Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Side by Side Comparison of Synthetic Methodologies
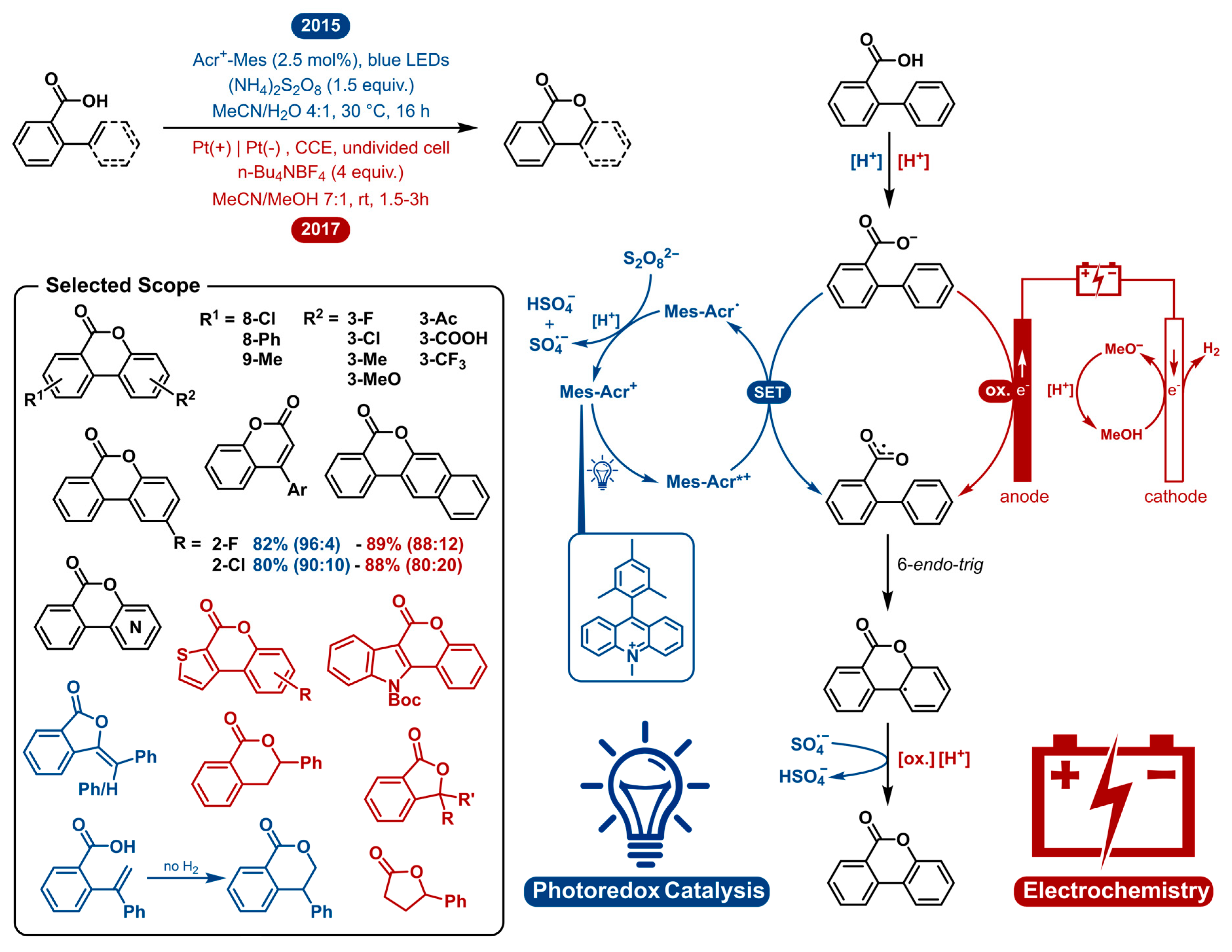
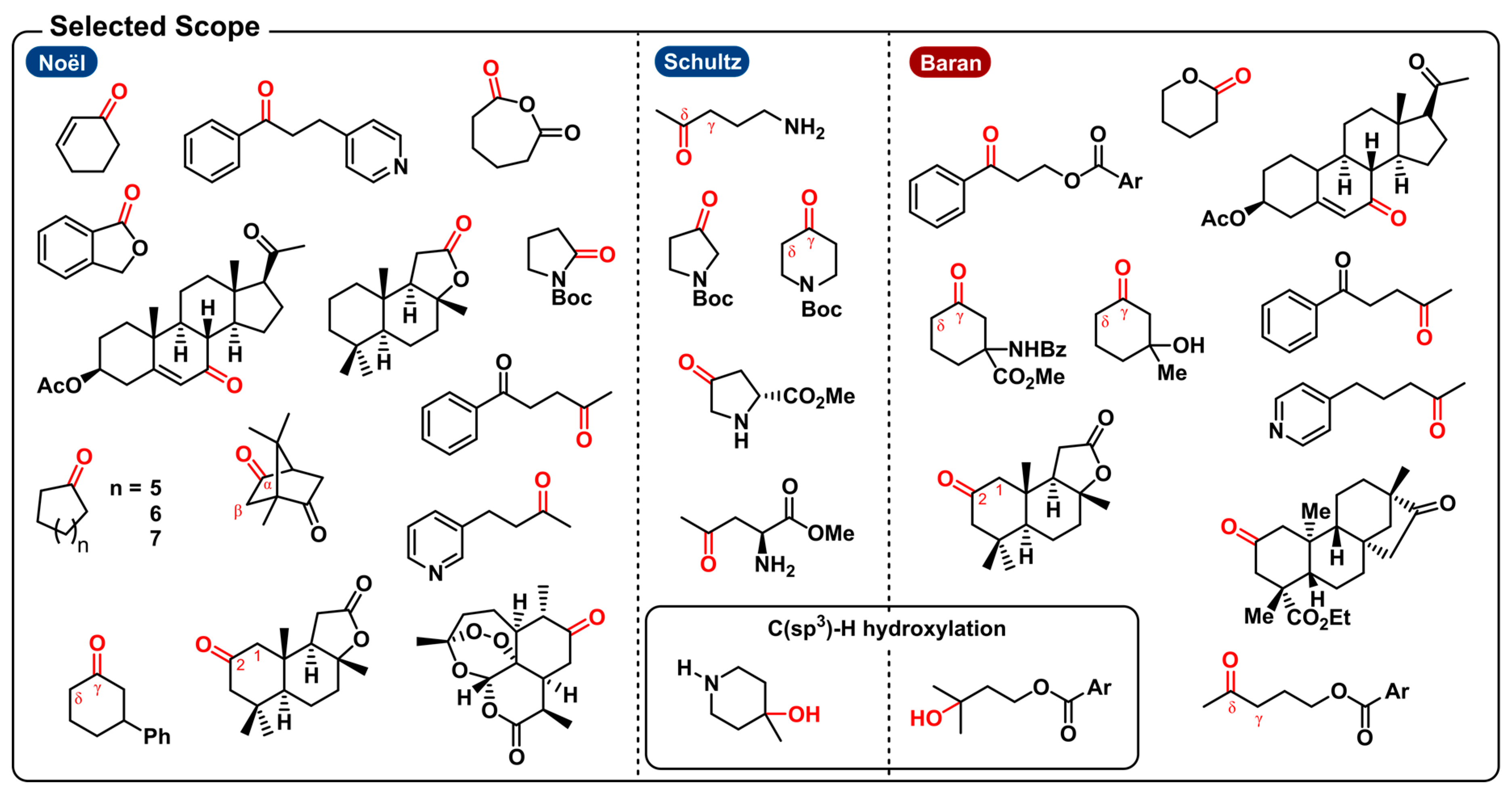
2.1. Dehydrogenative Lactonization of C(sp2/sp3)-H Bonds
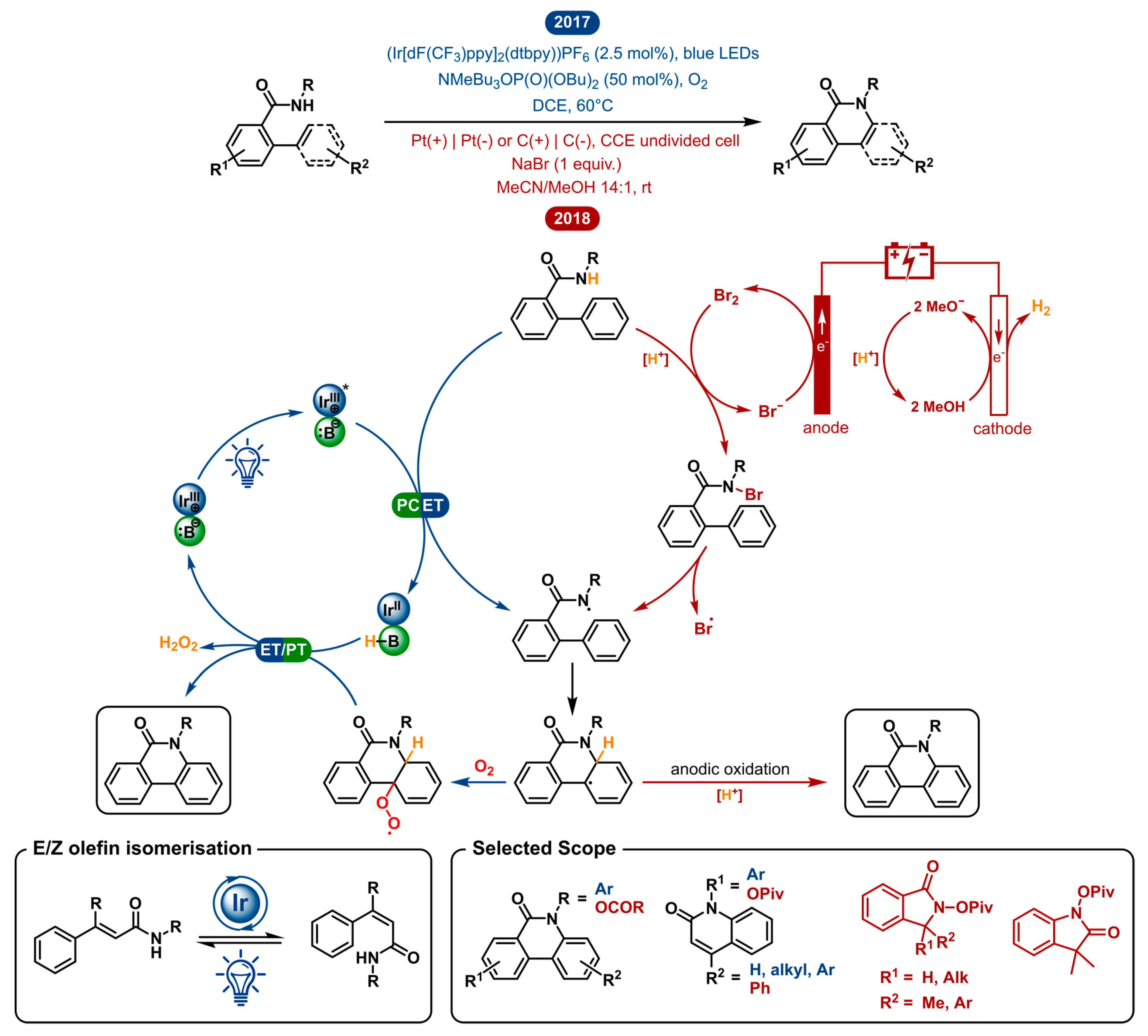
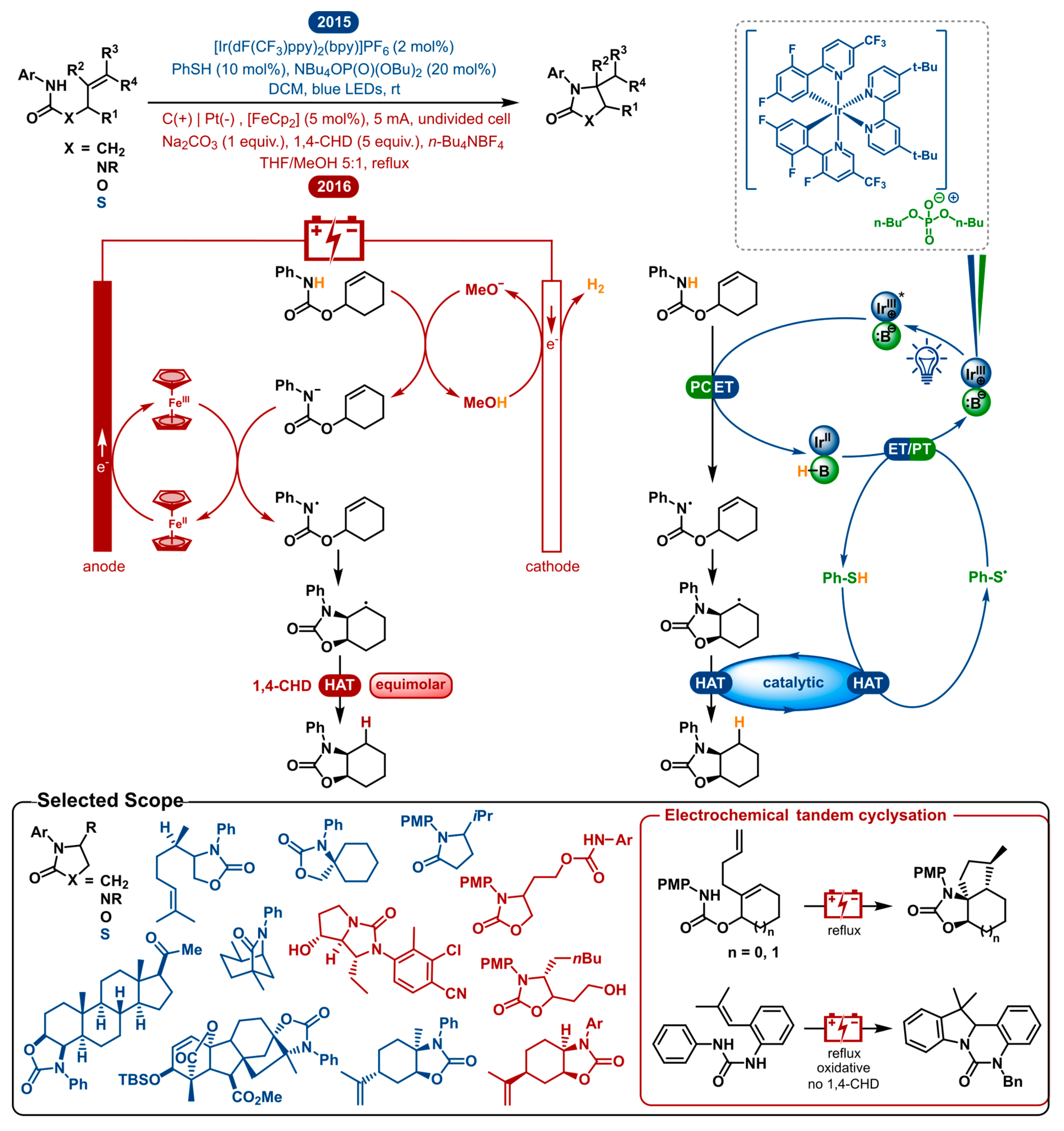
2.2. Dehydrogenative Lactamization
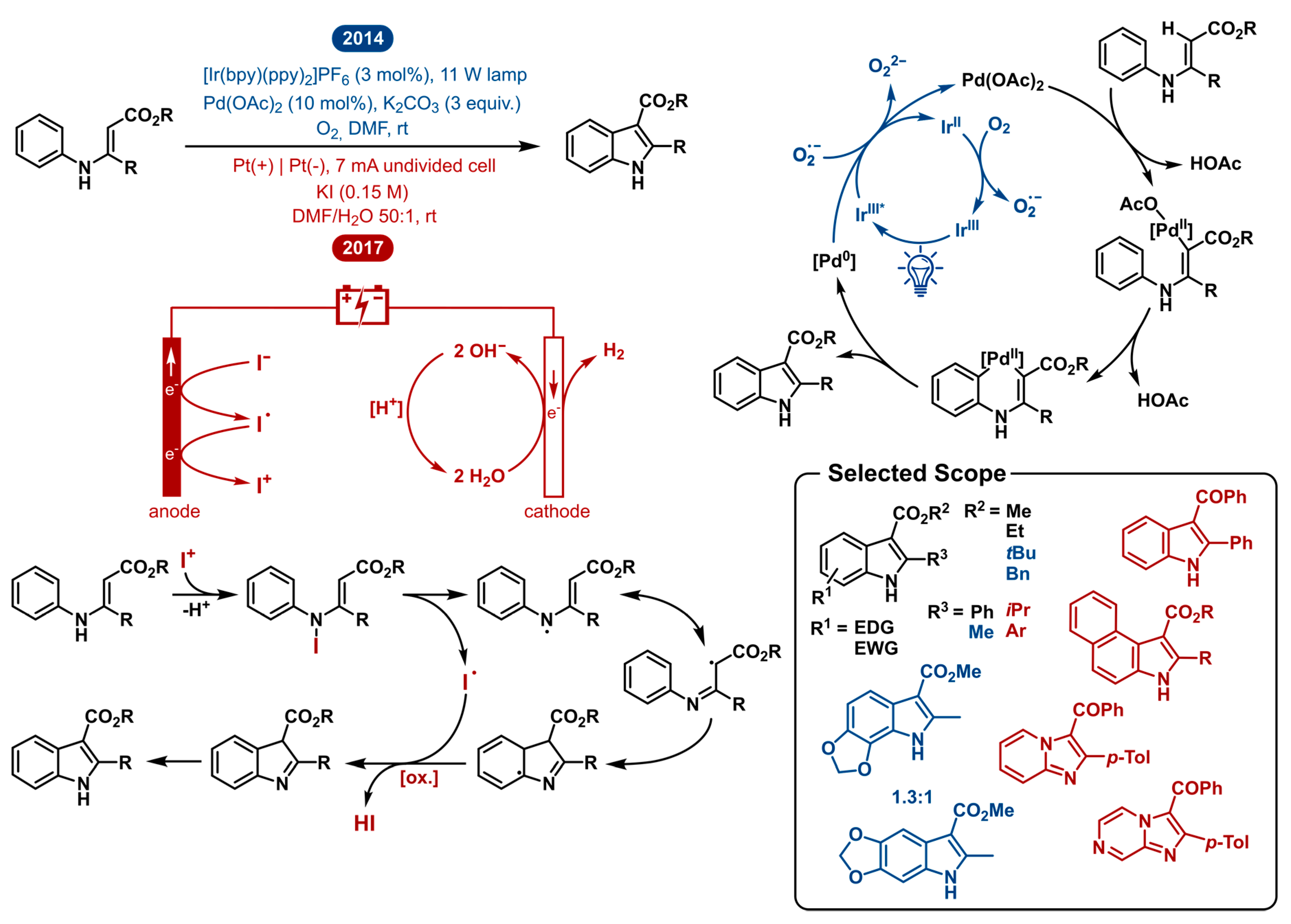
2.3. Intramolecular Oxidative Annulation of N-Aryl Enamines: Indole Synthesis
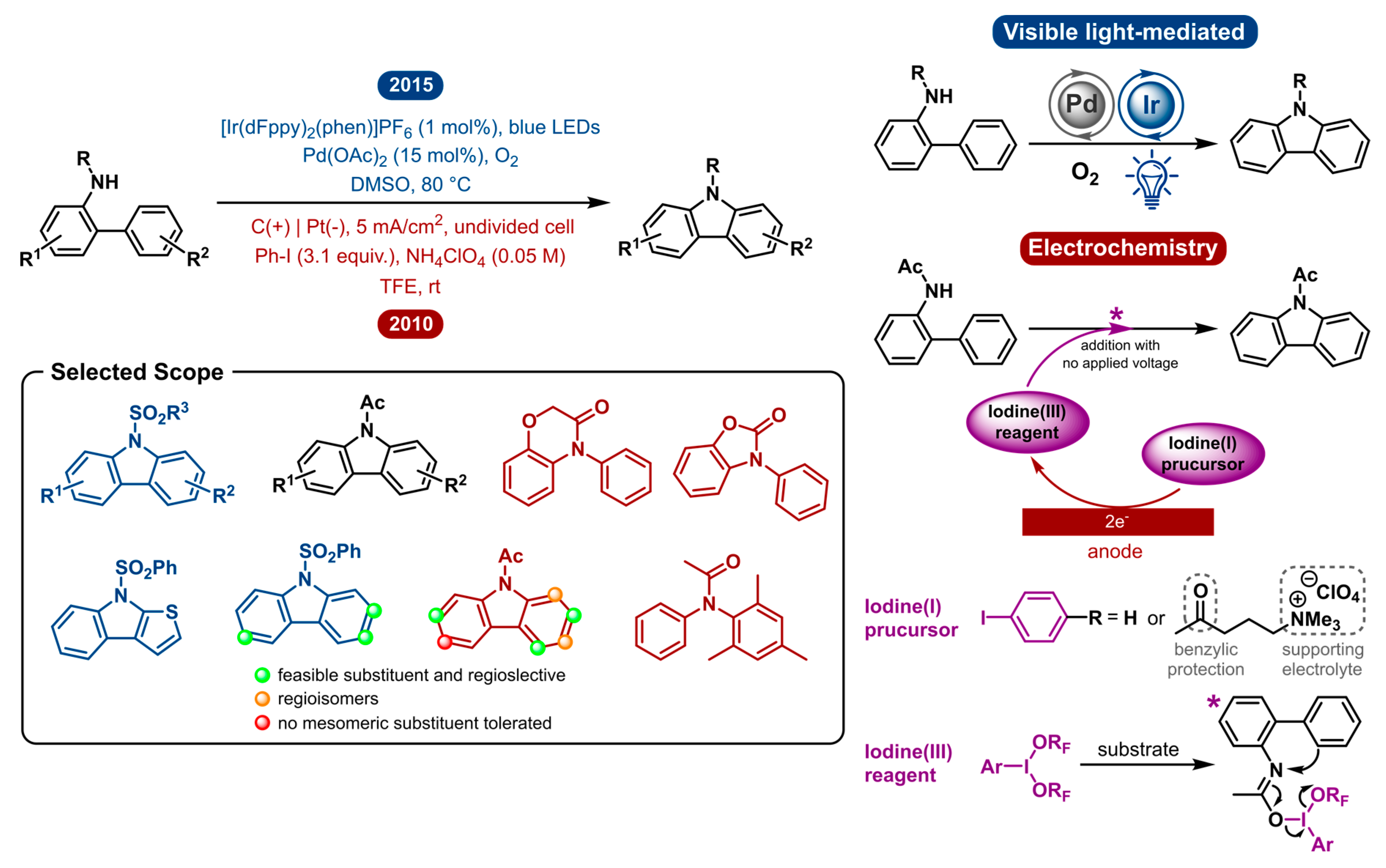
2.4. Carbazole Synthesis
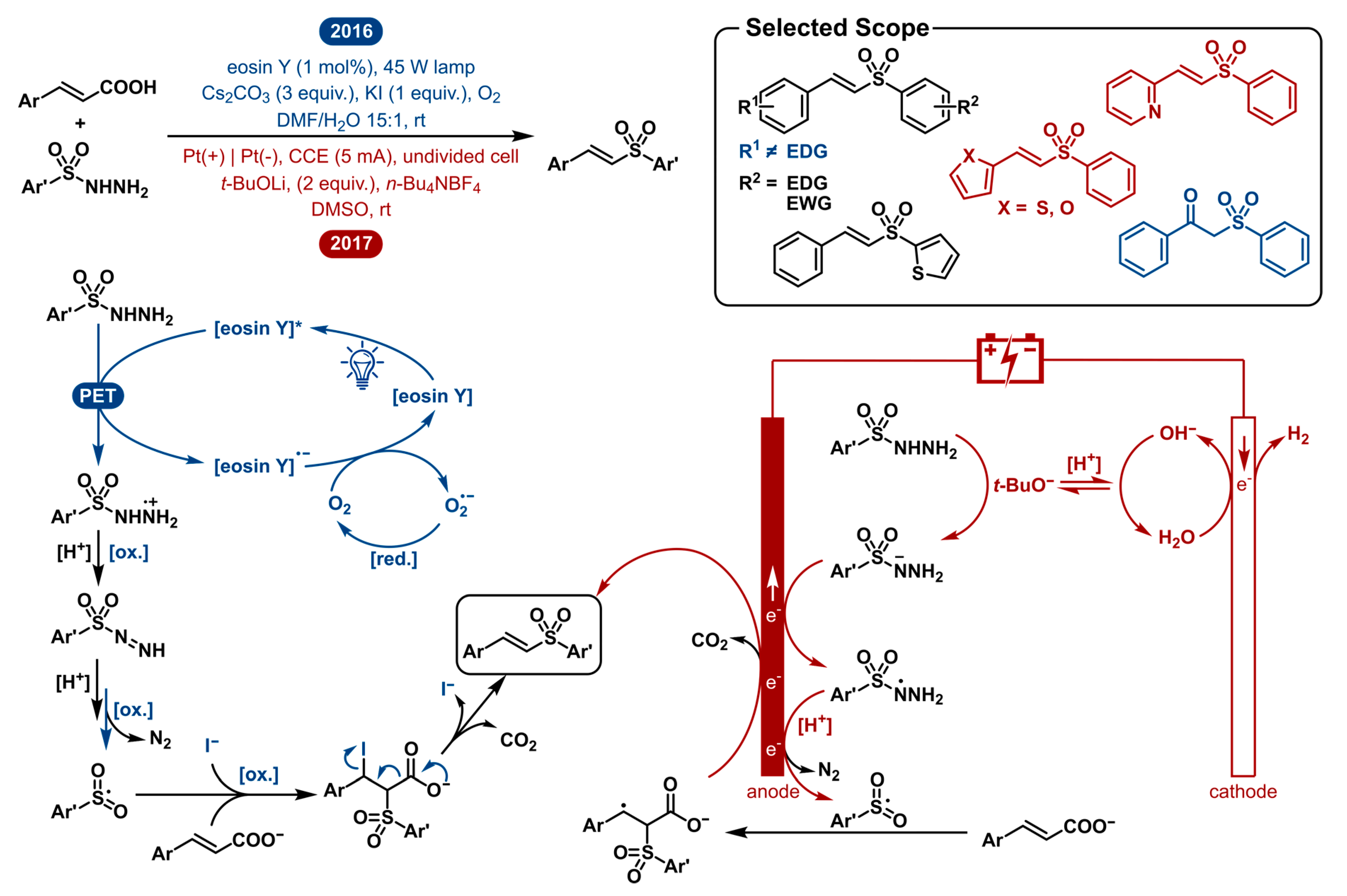
2.5. Decarboxylative Sulfonylation of Cinnamic Acids with Aromatic Sulfonylhydrazides to Vinyl Sulfones
2.6. Hydroamination of Olefins
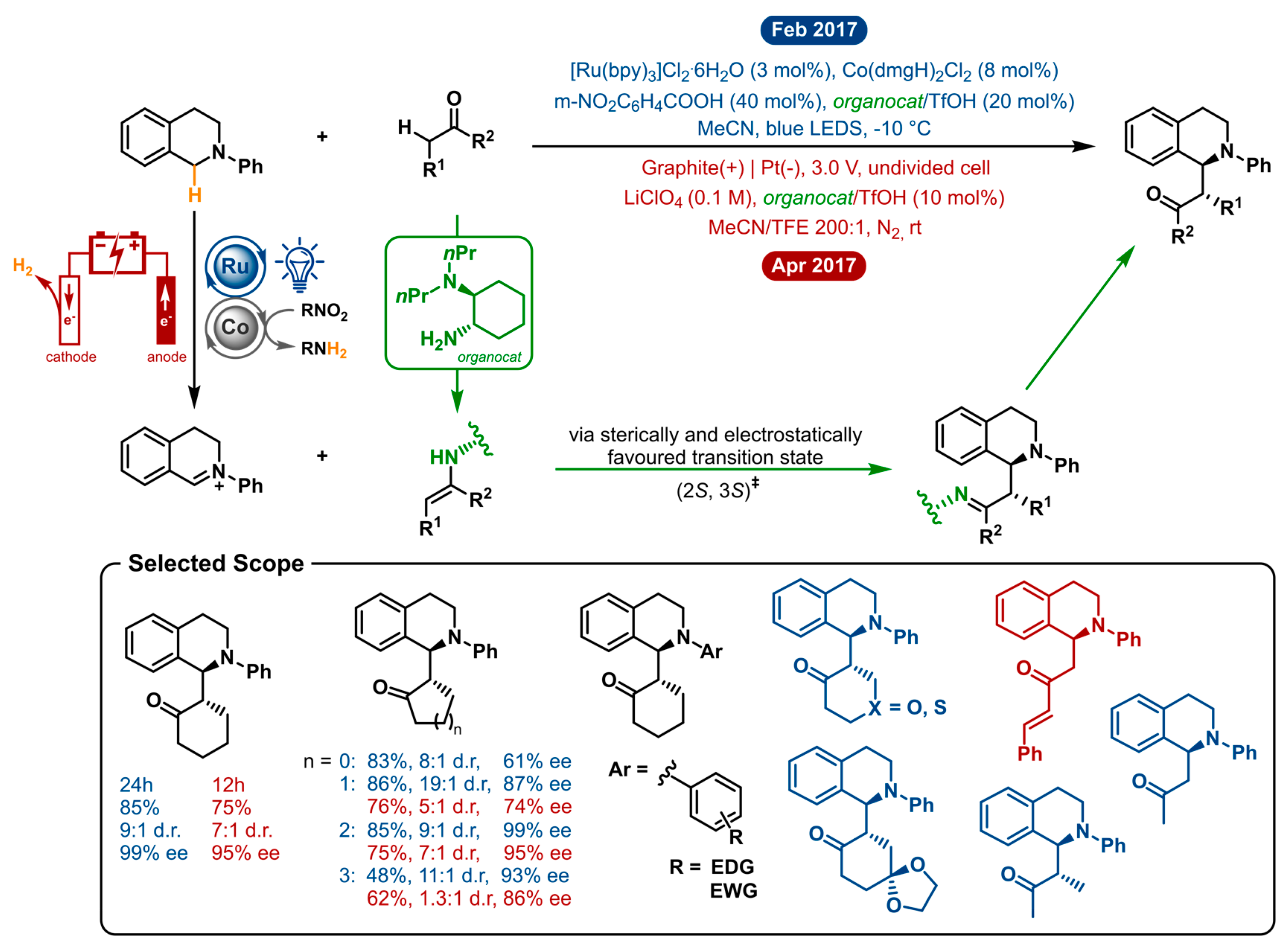
2.7. Cross-Dehydrogenative Coupling of Tertiary Amines with Ketones
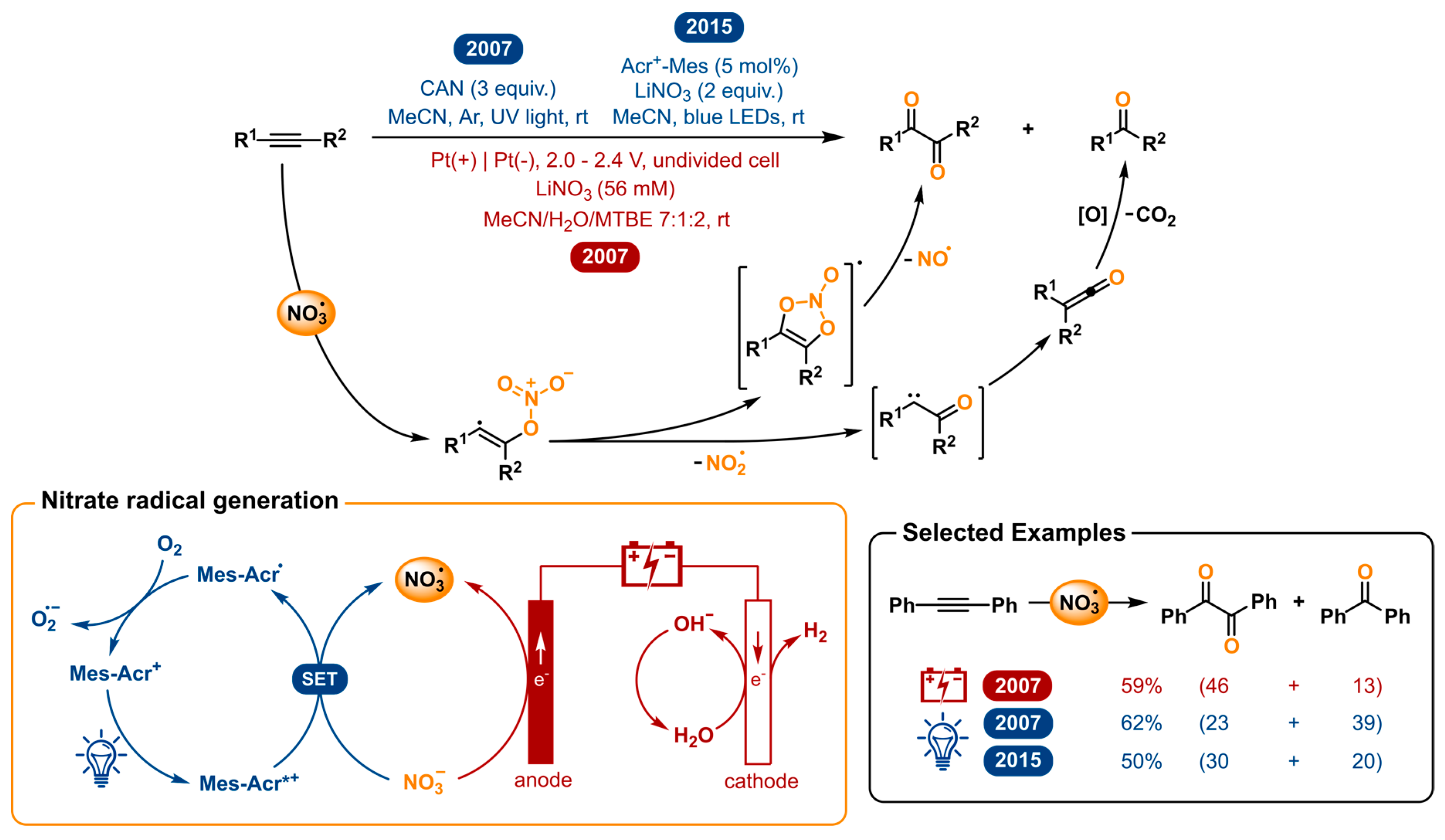
2.8. Nitrate Radical Induced Alkyne Oxygenation to 1,2-Diketones and Other Transformations
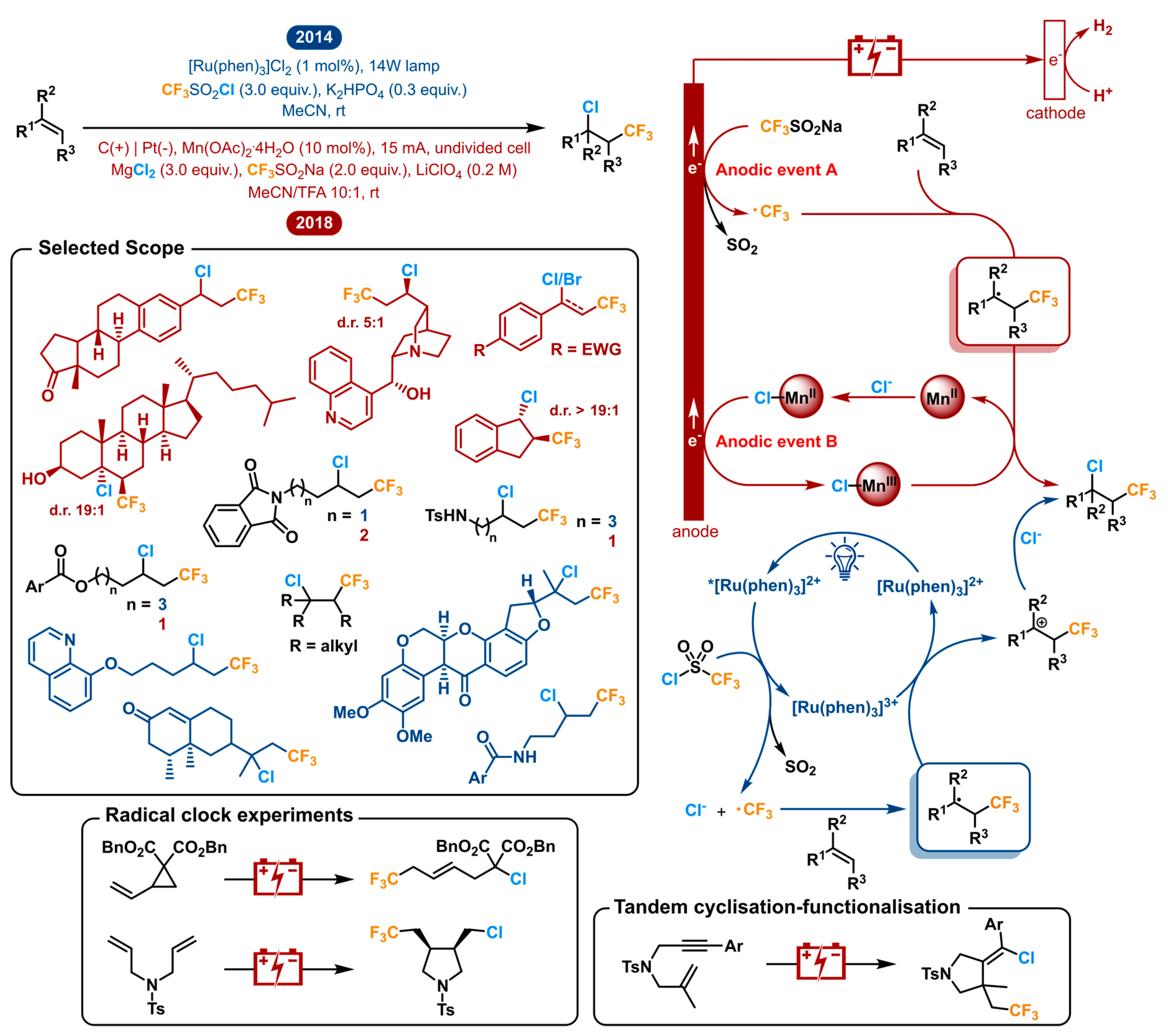
2.9. Vicinal Difunctionalization of Alkenes: Chlorotrifluoromethylation
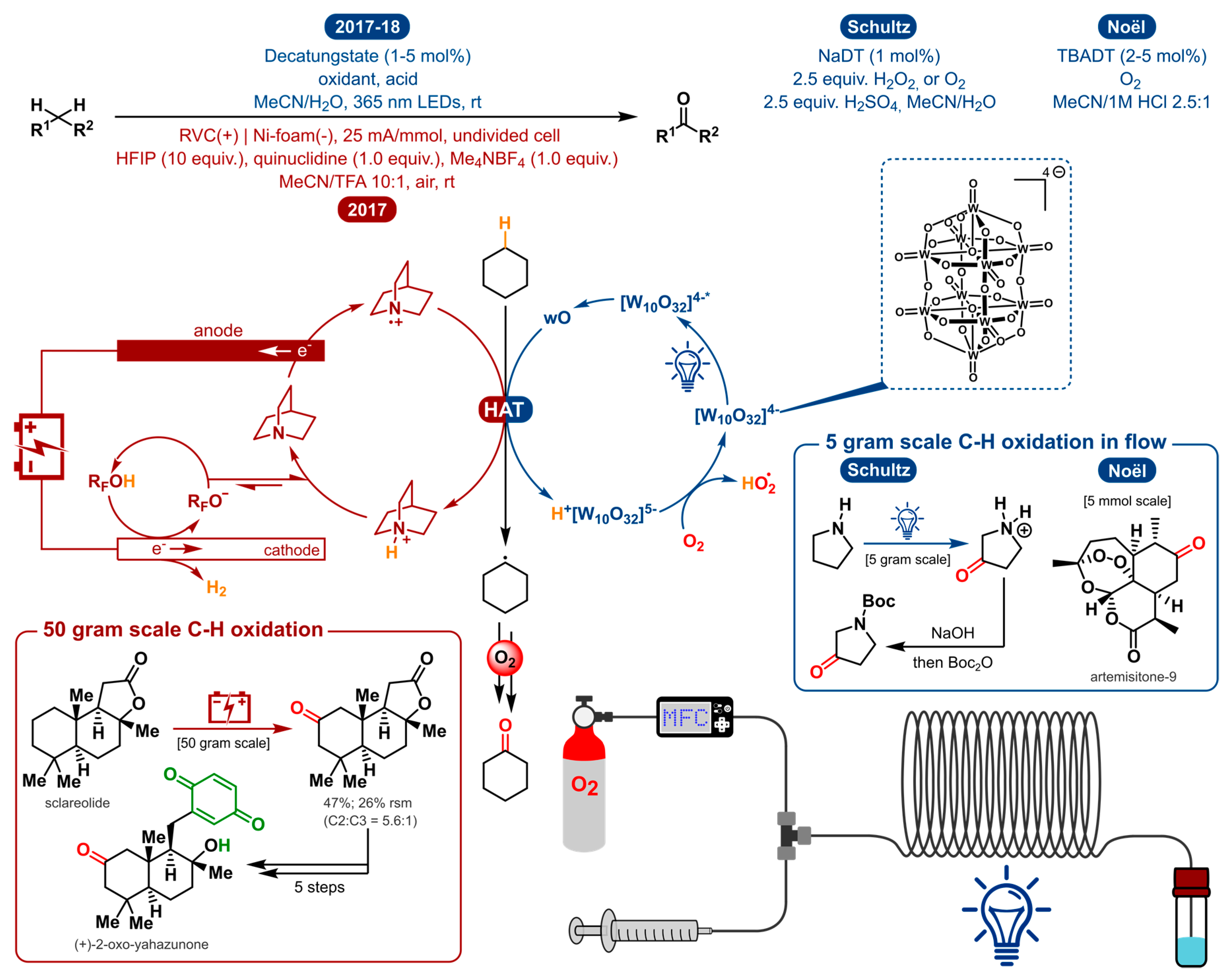
2.10. Oxygenation of Unactivated C-H Bonds
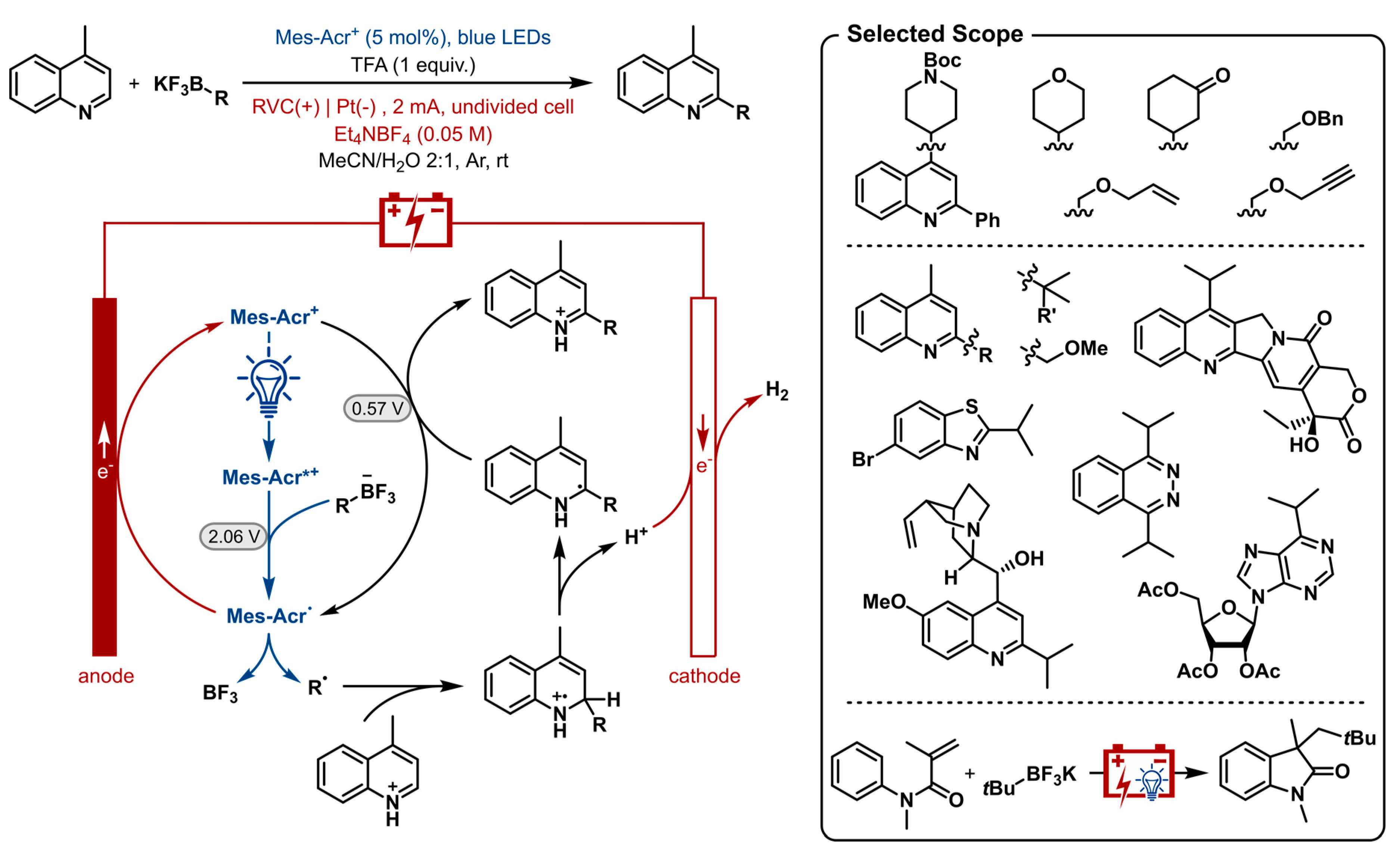
2.11. Photoelectrochemical Chemical Oxidant-Free Minisci-Type C-H Alkylation of Heterocycles
3. Discussion
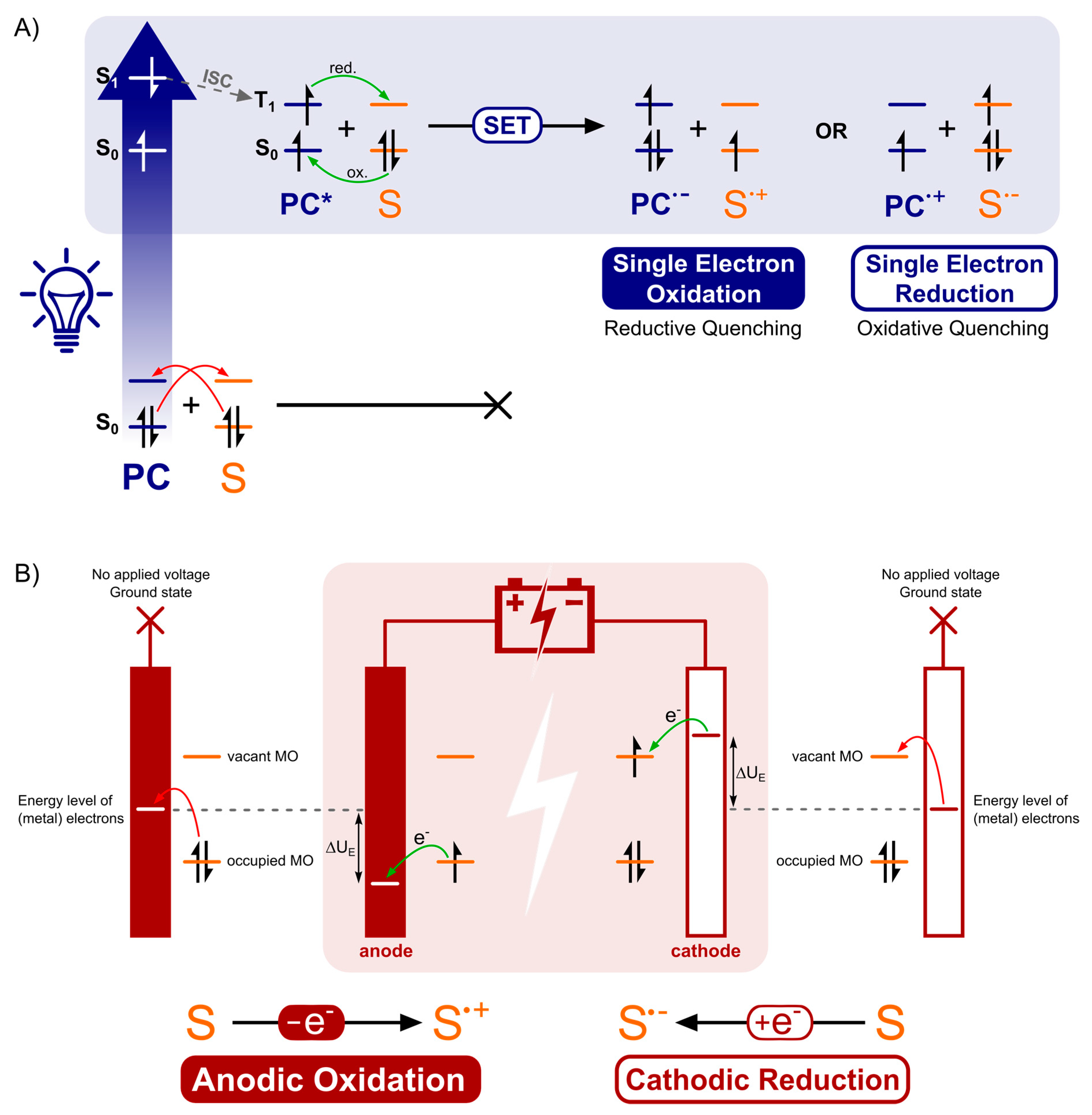
3.1. Underlying Physicochemical Structure and Mode of Activation
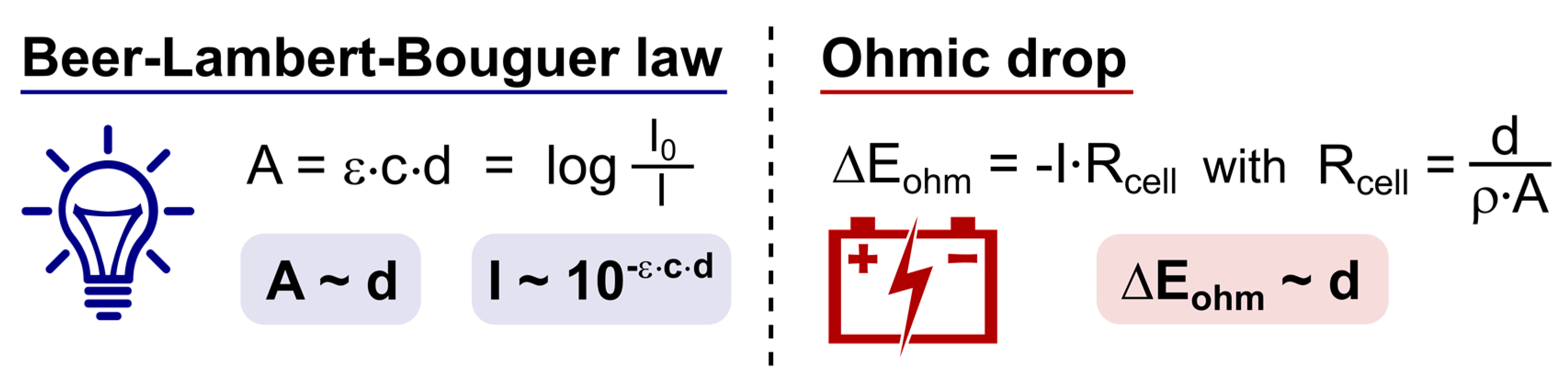
3.2. Tunability, Advantages and Restrictions
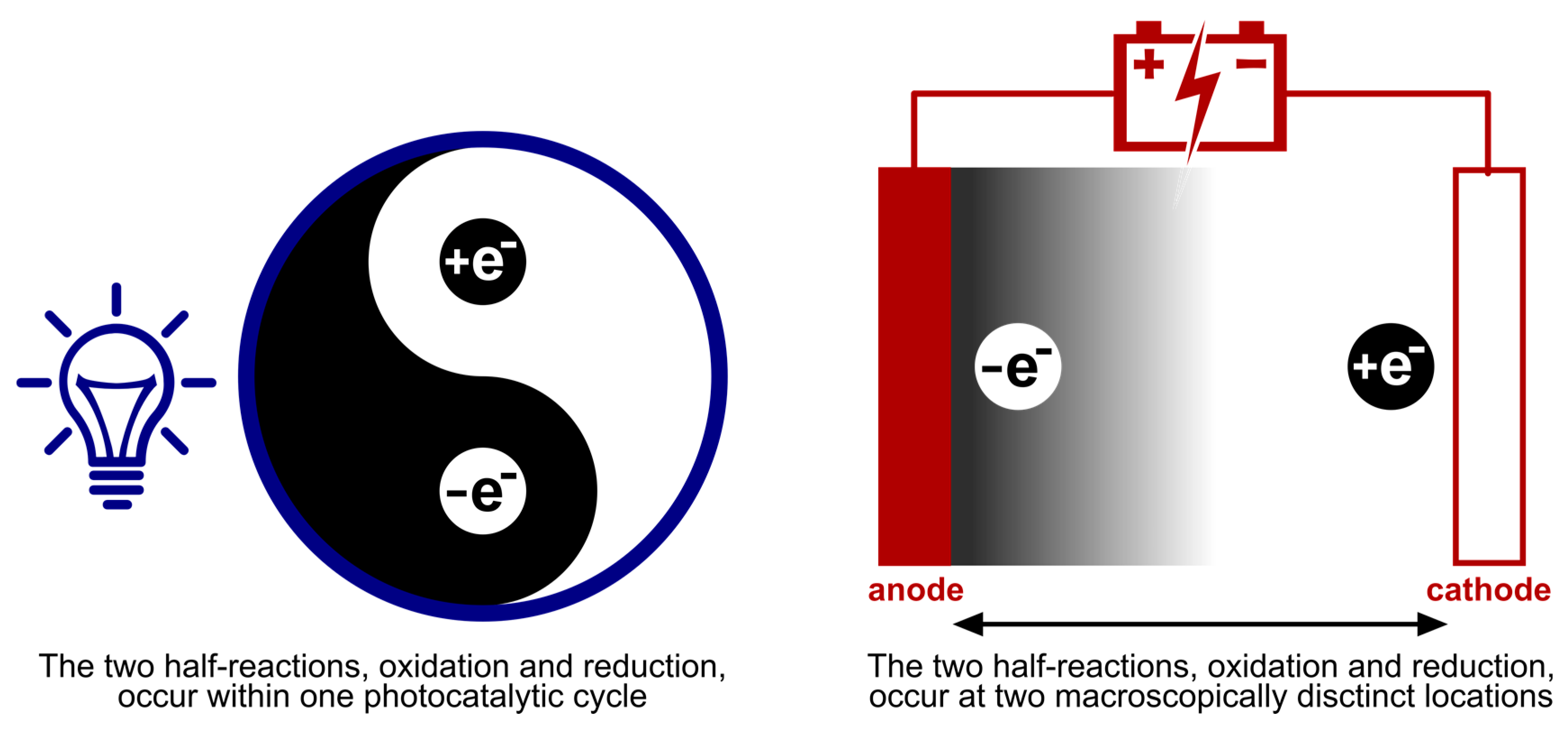
3.3. Remoteness of Redox Events and Overall Redox Demand of Transformations
3.4. Product Scope, Reactivity, Selectivity and Functional Group Tolerance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, H.J. Anodic and Cathodic CC-Bond Formation. Angew. Chem. Int. Ed. 1981, 20, 911–934. [Google Scholar] [CrossRef]
- Sperry, J.B.; Wright, D.L. The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev. 2006, 35, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef] [PubMed]
- Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S.R. Electrifying Organic Synthesis. Angew. Chem. Int. Ed. 2018, 57, 5594–5619. [Google Scholar] [CrossRef] [PubMed]
- Lund, H. A century of organic electrochemistry. J. Electrochem. Soc. 2002, 149, S21–S33. [Google Scholar] [CrossRef]
- Weinberg, N.L.; Weinberg, H.R. Electrochemical oxidation of organic compounds. Chem. Rev. 1968, 68, 449–523. [Google Scholar] [CrossRef]
- Degner, D. Organic electrosyntheses in industry. In Electrochemistry III. Topics in Current Chemistry; Stekchan, E., Ed.; Springer: Berlin/Heidelberg, Germany, 1988; Volume 148, pp. 1–95. [Google Scholar]
- Fichter, F. Organische elektrochemie; T. Steinkopff: Dresden/Leipzig, Germany, 1942; Volume 6, p. 359. [Google Scholar]
- Hammerich, O.; Speiser, B. Organic Electrochemistry, 5th ed.; CRC Press: Boca Raton, FL, USA, 2015; p. 1736. [Google Scholar]
- Alo, B.I.; Kandil, A.; Patil, P.A.; Sharp, M.J.; Siddiqui, M.A.; Snieckus, V.; Josephy, P.D. Sequential directed ortho metalation-boronic acid cross-coupling reactions. A general regiospecific route to oxygenated dibenzo[b,d]pyran-6-ones related to ellagic acid. J. Org. Chem. 1991, 56, 3763–3768. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Abe, T.; Tanaka, T.; Yang, C.-R.; Kouno, I. Phyllanemblinins A−F, New Ellagitannins from Phyllanthus emblica. J. Nat. Prod. 2001, 64, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Trend, R.M.; Ramtohul, Y.K.; Ferreira, E.M.; Stoltz, B.M. Palladium-Catalyzed Oxidative Wacker Cyclizations in Nonpolar Organic Solvents with Molecular Oxygen: A Stepping Stone to Asymmetric Aerobic Cyclizations. Angew. Chem. Int. Ed. 2003, 42, 2892–2895. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.J.; Chou, S.-C. The Structural Diversity of Phthalides from the Apiaceae. J. Nat. Prod. 2007, 70, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Vishnumurthy, K.; Makriyannis, A. Novel and efficient one-step parallel synthesis of dibenzopyranones via Suzuki-Miyaura cross coupling. J. Comb. Chem. 2010, 12, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Moschitto, M.J.; Anthony, D.R.; Lewis, C.A. Syntheses of Arnottin I and Arnottin II. J. Org. Chem. 2015, 80, 3339–3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo-Donaire, J.; Martin, R. Cu-Catalyzed Mild C(sp2)–H Functionalization Assisted by Carboxylic Acids en Route to Hydroxylated Arenes. J. Am. Chem. Soc. 2013, 135, 9350–9353. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ding, Y.-J.; Wang, J.-Y.; Su, Y.-M.; Wang, X.-S. Pd-Catalyzed C–H Lactonization for Expedient Synthesis of Biaryl Lactones and Total Synthesis of Cannabinol. Org. Lett. 2013, 15, 2574–2577. [Google Scholar] [CrossRef]
- Wang, Y.; Gulevich, A.V.; Gevorgyan, V. General and Practical Carboxyl-Group-Directed Remote C-H Oxygenation Reactions of Arenes. Chem. Eur. J. 2013, 19, 15836–15840. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gallardo-Donaire, J.; Martin, R. Mild ArI-Catalyzed C(sp2)–H or C(sp3)–H Functionalization/C-O Formation: An Intriguing Catalyst-Controlled Selectivity Switch. Angew. Chem. Int. Ed. 2014, 53, 11084–11087. [Google Scholar] [CrossRef]
- Dai, J.-J.; Xu, W.-T.; Wu, Y.-D.; Zhang, W.-M.; Gong, Y.; He, X.-P.; Zhang, X.-Q.; Xu, H.-J. Silver-Catalyzed C(sp2)–H Functionalization/C–O Cyclization Reaction at Room Temperature. J. Org. Chem. 2015, 80, 911–919. [Google Scholar] [CrossRef]
