as shown in SI
Tables S1–S3. All such comparisons indicate the adequacy of the adopted B3LYP/mixed-PP model and the extremal RPH
2···IF versus RPH
2···FI species for describing the full range of RPH
2···XY complexation (X, Y = F, Cl, Br, I; R = CH
3, OH, CF
3, CN, NO
2), including general consistency with previous MP2/aug-cc-pVTZ-level calculations.
Figure 1 depicts the numerical structural data of
Table 1 in graphical form, showing the distinct structural motifs that characterize each extremal “type” of association. Though the graphical imagery primarily allows for the visualization of the angular features of each complex, the progressive shortening of phosphorus–halogen bond length
RP···F or
RP···I (as well as concomitant elongation of dihalogen bond length
RIF) with increasing binding energy Δ
Ebind is also discernable in the details of the figure panels. From both
Table 1 and
Figure 1, one can recognize that the structural effects of different R-substituents are modest compared to that of reversing dihalogen polarity.
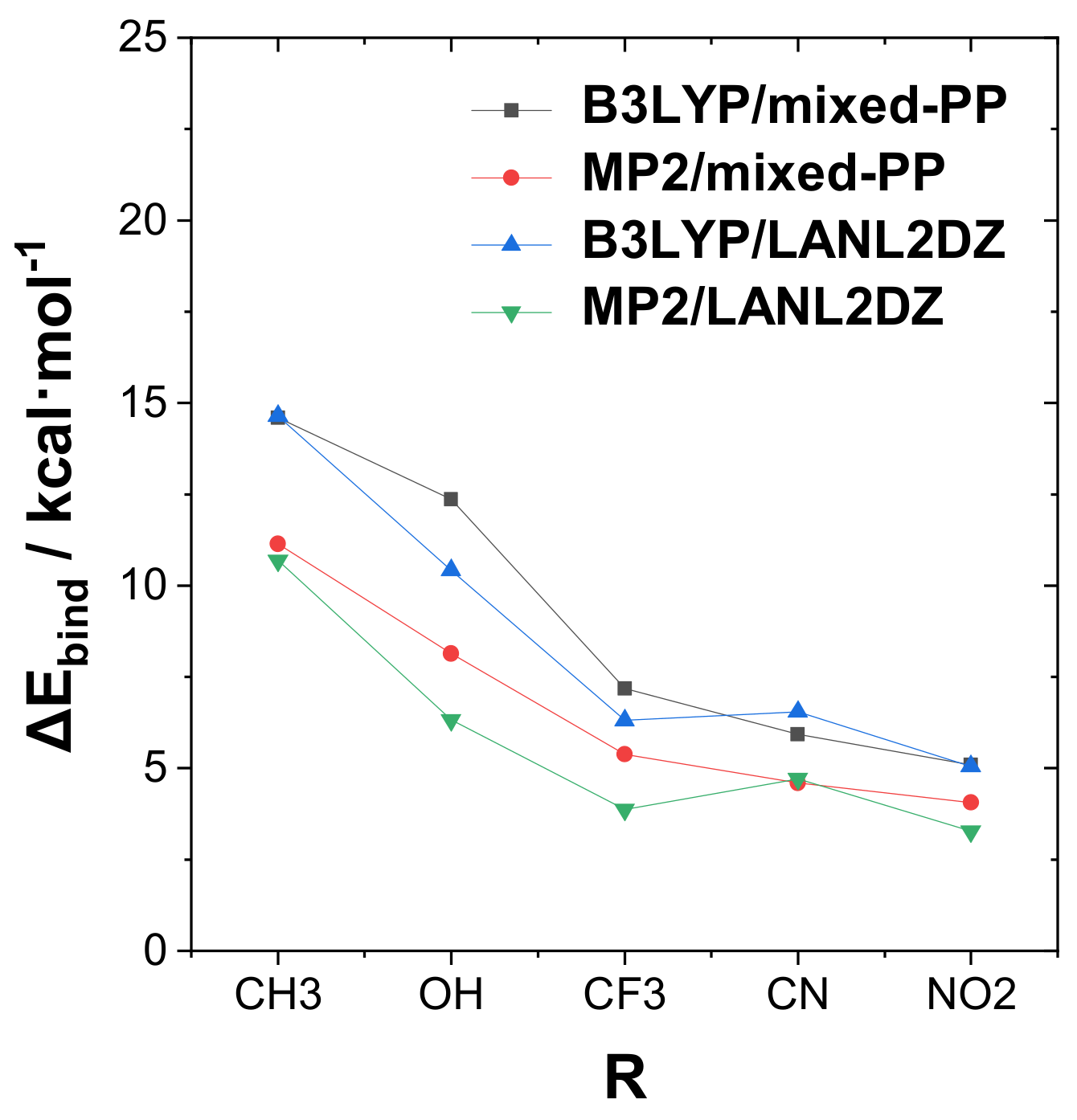
Figure 2 presents a graphical display of the trends in tuning binding energy Δ
