4. Experimental Section
General Experimental Procedures. All reactions were performed under a nitrogen atmosphere using anhydrous techniques unless otherwise noted. 1H and 13C-NMR on a Varian Mercury 500 spectrometer were recorded in DMSO-d6 or CDCl3. Chemical shifts were reported in δ(ppm) units relative to the internal standard tetramethylsilane (TMS). All the reactions were monitored by thin layer chromatography (TLC) analysis on pre-coated silica gel G plates at 254 nm under UV lamp or HPLC analysis.
Chromatographic Methods. Analysis for fragments B, C, and D was achieved on the Agilent model 1260-DAD high-performance liquid chromatographsystem along with the detective wavelength of 220, 225, and 280 nm. The mobile phase consisted of acetonitrile (A) and deionized water (B), both with 0.1% TFA (trifluoroacetic acid) (vol./vol.), using gradient elution. The flow rate was 1.0 mL·min
−1, and the column temperature was maintained at 25 °C. The concrete gradient elution conditions are displayed in
Table S2. Analysis for compound
10 was achieved on the Waters UPLC, using gradient elution. The preparative column chromatography for crude pasireotide was performed on SHIMADZU Shim-pack PRC-ODS (H) (20 × 250 mm), using gradient elution.
Materials. Protected amino acids, PyBop, and anhydrous HOBt were purchased from Aldrich. All other reagents and chemicals were purchased from Beijing Chemistry Works (Beijing, Chnia), and were used without further purification. Anhydrous organic solvents were obtained either from Aldrich or distilled from the drying agents: Na or CaH2. All reactions were carried out under a N2 atmosphere employing oven- or flame-dried glassware. All solvents were either distilled or obtained from passing through activated alumina. Acetonitrile and trifluoroacetic acid (HPLC grade) were purchased from Sigma-Aldrich (Milwaukee, Germany). Ultrapure water was purchased from Hangzhou Wahaha Co., Ltd. (Hangzhou, China)
Synthesis of Compound 1. To a stirred solution of N-Cbz-d-Trp (10.0 g, 29.6 mmol) in THF (100 mL) was added N-hydroxysuccinimide (3.4 g, 29.6 mmol) at 0 °C. The resulting solution was then added dropwise to a solution of N,N’-dicyclohexylcarbodiimide (DCC) (6.7 g, 32.6 mmol) in THF (20 mL). The mixture was stirred at room temperature for 10 h. After the reaction was completed, DCU was removed by filtration through celite and the crude product was obtained by removing the solvent under vacuum. Pure compound 1, as a slight yellow solid, was obtained after recrystallization in isopropyl alcohol (80% yield). HRMS: m/z Calcd. 436.1509 For C23H22N3O6, Found 436.1504; 1H-NMR(500 MHz, DMSO-d6) δ/ppm m/z 10.94(s, 1H), 8.14(d, J = 8.0 Hz, 1H), 7.56(d, J = 8.0 Hz, 1H), 7.38–7.26(m, 6H), 7.09(t, J = 16.0 Hz, 2H), 7.04(t, J = 12.0 Hz, 1H), 4.99(q, J = 12.6 Hz, 2H), 4.69–4.63(m, 1H), 3.35(dd, J = 4.0, 16.0 Hz, 1H), 3.17(dd, J = 12.0, 8.0 Hz, 1H), 2.84(s, 4H); 13C-NMR(125 MHz, DMSO-d6) δ/ppm = 170.74, 169.08,156.55,137.28, 136.80, 129.02, 128.50, 128.27, 127.45, 124.97, 121.78, 119.31, 118.51, 112.25, 109.38, 60.44, 66.36, 27.71, 26.18.
Synthesis of Compound 2. To a solution of Ne-Boc-l-lysine (5.0 g, 20.3 mmol) in CH2Cl2 (50 mL) was added, dropwise, BSA (7.2 g, 35.5 mmol) at 0 °C followed by compound 1 (7.3 g, 16.8 mmol) in CH2Cl2 (50 mL). The mixture was stirred at room temperature for 12 h and then concentrated in vacuo. The residue was dissolved in ether (150 mL) and extracted with 5% sodium hydrocarbonate (NaHCO3) solution (50 mL × 3). The combined water layer was acidified with citric acid to pH 6–7 and extracted with EtOAc (100 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, and the solvent was removed by a rotary evaporator, under reduced pressure, to yield 2 as a yellow power in 82% yield. HRMS: m/z Calcd. 567.2918 For C30H39N4O7, Found 567.2915; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 12.58(s, 1H), 10.79(s, 1H), 8.24(d, J = 8.0 Hz, 1H), 7.66(d, J = 8.0 Hz, 1H), 7.35–7.28(m, 4H), 7.25–7.24(d, J = 4.0 Hz, 1H), 7.15(s, 1H), 7.05(t, J = 4.0 Hz, 1H), 6.96(t, J = 7.5 Hz, 1H), 6.74(s, 1H), 4.94(s, 2H), 4.38(td, J = 8.0, 12.0 Hz, 1H), 4.15(q, J = 7.6 Hz, 1H), 3.07(dd, J = 8.0, 16.0 Hz, 1H), 2.94–2.88(m, 1H), 2.85(d, J = 4.0 Hz, 2H), 1.65–1.63(m, 1H), 1.55–1.51(m, 1H), 1.35–1.30(m, 11H), 1.17(t, J = 8.0 Hz, 2H); 13C-NMR(125 MHz, DMSO-d6) δ/ppm = 174.31, 172.36, 156.21, 137.68, 136.68, 128.95, 128.30, 128.08, 127.92, 124.55, 121.44, 119.27, 118.79, 111.89, 110.74, 77.99, 65.83, 60.43, 56.06, 31.68, 29.77, 28.91, 23.19, 21.43, 14.74.