- Ramirez, N.P.; Bosque, I.; Gonzalez-Gomez, J.C. Photocatalytic Dehydrogenative Lactonization of 2-Arylbenzoic Acids. Org. Lett. 2015, 17, 4550–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yang, Q.; Jia, Z.; Luo, S. Organocatalytic Electrochemical C–H Lactonization of Aromatic Carboxylic Acids. Synthesis 2018, 50, 2924–2929. [Google Scholar] [CrossRef]
- Tao, X.-Z.; Dai, J.-J.; Zhou, J.; Xu, J.; Xu, H.-J. Electrochemical C−O Bond Formation: Facile Access to Aromatic Lactones. Chem. Eur. J. 2018, 24, 6932–6935. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jia, Z.; Li, L.; Zhang, L.; Luo, S. Visible-light promoted arene C–H/C–X lactonization via carboxylic radical aromatic substitution. Org. Chem. Front. 2018, 5, 237–241. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Wang, H.; Li, Q.; Liu, W.; Xu, K.; Zeng, C. Scalable Electrochemical Dehydrogenative Lactonization of C(sp2/sp3)–H Bonds. Org. Lett. 2018, 20, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Fichter, F.; Stenzl, H.; Beglinger, E. Elektrolyse der 2,4-Dimethyl-benzoesäure zusammen mit ihrem Natriumsalz in Methylalkohol. Helv. Chim. Acta 1938, 21, 375–380. [Google Scholar] [CrossRef]
- Hayrapetyan, D.; Shkepu, V.; Seilkhanov, O.T.; Zhanabil, Z.; Lam, K. Electrochemical synthesis of phthalides via anodic activation of aromatic carboxylic acids. Chem. Commun. 2017, 53, 8451–8454. [Google Scholar] [CrossRef] [PubMed]
- Kenner, G.W.; Murray, M.A.; Tylor, C.M.B. Oxidative cyclisation of diphenyl-2 carboxylic acid. Tetrahedron 1957, 1, 259–268. [Google Scholar] [CrossRef]
- Moon, Y.; Jang, E.; Choi, S.; Hong, S. Visible-Light-Photocatalyzed Synthesis of Phenanthridinones and Quinolinones via Direct Oxidative C–H Amidation. Org. Lett. 2018, 20, 240–243. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Xue, M.; Zhang, R.; Xu, K.; Zeng, C. Electrochemical Formation of N-Acyloxy Amidyl Radicals and Their Application: Regioselective Intramolecular Amination of sp2 and sp3 C–H Bonds. Org. Lett. 2018, 20, 3443–3446. [Google Scholar] [CrossRef]
- Horner, J.H.; Musa, O.M.; Bouvier, A.; Newcomb, M. Absolute Kinetics of Amidyl Radical Reactions. J. Am. Chem. Soc. 1998, 120, 7738–7748. [Google Scholar] [CrossRef]
- Hazelard, D.; Nocquet, P.-A.; Compain, P. Catalytic C–H amination at its limits: Challenges and solutions. Org. Chem. Front. 2017, 4, 2500–2521. [Google Scholar] [CrossRef]
- Singh, K.; Staig, S.J.; Weaver, J.D. Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc. 2014, 136, 5275–5278. [Google Scholar] [CrossRef] [PubMed]
- Metternich, J.B.; Gilmour, R. A Bio-Inspired, Catalytic E → Z Isomerization of Activated Olefins. J. Am. Chem. Soc. 2015, 137, 11254–11257. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xue, M.; Yan, X.; Liu, W.; Xu, K.; Zhang, S. Electrochemical Hofmann rearrangement mediated by NaBr: Practical access to bioactive carbamates. Org. Biomol. Chem. 2018, 16, 4615–4618. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.C.; Tarantino, K.T.; Knowles, R.R. Proton-Coupled Electron Transfer in Organic Synthesis: Fundamentals, Applications, and Opportunities. Top. Curr. Chem. 2016, 374, 30. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Pangu, A.; Lévesque, F.; Roth, H.G.; Oliver, S.F.; Campeau, L.-C.; Nicewicz, D.; DiRocco, D.A. Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem. 2016, 81, 7244–7249. [Google Scholar] [CrossRef]
- Gribble, G.W. Recent developments in indole ring synthesis—methodology and applications. J. Chem. Soc. Perkin Trans. 1 2000, 1045–1075. [Google Scholar] [CrossRef]
- Tokuyama, H.; Fukuyama, T. Indole synthesis by radical cyclization of o-alkenylphenyl isocyanides and its application to the total synthesis of natural products. Chem. Rec. 2002, 2, 37–45. [Google Scholar] [CrossRef]
- Cacchi, S.; Fabrizi, G. Synthesis and Functionalization of Indoles Through Palladium-catalyzed Reactions. Chem. Rev. 2005, 105, 2873–2920. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, G.R.; Kuethe, J.T. Practical Methodologies for the Synthesis of Indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef] [PubMed]
- Taber, D.F.; Tirunahari, P.K. Indole synthesis: A review and proposed classification. Tetrahedron 2011, 67, 7195–7210. [Google Scholar] [CrossRef]
- Shiri, M. Indoles in Multicomponent Processes (MCPs). Chem. Rev. 2012, 112, 3508–3549. [Google Scholar] [CrossRef] [PubMed]
- Corsello, M.A.; Kim, J.; Garg, N.K. Indole diterpenoid natural products as the inspiration for new synthetic methods and strategies. Chem. Sci. 2017, 8, 5836–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Chen, D.-F.; Gong, L.-Z. Recent progress in organocatalytic asymmetric total syntheses of complex indole alkaloids. Natl. Sci. Rev. 2017, 4, 381–396. [Google Scholar] [CrossRef]
- Fischer, E.; Jourdan, F. Ueber die Hydrazine der Brenztraubensäure. Ber. Dtsch. Chem. Ges. 1883, 16, 2241–2245. [Google Scholar] [CrossRef]
- Van Order, R.B.; Lindwall, H.G. Indole. Chem. Rev. 1942, 30, 69–96. [Google Scholar] [CrossRef]
- Robinson, B. The Fischer Indole Synthesis. Chem. Rev. 1963, 63, 373–401. [Google Scholar] [CrossRef]
- Robinson, B. Recent Studies on Fischer Indole Synthesis. Chem. Rev. 1969, 69, 227–250. [Google Scholar] [CrossRef]
- Hughes, D.L. Progress in the Fischer Indole Reaction. A Review. Org. Prep. Proced. Int. 1993, 25, 607–632. [Google Scholar] [CrossRef]