Ebind with various R-substituents for the strongly bound complexes of the RPH
2···IF type. The graph includes comparison values for the alternative theory levels given in
Tables S1–S3 of SI. From
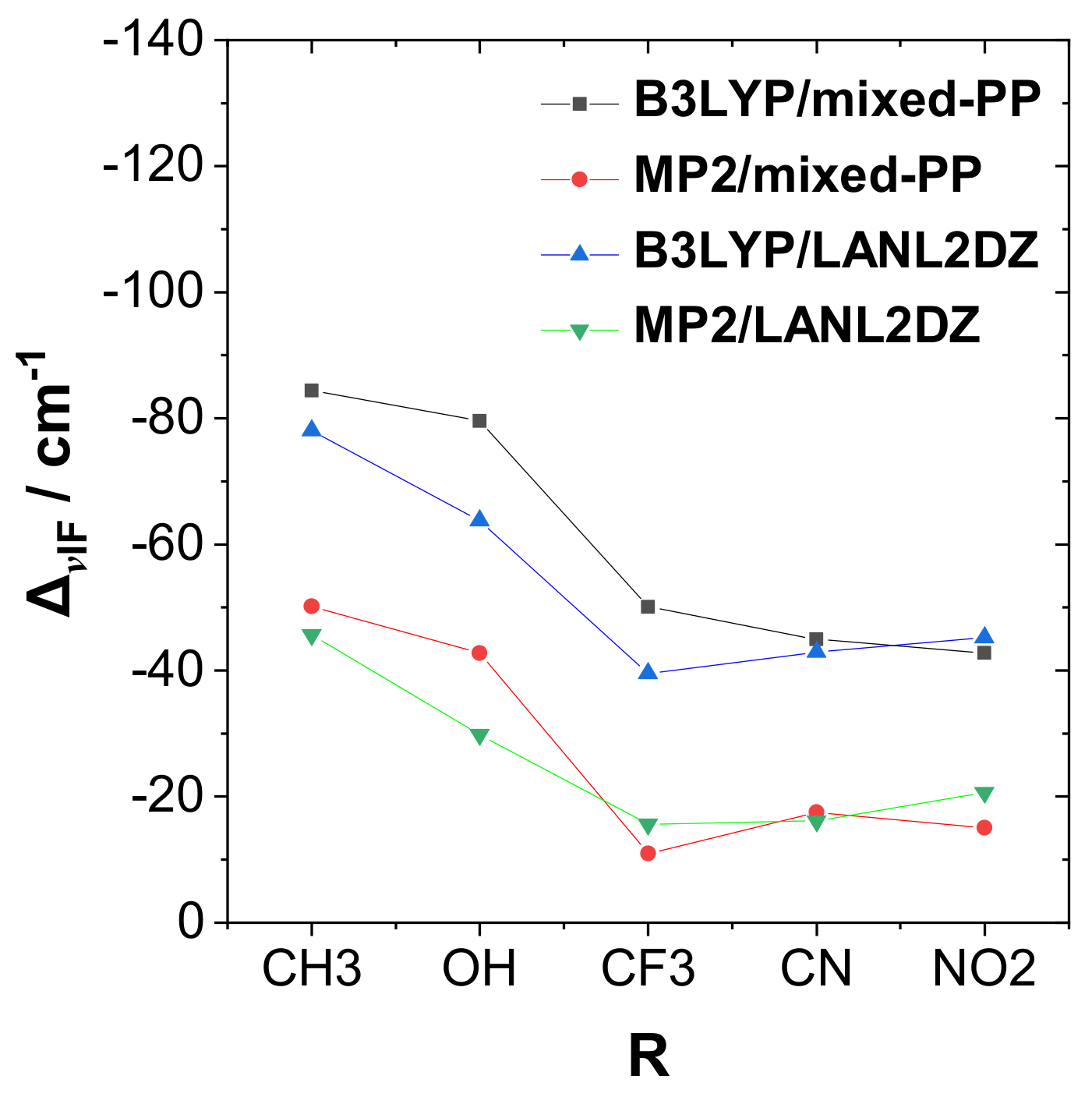
Figure 2, one can observe that significant quantitative differences in binding energies are calculated at the various theory levels, but the general trend with respect to the R-substituent variation is reasonably consistent in all cases. A slight exception is seen in the reduced cyano substituent effect in the “unmixed” B3LYP/LANL2DZ, MP2/LANLDZ theory levels, which may reflect the need for diffuse functions (present in the mixed-PP model but absent in uniform LANL2DZ) to better describe the strong electron-withdrawing effects of this substituent.
Figure 3 shows a corresponding plot of dihalogen frequency shift Δν
IF with respect to the R-substituent (including comparison results from alternative methods). As shown in the figure, Δν
IF is strongly red-shifted for the most strongly bound complexes, similar to the well-known spectroscopic signature of H-bonding.