- Nagaraju, K.; Ma, D. Oxidative coupling strategies for the synthesis of indole alkaloids. Chem. Soc. Rev. 2018, 47, 8018–8029. [Google Scholar] [CrossRef] [PubMed]
- Bariwal, J.; Voskressensky, L.G.; Van der Eycken, E.V. Recent advances in spirocyclization of indole derivatives. Chem. Soc. Rev. 2018, 47, 3831–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barluenga, J.; Rodriguez, F.; Fananas, F.J. Recent advances in the synthesis of indole and quinoline derivatives through cascade reactions. Chem. Asian J. 2009, 4, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Würtz, S.; Rakshit, S.; Neumann, J.J.; Dröge, T.; Glorius, F. Palladium-Catalyzed Oxidative Cyclization of N-Aryl Enamines: From Anilines to Indoles. Angew. Chem. Int. Ed. 2008, 47, 7230–7233. [Google Scholar] [CrossRef]
- Guan, Z.-H.; Yan, Z.-Y.; Ren, Z.-H.; Liu, X.-Y.; Liang, Y.-M. Preparation of indoles via iron catalyzed direct oxidative coupling. Chem. Commun. 2010, 46, 2823–2825. [Google Scholar] [CrossRef]
- Neumann, J.J.; Rakshit, S.; Dröge, T.; Würtz, S.; Glorius, F. Exploring the Oxidative Cyclization of Substituted N-Aryl Enamines: Pd-Catalyzed Formation of Indoles from Anilines. Chem. Eur. J. 2011, 17, 7298–7303. [Google Scholar] [CrossRef]
- Yu, W.; Du, Y.; Zhao, K. PIDA-Mediated Oxidative C−C Bond Formation: Novel Synthesis of Indoles from N-Aryl Enamines. Org. Lett. 2009, 11, 2417–2420. [Google Scholar] [CrossRef]
- Liu, J.; Wei, W.; Zhao, T.; Liu, X.; Wu, J.; Yu, W.; Chang, J. Iodine/Copper Iodide-Mediated C–H Functionalization: Synthesis of Imidazo[1,2-a]pyridines and Indoles from N-Aryl Enamines. J. Org. Chem. 2016, 81, 9326–9336. [Google Scholar] [CrossRef]
- He, Z.; Liu, W.; Li, Z. I2-Catalyzed Indole Formation via Oxidative Cyclization of N-Aryl Enamines. Chem. Asian J. 2011, 6, 1340–1343. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Reddy, K.R. Isolation and Synthesis of Biologically Active Carbazole Alkaloids. Chem. Rev. 2002, 102, 4303–4428. [Google Scholar] [CrossRef] [PubMed]
- Grazulevicius, J.V.; Strohriegl, P.; Pielichowski, J.; Pielichowski, K. Carbazole-containing polymers: Synthesis, properties and applications. Prog. Polym. Sci. 2003, 28, 1297–1353. [Google Scholar] [CrossRef]
- Brunner, K.; van Dijken, A.; Börner, H.; Bastiaansen, J.J.A.M.; Kiggen, N.M.M.; Langeveld, B.M.W. Carbazole Compounds as Host Materials for Triplet Emitters in Organic Light-Emitting Diodes: Tuning the HOMO Level without Influencing the Triplet Energy in Small Molecules. J. Am. Chem. Soc. 2004, 126, 6035–6042. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, C.; Méndez, C.; Salas, J.A. Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006, 23, 1007–1045. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.-Y.; Ho, C.-L.; Gao, Z.-Q.; Mi, B.-X.; Chen, C.-H.; Cheah, K.-W.; Lin, Z. Multifunctional Iridium Complexes Based on Carbazole Modules as Highly Efficient Electrophosphors. Angew. Chem. Int. Ed. 2006, 45, 7800–7803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaupre, S.; Boudreault, P.L.; Leclerc, M. Solar-energy production and energy-efficient lighting: Photovoltaic devices and white-light-emitting diodes using poly(2,7-fluorene), poly(2,7-carbazole), and poly(2,7-dibenzosilole) derivatives. Adv. Mater. 2010, 22, E6–E27. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.W.; Reddy, K.R.; Knölker, H.-J. Occurrence, Biogenesis, and Synthesis of Biologically Active Carbazole Alkaloids. Chem. Rev. 2012, 112, 3193–3328. [Google Scholar] [CrossRef] [PubMed]
- Bashir, M.; Bano, A.; Ijaz, A.S.; Chaudhary, B.A. Recent Developments and Biological Activities of N-Substituted Carbazole Derivatives: A Review. Molecules 2015, 20, 13496–13517. [Google Scholar] [CrossRef] [Green Version]
- Sherer, C.; Snape, T.J. Heterocyclic scaffolds as promising anticancer agents against tumours of the central nervous system: Exploring the scope of indole and carbazole derivatives. Eur. J. Med. Chem. 2015, 97, 552–560. [Google Scholar] [CrossRef]
- Lissa, S.T.; Daniela, G.; Dianqing, S. Carbazole Scaffold in Medicinal Chemistry and Natural Products: A Review from 2010–2015. Curr. Top. Med. Chem. 2016, 16, 1290–1313. [Google Scholar] [CrossRef]
- Sathiyan, G.; Sivakumar, E.K.T.; Ganesamoorthy, R.; Thangamuthu, R.; Sakthivel, P. Review of carbazole based conjugated molecules for highly efficient organic solar cell application. Tetrahedron Lett. 2016, 57, 243–252. [Google Scholar] [CrossRef]
- Wex, B.; Kaafarani, B.R. Perspective on carbazole-based organic compounds as emitters and hosts in TADF applications. J. Mater. Chem. C 2017, 5, 8622–8653. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.C.P.; Zheng, N.; Buchwald, S.L. Combined C-H Functionalization/C−N Bond Formation Route to Carbazoles. J. Am. Chem. Soc. 2005, 127, 14560–14561. [Google Scholar] [CrossRef] [PubMed]
- Jordan-Hore, J.A.; Johansson, C.C.C.; Gulias, M.; Beck, E.M.; Gaunt, M.J. Oxidative Pd(II)-Catalyzed C-H Bond Amination to Carbazole at Ambient Temperature. J. Am. Chem. Soc. 2008, 130, 16184–16186. [Google Scholar] [CrossRef] [PubMed]