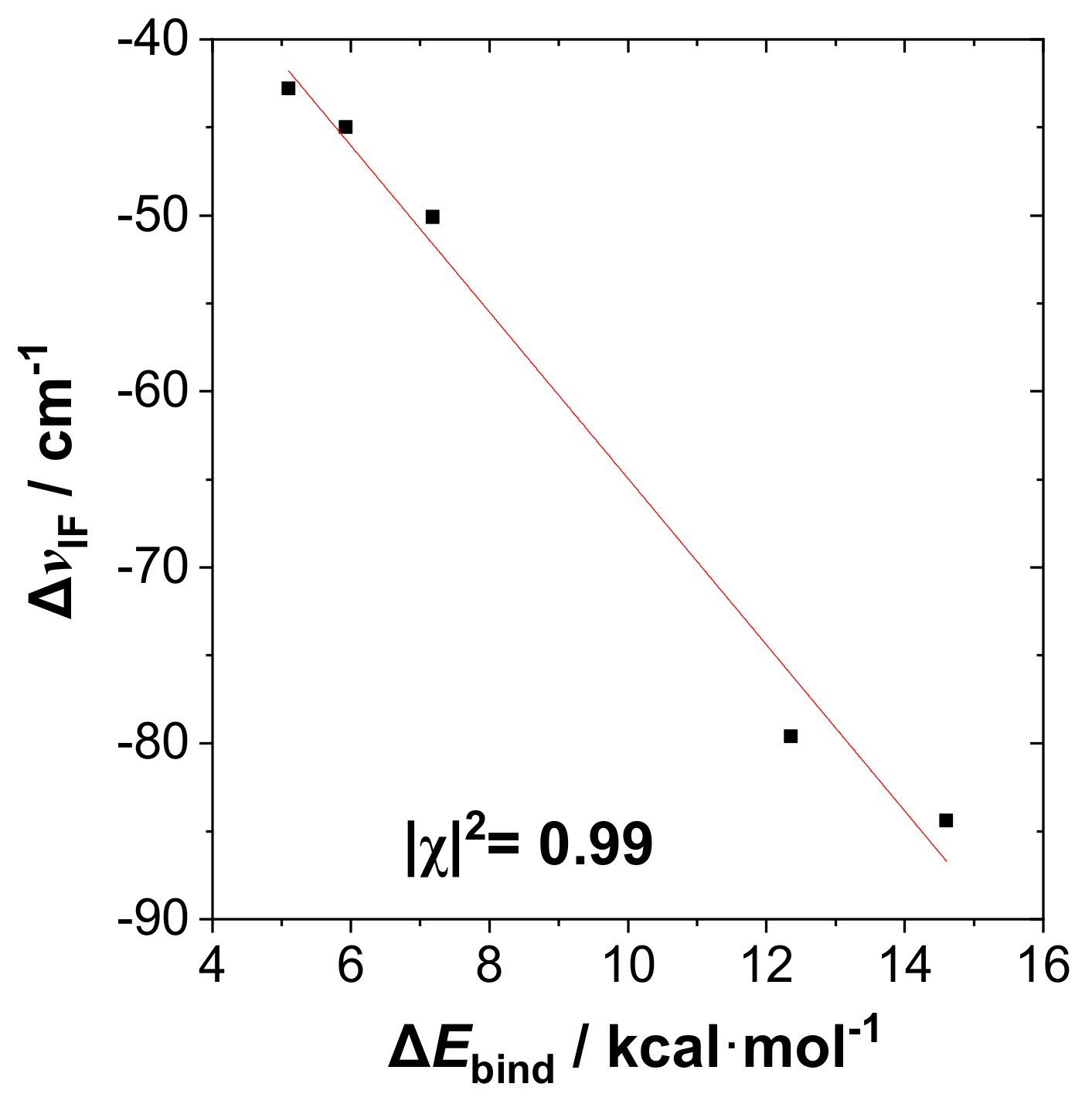
Figure 4 exhibits the correlation plot of Δν
IF (
Figure 3) versus Δ
Ebind (
Figure 2) in the RPH
2···IF species, showing the high correlation (Pearson |χ|
2 = 0.99) between these experimentally observable quantities that confirms the similarity to H-bonding.
Returning to the structural data of
Table 1 and
Figure 1 for the strongly bound RPH
2···IF complexes, one may note that the intermolecular P···I distances vary in the range 2.86–3.03Å.
Table 2 displays empirical van der Waals (vdW) radii (Pauling values [
59]) for the P, I, and F atoms of present interest, showing that the optimized
RP···I distances in these complexes are all more than 1Å inside nominal van der Waals contact. This deep penetration into the exponentially repulsive sub-vdW region implies the presence of powerful intermolecular attractive forces (also of exponentially increasing strength) to achieve the net binding energies displayed in
Table 1 and
Figure 2. In addition, the circa 0.2Å variation in penetration distance with the R-substituent testifies to the impressive strength of substituent effects that further modulate Δ
Ebind binding affinity and
RP···I separation in the deep sub-vdW region.
The computed structural and binding characteristics noted above identify the propensities and patterns to be explained and provide helpful clues to the nature of the underlying attractive forces. Even the more weakly bound complexes of the P···FI type exhibit optimized P···F distances (2.1–2.6Å) that lie significantly (>0.5Å) inside vdW contact (circa 3.2Å,
Table 2). Indeed, in both strong RPH
2···IF and weak RPH
2···FI complexes, the structural features hint at striking resemblances to familiar H-bonding phenomena, including deep sub-vdW penetration (with concomitant covalent bond
RIF elongation) and pronounced “linearization” tendency (albeit along different 3-center axes: P···IF in the RPH
2···IF case versus RP···F in the RPH
2···FI case). We take up NBO/NRT investigation of these apparent similarities and bonding relationships in the following sections.
3. NBO/NRT Descriptors
As suggested in (1a,b) above, an NBO/NRT analysis of B:···HA hydrogen-bonding phenomena reveals a host of characteristic correlations [
22,
23,
60] that implicate resonance-type
nB→σ*
AH “charge transfer” interactions as the fundamental electronic origin of H-bonding, consistent with the earliest results of NBO analysis [
61,
62,
63]. This conclusion also supports a considerable variety of other experimental and theoretical evidence [
64,
65,
66,
67,
68] for the charge–transfer nature of H-bonding. The question here is whether similar NBO/NRT diagnostics may provide analogous associations with
n→σ* (lone pair to valence antibond) donor–acceptor interactions of the present species.
The analogies of the preceding section suggest appropriate NBO/NRT descriptors for the corresponding ···X— (“X-ogen”) species that can be similarly visualized in simple orbital overlap terms. For RPH2···IF complexes, the key feature of envisioned P···I—F “halogen bonding” is the donor–acceptor delocalization of electronic charge from an occupied donor NBO of P (lone pair nP) to the favorably polarized acceptor NBO (antibond σ*IF) of the P···I—F triad. For RPH2···FI complexes, where the nP-σ*IF overlap is unfavorable, the analogous feature of envisioned F···P—R “pnicogen bonding” is the donation from a fluorine lone pair (nF) to the proximal antibond (σ*PR) of the phosphine monomer. Each such n→σ* delocalization can be alternatively quantified in terms of energetic (perturbative ΔE(2)nσ* or variational deletion ΔE($DEL)nσ*), charge transfer (Qnσ*), or fractional NRT bond order (bP···I or bP···F) descriptors.
3.1. Energy Descriptors
Table 3 summarizes estimates of the Δ
Enσ* stabilization energy associated with each type of delocalization in the strongly bound RPH
2···IF species. The first two columns display 2
nd-order perturbative Δ
E(2)nσ* estimates for
nP→σ*
IF (“halogen bond”) and
nI→σ*
PR (“pnicogen bond”) contributions. The final two columns of
Table 3 display corresponding Δ
E($DEL)nσ* variational deletion estimates, obtained as the variational energy raising when the specific
n→σ* NBO interaction is deleted from the total energy evaluation. The two estimates are seen to be in reasonable qualitative agreement for each interaction type. The Δ
E(2)n(P)→σ*(IF) values range from 19 to 37 kcal/mol (in the same order as Δ
Ebind), whereas Δ
E(2)n(I)→σ*(FI) is relatively negligible in each case, and a similar pattern is seen in the Δ
E($DEL)nσ* values. We therefore expect that the stabilization energy of the
nP→σ*
IF interaction is the dominant attractive contribution to the structure and binding of RPH
2···IF complexes, similar to the dominance of the
nB→σ*
AH interaction in H-bonding.
For the weakly bound RPH2···FI species, the situation is more complex. Due to the unfavorable polarization of σ*FI away from the nP lone pair, the single nP→σ*IF interaction that dominates bonding in RPH2···IF species becomes only one of several such competing contributions to net binding. Specifically, for the opposed F···P—R (pnicogen bonding) motif, three possible nF lone pairs (one of sigma type and two of pi type) and three σ*PY acceptors (one σ*PR and two σ*PH) are within proximal interaction range as potential contributors to a complex resonance mixture. A particular interaction such as nF(σ)→σ*PR therefore represents only one of the nine related n→σ* interactions that may exert leverage on angular structure and binding energy. In such a case, only a more nuanced resonance-type description (see the discussion of NRT bond orders below) can describe the complete bonding picture that provides useful correlations with experimentally measurable properties. We therefore defer further discussion of RPH2···FI complexes to a later subsection.
3.2. Graphical Orbital Overlap Imagery
A powerful aspect of NBO energetic descriptors is their intimate relationship to the intuitive visual imagery of the orbital overlap, the NBO counterpart of the well-known “Mulliken approximation” [
69,
70,
71,
72] that underlies semi-empirical MO theory. For such visualization purposes, “pre-orthogonal” NBOs (PNBOs) [
49] are employed that maintain free-space symmetries (cartoon-like “textbook” shapes) of non-interacting NBOs prior to the actual distortions of the molecular environment.
Table 4 displays such PNBO visualizations for the various donor–acceptor
n→σ* interactions of halogen (
nP→σ*
IF; first column) or pnicogen (
nI(π)→σ*
PR,
nI(σ)→σ*
PR; final two columns) types, with corresponding numerical Δ
E(2)nσ* values (
Table 3) inset in the panels.
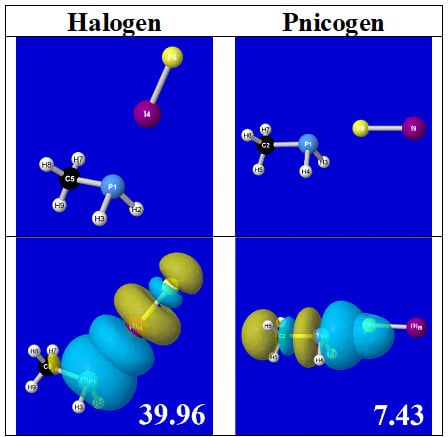
For example, in the three panels for CH
3PH
2···IF (first row of
Table 4) one can see that the “end-on” (sigma-type) overlap of σ*
IF with the nitrogen lone pair
nP (left panel) is much stronger than the “edge-on” (pi-type) overlap of σ*
PR with the in-plane iodine
nI(π) (middle panel) or
nI(σ) (right panel) lone pairs, in accordance with the Δ
E(2)nσ* values shown in each panel. Even a casual glance at the overall patterns of the orbital overlap in
Table 4 can therefore make clear why the slight variations of orbital shape with R-substitution do not materially alter the strong propensity for halogen-type (
nP→σ*
IF) rather than pnicogen-type (
nI(π)→σ*
PR,
nI(σ)→σ*
PR) bonding throughout the RPH
2···IF series, as numerically displayed in
Table 3.
Though the donor–acceptor Δ
Enσ* stabilization energies of
Table 3 display a qualitative ordering pattern similar to that of the Δ
Ebind binding energies of
Table 1, it is evident that these estimates significantly “overshoot” the final net binding energy. This is to be expected, because each stabilizing donor–acceptor interaction is opposed by steric repulsions of the corresponding Δ
Enσ donor–donor interaction.