- Antonchick, A.P.; Samanta, R.; Kulikov, K.; Lategahn, J. Organocatalytic, Oxidative, Intramolecular C-H Bond Amination and Metal-free Cross-Amination of Unactivated Arenes at Ambient Temperature. Angew. Chem. Int. Ed. 2011, 50, 8605–8608. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Yoon, J.; Chang, S. Intramolecular Oxidative C−N Bond Formation for the Synthesis of Carbazoles: Comparison of Reactivity between the Copper-Catalyzed and Metal-Free Conditions. J. Am. Chem. Soc. 2011, 133, 5996–6005. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.W.; Bihn, J.H.; Kim, B.S. Pd-Catalyzed Intramolecular Oxidative C–H Amination: Synthesis of Carbazoles. Org. Lett. 2011, 13, 3738–3741. [Google Scholar] [CrossRef]
- Chng, L.L.; Yang, J.; Wei, Y.; Ying, J.Y. Palladium nanomaterials in catalytic intramolecular C–H amination reactions. Chem. Commun. 2014, 50, 9049–9052. [Google Scholar] [CrossRef]
- Takamatsu, K.; Hirano, K.; Satoh, T.; Miura, M. Synthesis of Carbazoles by Copper-Catalyzed Intramolecular C–H/N–H Coupling. Org. Lett. 2014, 16, 2892–2895. [Google Scholar] [CrossRef]
- Suzuki, C.; Hirano, K.; Satoh, T.; Miura, M. Direct Synthesis of N-H Carbazoles via Iridium(III)-Catalyzed Intramolecular C–H Amination. Org. Lett. 2015, 17, 1597–1600. [Google Scholar] [CrossRef]
- Kajiyama, D.; Inoue, K.; Ishikawa, Y.; Nishiyama, S. A synthetic approach to carbazoles using electrochemically generated hypervalent iodine oxidant. Tetrahedron 2010, 66, 9779–9784. [Google Scholar] [CrossRef]
- Amano, Y.; Nishiyama, S. Oxidative synthesis of azacyclic derivatives through the nitrenium ion: Application of a hypervalent iodine species electrochemically generated from iodobenzene. Tetrahedron Lett. 2006, 47, 6505–6507. [Google Scholar] [CrossRef]
- Amano, Y.; Inoue, K.; Nishiyama, S. Oxidative Access to Quinolinone Derivatives with Simultaneous Rearrangement of Functional Groups. Synlett 2008, 2008, 134–136. [Google Scholar] [CrossRef]
- Amano, Y.; Nishiyama, S. Effects of Aromatic Substituents of Electrochemically Generated Hypervalent Iodine Oxidant on Oxidation Reactions. Heterocycles 2008, 75, 1997. [Google Scholar] [CrossRef]
- Ates, M.; Uludag, N. Carbazole derivative synthesis and their electropolymerization. J. Solid State Electrochem. 2016, 20, 2599–2612. [Google Scholar] [CrossRef]
- Karon, K.; Lapkowski, M. Carbazole electrochemistry: a short review. J. Solid State Electrochem. 2015, 19, 2601–2610. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, J.F.; Nelson, R.F. Anodic Oxidation Pathways of Carbazoles: I. Carbazole and N-Substituted Derivatives. J. Electrochem. Soc. 1968, 115, 1159–1164. [Google Scholar] [CrossRef]
- Broese, T.; Francke, R. Electrosynthesis Using a Recyclable Mediator–Electrolyte System Based on Ionically Tagged Phenyl Iodide and 1,1,1,3,3,3-Hexafluoroisopropanol. Org. Lett. 2016, 18, 5896–5899. [Google Scholar] [CrossRef]
- Choi, S.; Chatterjee, T.; Choi, W.J.; You, Y.; Cho, E.J. Synthesis of Carbazoles by a Merged Visible Light Photoredox and Palladium-Catalyzed Process. ACS Catal. 2015, 5, 4796–4802. [Google Scholar] [CrossRef]
- Baudoin, O. New Approaches for Decarboxylative Biaryl Coupling. Angew. Chem. Int. Ed. 2007, 46, 1373–1375. [Google Scholar] [CrossRef]
- Gooßen, L.J.; Rodríguez, N.; Gooßen, K. Carboxylic Acids as Substrates in Homogeneous Catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C–C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Liu, L. Transition metal-catalyzed decarboxylative cross-coupling reactions. Sci. China Chem. 2011, 54, 1670–1687. [Google Scholar] [CrossRef]
- Weaver, J.D.; Recio, A.; Grenning, A.J.; Tunge, J.A. Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions. Chem. Rev. 2011, 111, 1846–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornella, J.; Larrosa, I. Decarboxylative Carbon-Carbon Bond-Forming Transformations of (Hetero)aromatic Carboxylic Acids. Synthesis 2012, 44, 653–676. [Google Scholar] [CrossRef]
- Dzik, W.I.; Lange, P.P.; Gooßen, L.J. Carboxylates as sources of carbon nucleophiles and electrophiles: Comparison of decarboxylative and decarbonylative pathways. Chem. Sci. 2012, 3, 2671–2678. [Google Scholar] [CrossRef]
- Park, K.; Lee, S. Transition metal-catalyzed decarboxylative coupling reactions of alkynyl carboxylic acids. RSC Adv. 2013, 3, 14165–14182. [Google Scholar] [CrossRef]
- Borah, A.J.; Yan, G. Decarboxylative functionalization of cinnamic acids. Org. Biomol. Chem. 2015, 13, 8094–8115. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T.S.A.; Liu, X. Recent advances in C–S bond formation via C–H bond functionalization and decarboxylation. Chem. Soc. Rev. 2015, 44, 291–314. [Google Scholar] [CrossRef]
- Xuan, J.; Zhang, Z.-G.; Xiao, W.-J. Visible-Light-Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641. [Google Scholar] [CrossRef]
- Jin, Y.; Fu, H. Visible-Light Photoredox Decarboxylative Couplings. Asian J. Org. Chem. 2017, 6, 368–385. [Google Scholar] [CrossRef]
- Singh, R.; Allam, B.K.; Singh, N.; Kumari, K.; Singh, S.K.; Singh, K.N. A Direct Metal-Free Decarboxylative Sulfono Functionalization (DSF) of Cinnamic Acids to α,β-Unsaturated Phenyl Sulfones. Org. Lett. 2015, 17, 2656–2659. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Xu, Y.; Chen, D.; Li, L.; Chen, Q.; Huang, M.; Weng, W. Visible-Light-Enabled Decarboxylative Sulfonylation of Cinnamic Acids with Sulfonylhydrazides under Transition-Metal-Free Conditions. Org. Lett. 2016, 18, 2990–2993. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lai, Y.-L.; Du, K.-S.; Lin, D.-Z.; Huang, J.-M. Electrochemical Decarboxylative Sulfonylation of Cinnamic Acids with Aromatic Sulfonylhydrazides to Vinyl Sulfones. J. Org. Chem. 2017, 82, 9655–9661. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, A.L.; Hultzsch, K.C. Hydroamination of Alkenes. In Organic Reactions; Wiley-VCH: Weinheim, Germany, 2015; Volume 88, pp. 1–554. [Google Scholar]