Table 5 displays orbital overlap diagrams and inset values of the pairwise estimates Δ
E(pw) n-σ for repulsive “steric exchange energy” [
54] between occupied NBOs of the RPH
2···IF series. As shown in the panels, the two leading intermolecular repulsions refer to the
nP-
nI steric clash between lone pairs of proximal P and I atoms (left) and
nP-σ
IF clash between a lone pair and bond of the halogen bond P···I—F triad. However, weaker
nI-σ
PR,
nI-σ
PH clashes of the alternative pnicogen bond triads (I···P—R, I···P—H) also contribute to sub-vdW steric opposition, thereby further offsetting the apparently “excessive” NBO estimate of donor–acceptor attraction needed to achieve the final equilibrium geometry and net binding energy.
PNBO visualizations for occupied
nP,
nI, and σ
IF NBOs (not shown) similarly serve to rationalize the trends in repulsive donor–donor interactions (
Table 5). Furthermore, it becomes evident from graphical comparisons that all such orbital visualizations for the presently considered species are highly analogous to those for the corresponding interactions of H-bonding [
22], which is in accordance with the patterns and relationships inferred from numerical descriptors.
3.3. Structural, Energetic, and Spectroscopic Effects of Donor–Acceptor Deletion
Still another widget from the NBO toolbox can be used to exhibit the profound effects of
nP→σ*
IF delocalization on supramolecular bonding properties. Similar to the variational
$DEL-deletion Δ
E($DEL)n(P)→σ*(IF) evaluations of
n→σ* donor–acceptor strength in the equilibrium species (
Table 3), one can variationally reoptimize the structure for the associated
E($DEL) n(P)→σ*(IF) potential energy surface in which
n→σ* delocalization is absent, as though nature (in accordance with common textbook presentations) failed to include such interactions.
Table 6 displays results for the variational reoptimization of a simple 2D model of RPH
2···IF dissociation, with the
nP→σ*
IF interaction deleted. Starting from the equilibrium values of the actual RPH
2···IF species, the distance variables
RP···I,
RIF (for fixed idealized angular geometry) were optimized to find the new binding energy (Δ
Ebind) and shifts (Δ
RP···I, Δ
RIF, ΔΔ
Ebind) that describe the hypothetical “resonance-free” RPH
2···IF that lacks
nP→σ*
IF donor–acceptor attraction. From the resulting values, one sees that characteristic structural and energetic signatures of RPH
2···IF bonding are lost when the
nP→σ*
IF interaction is “turned off;” the RPH
2 and IF monomers retreat (by circa 1.3Å) to beyond-vdW separation, the characteristic
RIF elongation (associated with vibrational red-shifting) from isolated monomer geometry essentially disappears, and the net binding energy drops precipitously (by >80%) to a remnant value that might plausibly be associated with residual “dipole–dipole” forces. Note that the monomer dipole moments or other aspects of electron density distribution are scarcely altered in the
$DEL-reoptimized geometry, and the “lost” energy of
nP→σ*
IF stabilization is negligibly small (1.59 kcal/mol) at the reoptimized separation distance. From the results of such
$DEL-deletion calculations, one recognizes
nP→σ*
IF stabilization to be the unique “smoking gun” that is both necessary and sufficient to bring the supramolecular complex to the actual short-range geometry and other signatures of halogen bonding, which is in direct correspondence to the analogous
nB→σ*
HA role in H-bonding.
Still another use of
$DEL-deletion techniques is to prepare relaxed-scan potential curves that illustrate how intermolecular
n→σ* interactions lead to net binding against the essentially repulsive potential curve for hypothetical monomers that interact without benefit of resonance-type stabilization.
Figure 5 illustrates such potential curves for CH
3PH
2···IF dissociation, showing the potential curve for the fully interacting monomers (squares, black line, “Δ
Ebind”; cf.
Table 1) versus that for “resonance-free” monomers that lack intermolecular
n→σ* interactions (triangles, blue line, repulsive “Δ
Er”). The “missing” resonance-type (purely attractive) interactions are shown as the difference curve (diamonds, red line, attractive “Δ
Ea”). The blue curve exhibits a feeble attractive well beyond vdW contact at
RP···I ≈ 4.0Å (presumably due to “dipole–dipole” attraction) but rises steeply as a repulsive steric wall in the sub-vdW region. Already in the region of the initial sub-vdW penetration, the red curve (
n→σ* resonance attraction) has achieved sufficient stabilization to oppose thermal fluctuations (
kT ≈ 0.6 kcal/mol at 300K) and yield significant sub-vdW binding in the black (full potential) curve down to
RP···I ≈ 2.9 equilibrium separation. Inside this equilibrium distance, the intermolecular
n-σ steric repulsion overcomes
n-σ* attraction to give the steeply repulsive inner wall of the full potential, resonance-shifted inside circa 2.5Å. Where such
nP-σ
IF versus
nP-σ*
IF “cross-over” occurs will evidently depend on the polarization of the dihalogen bond and is therefore expected to shift to a progressively larger
RP···I separation (and weakened Δ
Ebind) for other members of the dihalogen series, thereby appearing as a secondary “electrostatic” modulating effect on the overall halogen bonding phenomenon.
3.4. Charge and Polarity Descriptors
Table 7 displays various NBO charge/polarization descriptors for the RPH
2···IF species (net intermolecular charge transfer
QCT, atomic charges
qP,
qI, and
qF of the P···I—F triad, IF bond ionicity
iIF [
73]), compared with those for isolated IF monomer. As shown for the dihalogen monomer in the final column of the table, I–F bond ionicity (−1 ≤
iIF ≤ +1, related to dipole moment direction from I to F) tends to increase steadily with strength of RPH
2···IF binding or net
QCT. This trend reflects the expected dominance of the
nP→σ*
IF interaction, which is evidently enhanced (cf. overlap diagrams of
Table 5) if σ
IF bond ionicity increases toward F, thereby polarizing the σ*
IF antibond toward I (and adjacent
nP). The combined effect of repolarization (
iIF) and net charge transfer (
QCT) is to give a somewhat irregular pattern to individual atomic charges
qI,
qF, but all such IF charge descriptors are seen to properly “add up,” as they must according to the strict logic of a natural population analysis.
In the RPH2 monomer, the phosphorus charge qP reflects the still more complex effects of sigma-type induction versus intra- and intermolecular lone pair delocalizations. One can see in the table the evident effect of bonding the phosphorus to more electronegative oxygen (qP ≈ 0.73) or nitrogen (qP ≈ 0.45) rather than carbon (qP ≈ 0.3–0.4). However, also important in the present context is the intra- versus intermolecular competition for charge donation from the “busy” phosphorus lone pair nP to various pi-type acceptor orbitals of the R-substituent (e.g., πCN, πNO) versus the intermolecular nP→σ*IF interaction of principal interest.
3.5. NRT Bond Order Descriptors
Perhaps the most useful and general descriptors of supramolecular bonding are NRT bond orders {
bAB}, which balance the nuances of multiple resonance structure contributions to give a single composite measure of A···B “connectivity.” As described elsewhere [
40,
41,
42,
43], NRT bond orders differ from those of the Pauling type, particularly in their close association with the first-order reduced density matrix [
74] (rather than assumed valence bond (VB) wavefunction) of the system. A vastly improved convex solver [
75] for the NRT variational objective function is a leading feature of the
NBO 7.0 program [
57].
The first three columns of
Table 8 show calculated NRT bond orders for the key P/I/F atoms in both the RPH
2···IF and RPH
2···FI series. The values vary rather continuously over the range 0 ≤
bAB ≤ 1, with no apparent “dividing line” between what should be described as the “covalent bond” (stick) versus the “noncovalent interaction” (dots). Most surprising in these species are the non-vanishing bond orders for the “long-bond” [
76,
77,
78] interactions of P^F (in P···I—F triads) or P^I type (in P···F—I triads). Though negligibly small in the most strongly bound RPH
2···IF species, certain P^I long bonds achieve bond orders that rival or exceed those of familiar
bP···F or
bIF linkages (e.g., in OHPH
2···FI).
The final two columns of
Table 8 show two-term or three-term bond order sums that test the validity of resonance-type “bond order conservation” [
48] rules (cf. (1a,b) for H-bonding). The two-term sum (column 4) exhibits significant deviations from the expected unit value in a simple two-resonance model for a mono-valent central atom of the 3c/4e triad. These deviations indicate the need for a three-term sum (three-resonance model) that includes long-bond contributions (column 5). Remaining deviations from unity in the three-term sums of column 5 indicate additional conjugative or hyperconjugative interactions with the R-substituent that require still higher-order resonance couplings. All these results suggest the close relationship of NRT bond orders to the familiar resonance-theoretic concepts of the organic chemist, as well as their usefulness in expected empirical correlations with experimentally measurable structural, energetic, and spectroscopic properties, as discussed in the following section.
4. Mutual Correlations of NBO/NRT Descriptors with Measurable Properties
The principal NBO descriptors of donor–acceptor interactions (such as ΔE(2)nσ*, ΔE($DEL)nσ*, Q nσ*,...) intrinsically focus on orbitals of a specific n→σ* “delocalization” from a specific Lewis structural bonding pattern. Such orbital-specific descriptors are valuable when a single “parent” Lewis structure and “child” n→σ* delocalization clearly dominate quantum mechanical descriptions of the structural, energetic, and spectroscopic properties of the chosen system. However, for many chemical systems of interest (including the RPH2···FI species considered here), multiple n→σ* interactions come into play, corresponding to contributions of alternative resonance–structural bonding patterns that may no longer be “child-like” compared to a reference parent pattern. Such cases demand the more nuanced NRT descriptors such as interatomic bond orders {bAB} that balance the many possible orbital interactions contributing to the overall resonance hybrid.
As could be anticipated from the empirical origins of bond order descriptors in the pre-quantum mechanical conceptions of “mesomerism” theory [
43], the “averaged” (fractional) character of NRT bond orders {
bAB} and their association with atoms (rather than orbitals) makes them better adapted to describe empirical correlations with measurable properties. Well-known examples include bond order–bond length (
bAB-
RAB) [
79], bond order–bond energy [
80], and bond order–bond frequency (Badger’s rule) [
81] relationships. Here we wish to briefly examine the mutual correlations of NBO/NRT descriptors in the single strong
n→σ* limit (where orbital-type descriptors may suffice) as well as the more general correlations of NRT bond orders with experimentally measurable properties, spanning the full set of RPH
2···IF/FI species considered in this work.