- Pirnot, M.T.; Wang, Y.-M.; Buchwald, S.L. Copper Hydride-Catalyzed Hydroamination of Alkenes and Alkynes. Angew. Chem. Int. Ed. 2016, 55, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Zheng, N. A Visible-Light-Mediated Oxidative C-N Bond Formation/Aromatization Cascade: Photocatalytic Preparation of N-Arylindoles. Angew. Chem. Int. Ed. 2012, 51, 9562–9566. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.R.; Uyeda, C.; Brotherton, C.A.; Jacobsen, E.N. Enantioselective Thiourea-Catalyzed Intramolecular Cope-Type Hydroamination. J. Am. Chem. Soc. 2013, 135, 6747–6749. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.-Q.; Chen, J.-R.; Wei, Q.; Liu, F.-L.; Deng, Q.-H.; Beauchemin, A.M.; Xiao, W.-J. Photocatalytic Generation of N-Centered Hydrazonyl Radicals: A Strategy for Hydroamination of β,γ-Unsaturated Hydrazones. Angew. Chem. Int. Ed. 2014, 53, 12163–12167. [Google Scholar] [CrossRef]
- Gesmundo, N.J.; Grandjean, J.-M.M.; Nicewicz, D.A. Amide and Amine Nucleophiles in Polar Radical Crossover Cycloadditions: Synthesis of γ-Lactams and Pyrrolidines. Org. Lett. 2015, 17, 1316–1319. [Google Scholar] [CrossRef]
- Gui, J.; Pan, C.-M.; Jin, Y.; Qin, T.; Lo, J.C.; Lee, B.J.; Spergel, S.H.; Mertzman, M.E.; Pitts, W.J.; La Cruz, T.E.; et al. Practical olefin hydroamination with nitroarenes. Science 2015, 348, 886–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Shi, S.-L.; Niu, D.; Liu, P.; Buchwald, S.L. Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science 2015, 349, 62–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.C.; Choi, G.J.; Orbe, H.S.; Knowles, R.R. Catalytic Olefin Hydroamidation Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2015, 137, 13492–13495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Xiong, P.; Mao, Z.-Y.; Wang, Y.-H.; Yan, X.; Lu, X.; Xu, H.-C. Electrocatalytic Generation of Amidyl Radicals for Olefin Hydroamidation: Use of Solvent Effects to Enable Anilide Oxidation. Angew. Chem. Int. Ed. 2016, 55, 2226–2229. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-M.; Guin, J.; Mück-Lichtenfeld, C.; Grimme, S.; Studer, A. Radical-Transfer Hydroamination of Olefins with N-Aminated Dihydropyridines. Chem. Asian J. 2011, 6, 1197–1209. [Google Scholar] [CrossRef]
- Guin, J.; Fröhlich, R.; Studer, A. Thiol-Catalyzed Stereoselective Transfer Hydroamination of Olefins with N-Aminated Dihydropyridines. Angew. Chem. Int. Ed. 2008, 47, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Kemper, J.; Studer, A. Stable Reagents for the Generation of N-Centered Radicals: Hydroamination of Norbornene. Angew. Chem. Int. Ed. 2005, 44, 4914–4917. [Google Scholar] [CrossRef]
- Tang, Y.; Li, C. Facile 5-Endo Amidyl Radical Cyclization Promoted by Vinylic Iodine Substitution. Org. Lett. 2004, 6, 3229–3231. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.L.; Barluenga, S.; Hunt, K.W.; Kranich, R.; Vega, J.A. Iodine(V) Reagents in Organic Synthesis. Part 3. New Routes to Heterocyclic Compounds via o-Iodoxybenzoic Acid-Mediated Cyclizations: Generality, Scope, and Mechanism. J. Am. Chem. Soc. 2002, 124, 2233–2244. [Google Scholar] [CrossRef]
- Boivin, J.; Callier-Dublanchet, A.-C.; Quiclet-Sire, B.; Schiano, A.-M.; Zard, S.Z. Iminyl, amidyl, and carbamyl radicals from O-benzoyl oximes and O-benzoyl hydroxamic acid derivatives. Tetrahedron 1995, 51, 6517–6528. [Google Scholar] [CrossRef]
- Newcomb, M.; Esker, J.L. Facile production and cyclizations of amidyl radicals. Tetrahedron Lett. 1991, 32, 1035–1038. [Google Scholar] [CrossRef]
- Tokuda, M.; Miyamoto, T.; Fujita, H.; Suginome, H. Stereoselective synthesis of 4- or 5-substituted 2-benzyl- and 2-benzoylpyrrolidines by means of anodic oxidation of δ-alkenylamines. Tetrahedron 1991, 47, 747–756. [Google Scholar] [CrossRef]
- Li, C.-J. Cross-Dehydrogenative Coupling (CDC): Exploring C−C Bond Formations beyond Functional Group Transformations. Acc. Chem. Res. 2009, 42, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, C.J. Beyond Traditional Cross Couplings: The Scope of the Cross Dehydrogenative Coupling Reaction. Chem. Asian J. 2010, 5, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, H.; Shi, W.; Lei, A. Bond Formations between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions. Chem. Rev. 2011, 111, 1780–1824. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Catalytic Dehydrogenative Cross-Coupling: Forming Carbon−Carbon Bonds by Oxidizing Two Carbon−Hydrogen Bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.A.; Knauber, T.; Li, C.-J. The Cross-Dehydrogenative Coupling of C-H Bonds: A Versatile Strategy for C-C Bond Formations. Angew. Chem. Int. Ed. 2014, 53, 74–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Oxidative Coupling between Two Hydrocarbons: An Update of Recent C–H Functionalizations. Chem. Rev. 2015, 115, 12138–12204. [Google Scholar] [CrossRef]
- Fu, N.; Li, L.; Yang, Q.; Luo, S. Catalytic Asymmetric Electrochemical Oxidative Coupling of Tertiary Amines with Simple Ketones. Org. Lett. 2017, 19, 2122–2125. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, L.; Ye, C.; Luo, S.; Wu, L.-Z.; Tung, C.-H. Visible-Light-Promoted Asymmetric Cross-Dehydrogenative Coupling of Tertiary Amines to Ketones by Synergistic Multiple Catalysis. Angew. Chem. Int. Ed. 2017, 56, 3694–3698. [Google Scholar] [CrossRef]