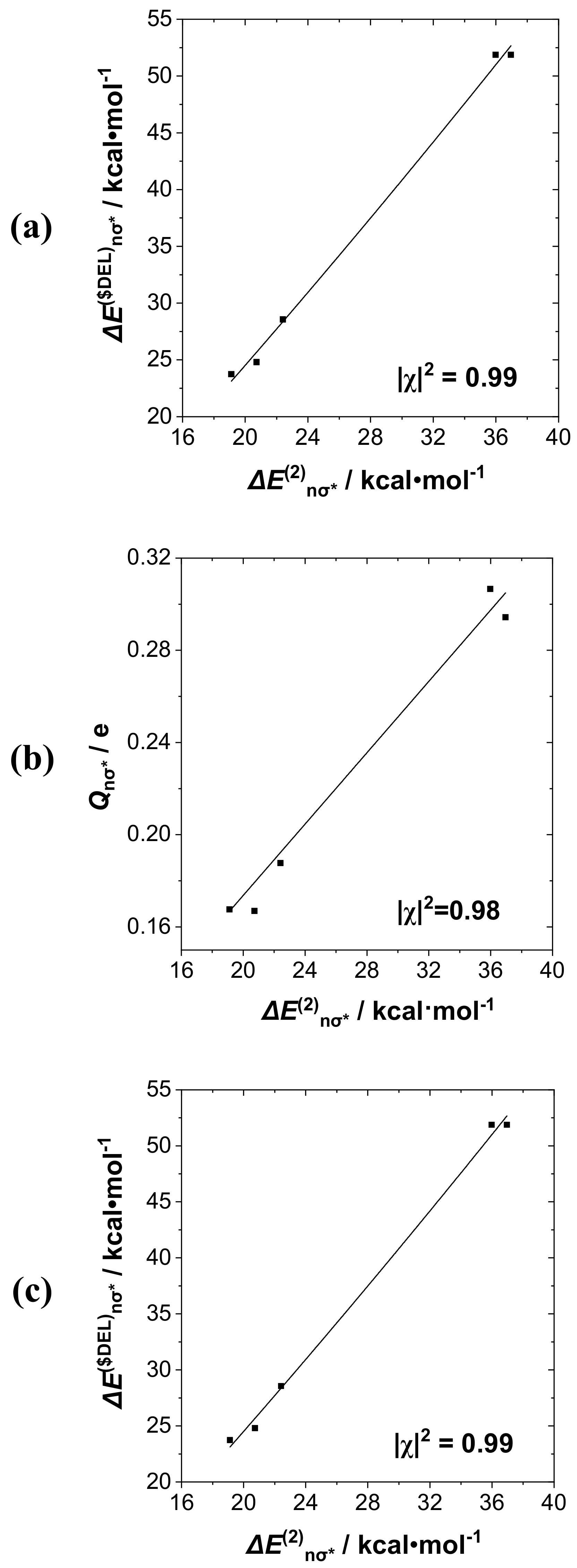
Starting with the RPH
2···IF series, in which the
nP→σ*
IF interaction plays a clearly dominant role, we first consider the mutual correlations of Δ
E(2)nσ*, Δ
E($DEL)nσ*,
Qnσ*,
bP···I descriptors for this interaction. Taking perturbative Δ
E(2)nσ* as the base descriptor for the group, we display (
a) Δ
E(2)nσ*-Δ
E($DEL)nσ*, (
b) Δ
E(2)nσ*-
Qnσ*, and (
c) Δ
E(2)nσ*-
bP···I correlation diagrams in successive panels (
a)–(
c) of
Figure 6. The displayed least-squares regression lines and Pearson |χ|
2 correlation coefficients demonstrate the high quality of these mutual correlations: In the range 0.94–0.99 for NBO-specific descriptors, but slightly lower (0.94) for correlation with the NRT bond order
bP···I (presumably due to the proper inclusion of secondary donor–acceptor interactions with R-substituents that are present only in the latter). The results show that descriptors of this group could be chosen rather interchangeably for correlations with measurable experimental properties when a single donor–acceptor interaction such as
nP→σ*
IF is clearly dominant.
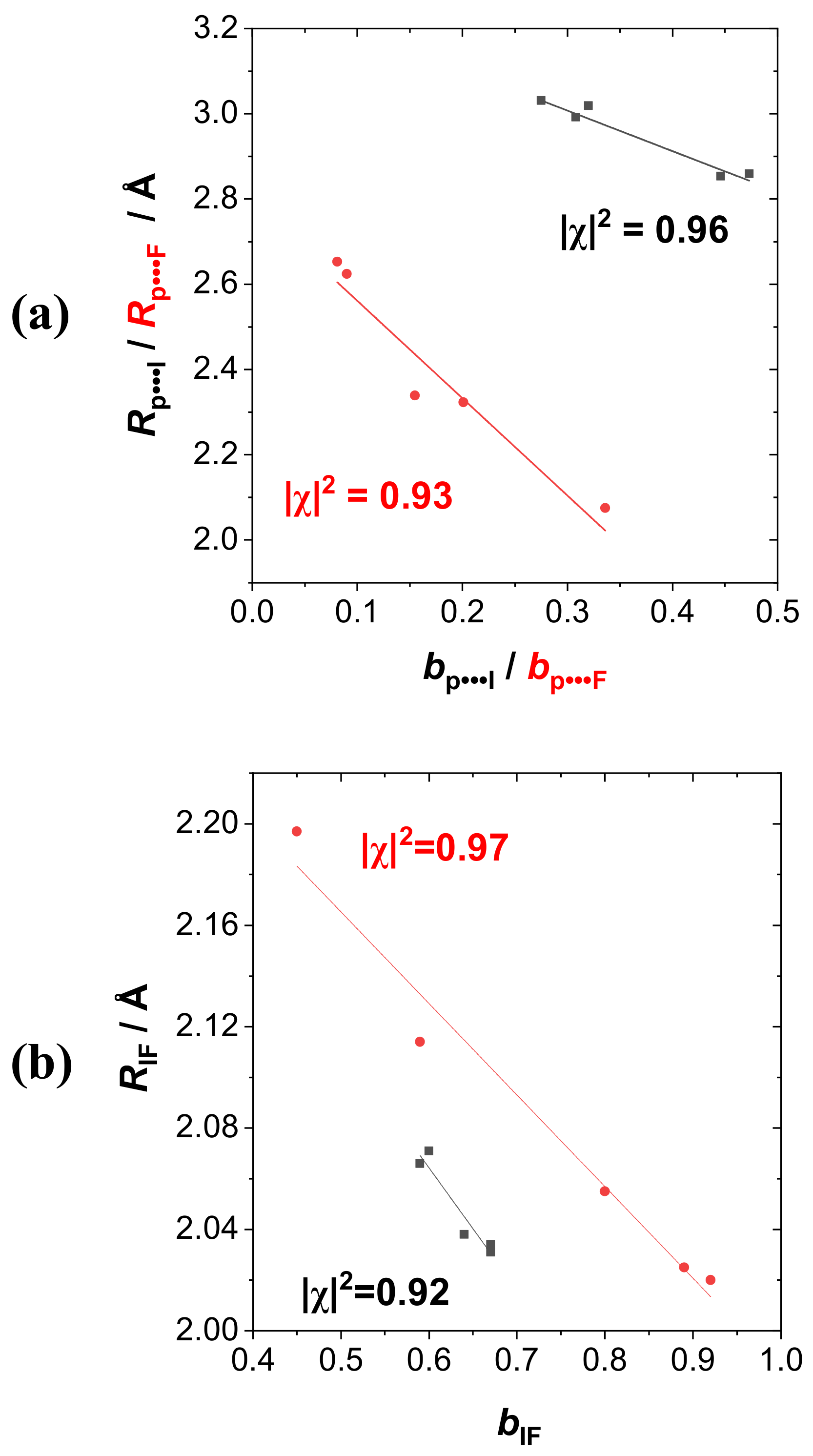
For more general correlations with measurable experimental properties such as (a) intermolecular
RP···I or
RP···F, (b) intramolecular
RIF, (c) binding energy Δ
Ebind, or (d) infrared stretching frequency ν
IF, we employed the NRT bond order
bAB (of specified atoms A, B) as base descriptor in the correlation diagrams of
Figure 7a–d, extending the correlations to both RPH
2···IF (black; approximate two-resonance) and RPH
2···FI (red: Multi-resonance) series. As examples of bond orderbond length correlations,
Figure 7a displays the intermolecular
bP···X-
RP···X correlations (X = I, F) for each series, with reasonably high correlations (|χ|
2 = 0.92) in each case.
Figure 7b similarly displays the intramolecular
bIF-
RIF correlation for the dihalogen monomer, with still higher correlation coefficients (|χ|
2 = 0.94 for RPH
2···IF, |χ|
2 = 0.98 for RPH
2···FI species). As examples of bond order–bond energy correlation,
Figure 7c displays the
bP···X-Δ
Ebind correlations (X = I, F), with similar correlation coefficients (|χ|
2 = 0.94) in each series. Finally, as bond order–bond frequency examples,
Figure 7d displays the
bIF-Δν
IF correlations, again with high correlation coefficients (|χ|
2 = 0.95 for RPH
2···IF, |χ|
2 = 0.98 for RPH
2···FI species). All these correlations suggest the high predictive utility of NRT bond orders for both the tuning effects of R-substituents and the polarity variations of the various dihalogen monomers that govern the broad range of binding energies in these supramolecular species.
5. Summary and Conclusions
We have computationally investigated the nature of supramolecular bonding in a series of R-substituted phosphine···dihalogen complexes (RPH2···IF/FI, R = CH3, OH, CF3, CN, NO2), focusing on the electronic origin of what might be identified as a “halogen bond,” “pnicogen bond,” or some combination of both. For this purpose, we employed a broad variety of natural bond orbital (NBO) and natural resonance theory (NRT) descriptors, searching for relationships to hydrogen bonding or other known forms of intermolecular attraction (often loosely termed “noncovalent”).
Our results show that resonance-type “
n→σ*” delocalization (leading to fractional intermolecular bond orders) is the dominant feature of halogen-type (e.g.,
nP→σ*
IF) or pnicogen-type (e.g.,
nF→σ*
PR) bonding motifs, just as for the dominant
nB→σ*
HA interaction of B:···H—A hydrogen bonding. We obtained evidence both for resonance-type “bond conservation” rules and the complete set of mutually consistent correlations of NBO/NRT descriptors with experimentally measurable properties of RPH
2···IF/FI complexes that closely parallel those previously demonstrated for H-bonded complexes. We also demonstrate that the removal of
n→σ* interactions obliterates the signature features of halogen/pnicogen bonding, whereas such features persist (albeit in muted form) even if envisioned “dipole–dipole” contributions are reversed (by reversing dihalogen monomer direction). Thus, our results establish that resonance-type
n→σ* stabilization is both necessary and sufficient for the characteristic structural, energetic, and spectroscopic features of halogen or pnicogen bonding, as was previously demonstrated for H-bonding [
22,
23]. The present results are fully consistent with previous studies of orbital-interaction effects in halogen or pnicogen bonding [
82,
83]. Connections can also be seen to more nuanced interpretation of the “σ-hole” in terms of chemical “lone pair” interactions with the “extension of one of the covalent bonds,” “dative sharing (coordinate covalence),” and other quantal phenomena [
84].
Based on these results, we anticipate that similar unique associations with resonance-type n→σ* “fractional bonding” will be found for chalcogen bonds, tetrel bonds, and all other such supramolecular bonding phenomena. Our conclusions thereby extend “resonance covalency” concepts to the entire supramolecular domain of sub-integer bond orders, challenging the common “electrostatic” assumptions that underlie current empirical force field modeling and textbook expositions of supramolecular chemistry.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
