- Zou, Y.-Q.; Hörmann, F.M.; Bach, T. Iminium and enamine catalysis in enantioselective photochemical reactions. Chem. Soc. Rev. 2018, 47, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Wille, U. Inorganic Radicals in Organic Synthesis. Chem. Eur. J. 2002, 8, 340–347. [Google Scholar] [CrossRef]
- Wayne, R.P.; Barnes, I.; Biggs, P.; Burrows, J.P.; Canosa-Mas, C.E.; Hjorth, J.; Le Bras, G.; Moortgat, G.K.; Perner, D.; Poulet, G.; et al. The nitrate radical: Physics, chemistry, and the atmosphere. Atmos. Environ. Part A 1991, 25, 1–203. [Google Scholar] [CrossRef]
- Atkinson, R.; Plum, C.N.; Carter, W.P.L.; Winer, A.M.; Pitts, J.N. Rate Constants for the Gas-Phase Reactions of Nitrate Radicals with a Series of Organics in Air at 298 +/− 1-K. J. Phys. Chem. 1984, 88, 1210–1215. [Google Scholar] [CrossRef]
- Japar, S.M.; Niki, H. Gas-phase reactions of the nitrate radical with olefins. J. Phys. Chem. A 1975, 79, 1629–1632. [Google Scholar] [CrossRef]
- Morris, E.; Niki, H. Reaction of the Nitrate Radical with Acetaldehyde and Propylene. J. Phys. Chem. A 1974, 78, 1337–1338. [Google Scholar] [CrossRef]
- Öhman, V. Darstellung von Salpetersäureestern auf elektrochemischem Wege. Z. Elektrochem. 1936, 42, 862–872. [Google Scholar] [CrossRef]
- Fichter, F.; Siegrist, W.; Buess, H. Elektrolysen von Mischungen von Propionaten und Nitraten. Helv. Chim. Acta 1935, 18, 18–25. [Google Scholar] [CrossRef]
- Wille, U.; Andropof, J. Oxidation of Aromatic Alkynes with Nitrate Radicals (NO3•): An Experimental and Computational Study on a Synthetically Highly Versatile Radical. Aust. J. Chem. 2007, 60, 420–428. [Google Scholar] [CrossRef]
- Hering, T.; Slanina, T.; Hancock, A.; Wille, U.; Konig, B. Visible light photooxidation of nitrate: The dawn of a nocturnal radical. Chem. Commun. 2015, 51, 6568–6571. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kotani, H.; Ohkubo, K.; Ogo, S.; Tkachenko, N.V.; Lemmetyinen, H. Electron-Transfer State of 9-Mesityl-10-methylacridinium Ion with a Much Longer Lifetime and Higher Energy Than That of the Natural Photosynthetic Reaction Center. J. Am. Chem. Soc. 2004, 126, 1600–1601. [Google Scholar] [CrossRef] [PubMed]
- Wille, U.; Lietzau, L. Diastereoselective formation of anellated tetrahydrofurans using a nitrate radical induced oxidative, self-terminating radical cyclization cascade. Tetrahedron 1999, 55, 10119–10134. [Google Scholar] [CrossRef]
- Wille, U.; Lietzau, L. Stereoselection in 5-exo radical cyclizations of polysubstituted 2-oxahex-5-enyl radicals: A systematic study of the combination substituent effect. Tetrahedron 1999, 55, 11465–11474. [Google Scholar] [CrossRef]
- Jensen, K.H.; Sigman, M.S. Mechanistic approaches to palladium-catalyzed alkene difunctionalization reactions. Org. Biomol. Chem. 2008, 6, 4083–4088. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.I.; Liu, G.; Stahl, S.S. Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev. 2011, 111, 2981–3019. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Liang, Y.; Jiao, N. Azidation in the Difunctionalization of Olefins. Molecules 2016, 21, 352. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.-W.; Wang, N.-X.; Xing, Y. Recent Advances in Radical Difunctionalization of Simple Alkenes. Eur. J. Org. Chem. 2017, 2017, 5821–5851. [Google Scholar] [CrossRef]
- Lin, J.; Song, R.-J.; Hu, M.; Li, J.-H. Recent Advances in the Intermolecular Oxidative Difunctionalization of Alkenes. Chem. Rec. 2019, 19, 440–451. [Google Scholar] [CrossRef]
- Oh, S.H.; Malpani, Y.R.; Ha, N.; Jung, Y.-S.; Han, S.B. Vicinal Difunctionalization of Alkenes: Chlorotrifluoromethylation with CF3SO2Cl by Photoredox Catalysis. Org. Lett. 2014, 16, 1310–1313. [Google Scholar] [CrossRef]
- Ye, K.-Y.; Pombar, G.; Fu, N.; Sauer, G.S.; Keresztes, I.; Lin, S. Anodically Coupled Electrolysis for the Heterodifunctionalization of Alkenes. J. Am. Chem. Soc. 2018, 140, 2438–2441. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-J.; Dolbier, W.R. Efficient Cu-catalyzed Atom Transfer Radical Addition Reactions of Fluoroalkylsulfonyl Chlorides with Electron-deficient Alkenes Induced by Visible Light. Angew. Chem. Int. Ed. 2015, 54, 4246–4249. [Google Scholar] [CrossRef] [PubMed]
- Bagal, D.B.; Kachkovskyi, G.; Knorn, M.; Rawner, T.; Bhanage, B.M.; Reiser, O. Trifluoromethylchlorosulfonylation of Alkenes: Evidence for an Inner-Sphere Mechanism by a Copper Phenanthroline Photoredox Catalyst. Angew. Chem. Int. Ed. 2015, 54, 6999–7002. [Google Scholar] [CrossRef] [PubMed]
- Shilov, A.E.; Shul’pin, G.B. Activation of C-H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Que Jr, L.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333. [Google Scholar] [CrossRef]
- Olivo, G.; Cussó, O.; Costas, M. Biologically Inspired C-H and C=C Oxidations with Hydrogen Peroxide Catalyzed by Iron Coordination Complexes. Chem. Asian J. 2016, 11, 3148–3158. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C–H activation. J. Biol. Inorg. Chem. 2017, 22, 185–207. [Google Scholar] [CrossRef]
- Hill, C.L. Introduction of Functionality into Unactivated Carbon-Hydrogen Bonds. Catalytic Generation and Nonconventional Utilization of Organic Radicals. Synlett 1995, 1995, 127–132. [Google Scholar] [CrossRef]
- Maldotti, A.; Molinari, A.; Bergamini, P.; Amadelli, R.; Battioni, P.; Mansuy, D. Photocatalytic oxidation of cyclohexane by (nBu4N)4W10O32/Fe(III) porphyrins integrated systems. J. Mol. Catal. A Chem. 1996, 113, 147–157. [Google Scholar] [CrossRef]
- Maldotti, A.; Amadelli, R.; Carassiti, V.; Molinari, A. Catalytic oxygenation of cyclohexane by photoexcited (nBu4N)4W10O32: The role of radicals. Inorg. Chim. Acta 1997, 256, 309–312. [Google Scholar] [CrossRef]
- Schultz, D.M.; Lévesque, F.; DiRocco, D.A.; Reibarkh, M.; Ji, Y.; Joyce, L.A.; Dropinski, J.F.; Sheng, H.; Sherry, B.D.; Davies, I.W. Oxyfunctionalization of the Remote C-H Bonds of Aliphatic Amines by Decatungstate Photocatalysis. Angew. Chem. Int. Ed. 2017, 56, 15274–15278. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, G.; Govaerts, S.; Wang, Y.; Ravelli, D.; Koolman, H.F.; Fagnoni, M.; Djuric, S.W.; Noël, T. Selective C(sp3)−H Aerobic Oxidation Enabled by Decatungstate Photocatalysis in Flow. Angew. Chem. Int. Ed. 2018, 57, 4078–4082. [Google Scholar] [CrossRef] [PubMed]
- Asensio, G.; Gonzalez-Nunez, M.E.; Bernardini, C.B.; Mello, R.; Adam, W. Regioselective oxyfunctionalization of unactivated tertiary and secondary carbon-hydrogen bonds of alkylamines by methyl(trifluoromethyl)dioxirane in acid medium. J. Am. Chem. Soc. 1993, 115, 7250–7253. [Google Scholar] [CrossRef]
- Tanielian, C. Decatungstate photocatalysis. Coord. Chem. Rev. 1998, 178–180, 1165–1181. [Google Scholar] [CrossRef]
- Duncan, D.C.; Netzel, T.L.; Hill, C.L. Early-Time Dynamics and Reactivity of Polyoxometalate Excited States. Identification of a Short-Lived LMCT Excited State and a Reactive Long-Lived Charge-Transfer Intermediate following Picosecond Flash Excitation of [W10O32]4− in Acetonitrile. Inorg. Chem. 1995, 34, 4640–4646. [Google Scholar] [CrossRef]
- Kawamata, Y.; Yan, M.; Liu, Z.; Bao, D.H.; Chen, J.; Starr, J.T.; Baran, P.S. Scalable, Electrochemical Oxidation of Unactivated C-H Bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. [Google Scholar] [CrossRef] [PubMed]
- Francke, R.; Little, R.D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Rafiee, M.; Stahl, S.S. Electrochemical Functional-Group-Tolerant Shono-type Oxidation of Cyclic Carbamates Enabled by Aminoxyl Mediators. Angew. Chem. Int. Ed. 2018, 57, 6686–6690. [Google Scholar] [CrossRef]
- Yan, H.; Hou, Z.-W.; Xu, H.-C. Photoelectrochemical C-H Alkylation of Heteroarenes with Organotrifluoroborates. Angew. Chem. Int. Ed. 2019, 131, 4640–4643. [Google Scholar] [CrossRef]
- Ohkubo, K.; Mizushima, K.; Iwata, R.; Souma, K.; Suzuki, N.; Fukuzumi, S. Simultaneous production of p-tolualdehyde and hydrogen peroxide in photocatalytic oxygenation of p-xylene and reduction of oxygen with 9-mesityl-10-methylacridinium ion derivatives. Chem. Commun. 2010, 46, 601–603. [Google Scholar] [CrossRef]
- Cismesia, M.A.; Yoon, T.P. Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Rotondo, D.M.; McCusker, J.K. The photophysics of photoredox catalysis: A roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. [Google Scholar] [CrossRef] [PubMed]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley: New York, NY, USA, 2001; p. 833. [Google Scholar]
- Mortimer, J.T. Electrochemistry of Stimulation Electrodes. Available online: https://case.edu/groups/ANCL/pages/04/04_04.htm (accessed on 28 March 2019).
- Zoski, C.G. Handbook of Electrochemistry; Elsevier Science Bv: Amsterdam, The Netherlands, 2007; p. 934. [Google Scholar]
- Pletcher, D.; Green, R.A.; Brown, R.C.D. Flow Electrolysis Cells for the Synthetic Organic Chemistry Laboratory. Chem. Rev. 2018, 118, 4573–4591. [Google Scholar] [CrossRef] [PubMed]
- Rybicka-Jasińska, K.; Shan, W.; Zawada, K.; Kadish, K.M.; Gryko, D. Porphyrins as Photoredox Catalysts: Experimental and Theoretical Studies. J. Am. Chem. Soc. 2016, 138, 15451–15458. [Google Scholar] [CrossRef] [PubMed]
- Theriot, J.C.; Lim, C.-H.; Yang, H.; Ryan, M.D.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed atom transfer radical polymerization driven by visible light. Science 2016, 352, 1082–1086. [Google Scholar] [CrossRef] [Green Version]
- Alfonzo, E.; Alfonso, F.S.; Beeler, A.B. Redesign of a Pyrylium Photoredox Catalyst and Its Application to the Generation of Carbonyl Ylides. Org. Lett. 2017, 19, 2989–2992. [Google Scholar] [CrossRef]
- Lim, C.-H.; Ryan, M.D.; McCarthy, B.G.; Theriot, J.C.; Sartor, S.M.; Damrauer, N.H.; Musgrave, C.B.; Miyake, G.M. Intramolecular Charge Transfer and Ion Pairing in N,N-Diaryl Dihydrophenazine Photoredox Catalysts for Efficient Organocatalyzed Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2017, 139, 348–355. [Google Scholar] [CrossRef]
- Antoni, P.W.; Hansmann, M.M. Pyrylenes: A New Class of Tunable, Redox-Switchable, Photoexcitable Pyrylium–Carbene Hybrids with Three Stable Redox-States. J. Am. Chem. Soc. 2018, 140, 14823–14835. [Google Scholar] [CrossRef]
- Margrey, K.A.; Czaplyski, W.L.; Nicewicz, D.A.; Alexanian, E.J. A General Strategy for Aliphatic C–H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 4213–4217. [Google Scholar] [CrossRef]
- McCarthy, B.G.; Pearson, R.M.; Lim, C.-H.; Sartor, S.M.; Damrauer, N.H.; Miyake, G.M. Structure–Property Relationships for Tailoring Phenoxazines as Reducing Photoredox Catalysts. J. Am. Chem. Soc. 2018, 140, 5088–5101. [Google Scholar] [CrossRef]
- Sartor, S.M.; McCarthy, B.G.; Pearson, R.M.; Miyake, G.M.; Damrauer, N.H. Exploiting Charge-Transfer States for Maximizing Intersystem Crossing Yields in Organic Photoredox Catalysts. J. Am. Chem. Soc. 2018, 140, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Speckmeier, E.; Fischer, T.G.; Zeitler, K. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Corrigan, N.; Lim, C.-H.; Jung, K.; Zhu, J.; Miyake, G.; Xu, J.; Boyer, C. Guiding the Design of Organic Photocatalyst for PET-RAFT Polymerization: Halogenated Xanthene Dyes. Macromolecules 2019, 52, 236–248. [Google Scholar] [CrossRef]
- Roth, H.G.; Romero, N.A.; Nicewicz, D.A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar] [CrossRef]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ. 2018, 95, 197–206. [Google Scholar] [CrossRef]
- Kratochvil, B.; Coetzee, J.F. Analytical Oxidation–Reduction Reactions in Organic Solvents. Crit. Rev. Anal. Chem. 1971, 1, 415–454. [Google Scholar] [CrossRef]
- Fuchigami, T.; Atobe, M.; Inagi, S. Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices; John Wiley & Sons: Chichester, UK, 2015; p. 226. [Google Scholar]
- Zakrzewski, A.; Neckers, D.C. Bleaching products of rose bengal under reducing conditions. Tetrahedron 1987, 43, 4507–4512. [Google Scholar] [CrossRef]
- Benniston, A.C.; Elliott, K.J.; Harrington, R.W.; Clegg, W. On the Photochemical Stability of the 9-Mesityl-10-methylacridinium Cation. Eur. J. Org. Chem. 2009, 2009, 253–258. [Google Scholar] [CrossRef]
- Yamashita, Y.; Ikeda, H.; Mukai, T. Organic photochemistry. 80. Photoinduced electron-transfer reactions of cage compounds. Novel pericyclic reactions involving a chain process. J. Am. Chem. Soc. 1987, 109, 6682–6687. [Google Scholar] [CrossRef]
- Banerjee, A.; Lei, Z.; Ngai, M.-Y. Acyl Radical Chemistry via Visible-Light Photoredox Catalysis. Synthesis 2019, 51, 303–333. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemistry: Calling All Engineers. Angew. Chem. Int. Ed. 2018, 57, 4149–4155. [Google Scholar] [CrossRef] [PubMed]
- Britz, D. iR elimination in electrochemical cells. J. Electroanal. Chem. 1978, 88, 309–352. [Google Scholar] [CrossRef]
- Tajima, T.; Fuchigami, T. Development of an Electrolytic System Using Solid-Supported Bases for in Situ Generation of a Supporting Electrolyte from Methanol as a Solvent. J. Am. Chem. Soc. 2005, 127, 2848–2849. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Fuchigami, T. Development of a Novel Environmentally Friendly Electrolytic System by Using Recyclable Solid-Supported Bases for In Situ Generation of a Supporting Electrolyte from Methanol as a Solvent: Application for Anodic Methoxylation of Organic Compounds. Chem. Eur. J. 2005, 11, 6192–6196. [Google Scholar] [CrossRef]
- Tajima, T.; Kurihara, H. Deprotonation in anodic methoxylation of fluoroethyl phenyl sulfides using site-isolated heterogeneous bases. Chem. Commun. 2008, 5167–5169. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Kurihara, H.; Shimizu, S.; Tateno, H. Anodic Alkoxylation of Lactams Followed by Reactions with Carbon Nucleophiles in a One-Pot Manner Using HFIP as a Solvent. Electrochemistry 2013, 81, 353–355. [Google Scholar] [CrossRef] [Green Version]
- Kathiresan, M.; Velayutham, D. Ionic liquids as an electrolyte for the electro synthesis of organic compounds. Chem. Commun. 2015, 51, 17499–17516. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Modern Strategies in Electroorganic Synthesis. Chem. Rev. 2008, 108, 2265–2299. [Google Scholar] [CrossRef] [PubMed]
- Atobe, M.; Tateno, H.; Matsumura, Y. Applications of Flow Microreactors in Electrosynthetic Processes. Chem. Rev. 2018, 118, 4541–4572. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.P.; Elliott, L.D.; Booker-Milburn, K.I. Flow photochemistry: Old light through new windows. Beilstein J. Org. Chem. 2012, 8, 2025–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambié, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noël, T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef] [PubMed]
- Straathof, N.J.W.; Noël, T. Accelerating Visible-Light Photoredox Catalysis in Continuous-Flow Reactors. Vis. Light Photocatal. Org. Chem. 2018, 389–413. [Google Scholar] [CrossRef]
- Su, Y.; Straathof, N.J.W.; Hessel, V.; Noël, T. Photochemical Transformations Accelerated in Continuous-Flow Reactors: Basic Concepts and Applications. Chem. Eur. J. 2014, 20, 10562–10589. [Google Scholar] [CrossRef] [PubMed]
- Garlets, Z.J.; Nguyen, J.D.; Stephenson, C.R.J. The Development of Visible-Light Photoredox Catalysis in Flow. Isr. J. Chem. 2014, 54, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Horn, E.J.; Rosen, B.R.; Baran, P.S. Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. Acs Cent. Sci. 2016, 2, 302–308. [Google Scholar] [CrossRef] [PubMed]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verschueren, R.H.; De Borggraeve, W.M. Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis. Molecules 2019, 24, 2122. https://doi.org/10.3390/molecules24112122
Verschueren RH, De Borggraeve WM. Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis. Molecules. 2019; 24(11):2122. https://doi.org/10.3390/molecules24112122
Chicago/Turabian StyleVerschueren, Rik H., and Wim M. De Borggraeve. 2019. "Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis" Molecules 24, no. 11: 2122. https://doi.org/10.3390/molecules24112122
APA StyleVerschueren, R. H., & De Borggraeve, W. M. (2019). Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis. Molecules, 24(11), 2122. https://doi.org/10.3390/molecules24112122