Synthesis of Compound 3. To a stirred solution of Fmoc-l-phenylglycine (15.0 g, 40.0 mmol) in THF (150 mL) was added N-hydroxysuccinimide (4.6 g, 40.0 mmol), dropwise, at 0 °C followed by DCC (9.1 g, 44.0 mmol) in THF dropwise at 5 °C. The mixture was stirred at room temperature for 6 h. After the reaction was completed, DCU was removed by filtration through celite and the crude product was obtained by removing the solvent under vacuum. Pure compound 3, as a white solid, was obtained after recrystallization in EtOAc (87% yield). HRMS: m/z Calcd. 470.1478 For C27H23N2O6, Found 470.1474; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.74–7.75(d, 2H, J = 5.0 Hz), 7.56(s, 2H), 7.37–7.46(m, 7H), 7.28(s, 2H), 5.79–5.80(d, 1H, J = 5.0 Hz), 5.68(s, 1H), 4.42–4.45(t, 2H, J = 7.5 Hz), 4.21(s, 1H), 2.78(s, 4H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 168.33, 166.91, 155.22, 143.76, 143.57, 141.30, 134.41, 129.40, 129.31, 127.75, 127.62, 127.12, 125.07, 120.00, 67.45, 56.44, 47.07, 25.55.
Synthesis of fragment B. To a stirred solution of 2 (5.0 g, 8.8 mmol) in MeOH (50 mL) was added Pd/C10% (0.5 g). A hydrogen atmosphere (1 atm) was applied and the mixture was stirred at room temperature for 5 h. The mixture was filtered through celite and the filtrate was concentrated in vacuo. Next, 5% NaHCO3 solution (30 mL) was added to dissolve the concentrated filtrate and then citric acid was added to adjust the pH to ≈ 6. After that, a white solid precipitated out and was collected by filtration to afford amine intermediate in 94%. HRMS: m/z Calcd. 433.2451 For C25H33N4O5, Found 433.2453. To a solution of the above amine (3.6 g, 8.3 mmol) in dry THF (30 mL) at 0 °C was added BSA (2.9 g, 14.5 mmol), dropwise, and the mixture was stirred for 2 h at room temperature, after which 3 (3.0 g, 6.9 mmol) in THF (20 mL) was added, dropwise, and the resulting mixture was stirred for 10 h at room temperature. After the reaction was completed, the solvents were evaporated. The residue was then dissolved in ether (50 mL) and extracted with 5% NaHCO3 solution (50 mL × 3). The combined water layer was acidified with citric acid to pH 6–7 and extracted with EtOAc (100 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, and filtered through celite. The organic solution was evaporated in vacuo and 4.57 g of fragment B was obtained after recrystallization in MeOH (84% yield). HRMS: m/z Calcd. 788.3659 For C45H50N5O8, Found 788.3650; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 12.53(s, 1H), 10.68(d, J = 2.4 Hz, 1H), 8.50(d, J = 8.1 Hz, 1H), 8.25(d, J = 7.8 Hz, 1H), 8.02(d, J = 8.5 Hz, 1H), 7.86(d, J = 7.5 Hz, 2H), 7.75(dd, J = 11.3, 7.6 Hz, 2H), 7.55(d, J = 7.9 Hz, 1H), 7.39(td, J = 7.4, 3.0 Hz, 2H), 7.31–7.17(m, 7H), 7.01(t, J = 7.4 Hz, 1H), 6.91(s, 1H), 6.71(t, J = 5.8 Hz, 1H), 5.36(d, J = 8.4 Hz, 1H), 4.57(q, J = 8.1 Hz, 1H), 4.22(dd, J = 13.5, 7.1 Hz, 2H), 3.04(dd, J = 14.5, 5.2 Hz, 1H), 2.88(d, J = 9.8 Hz, 1H), 2.82(q, J = 6.7, 6.1 Hz, 2H), 1.58(s, 1H), 1.49(t, J = 7.5 Hz, 1H), 1.33(s, 9H), 1.26(q, J = 7.2 Hz, 2H), 1.09(dt, J = 13.9, 7.6 Hz, 2H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 174.25, 171.87, 170.20, 156.42, 156.21, 144.60, 144.32, 141.32, 138.99, 136.62, 128.70, 128.29, 127.94, 127.72, 126.25, 126.12, 124.52, 121.36, 120.73, 119.08, 118.74, 111.81, 110.15, 77.98, 66.67, 58.49, 54.19, 52.61, 47.23, 31.59, 30.58, 29.74, 28.91, 23.19.
Synthesis of Compound 4. To a stirred solution of N-Boc-O-benzyl-d-tyrosine (10.0 g, 27.0 mmol) in dry THF (100 mL) was added N-hydroxysuccinimide (3.1 g, 27.0 mmol) at 0 °C. The resulting solution was then added a solution of DCC (6.1 g, 29.7 mmol) in THF, dropwise, at 5 °C. The mixture was stirred at room temperature for 4 h and monitored by TLC. After the reaction was completed, DCU was removed by filtration through celite. The solvent was evaporated off, after which 11.5 g of 4 was obtained after recrystallization in EtOAc in 91% yield. HRMS: m/z Calcd. 469.1975 For C25H29N2O7, Found 469.1971; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.41–7.42(d, 2H, J = 5.0 Hz), 7.37–7.39(t, 2H, J = 5.0 Hz), 7.20–7.21(d, 2H, J = 5.0 Hz), 6.92–6.94(d, 2H, J = 10.0 Hz), 5.04(s, 2H), 4.89(s, 1H), 3.13–3.25(m, 2H), 2.84(s, 4H), 1.42(s, 9H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 168.65, 167.77, 158.18, 154.62, 136.97, 130.82, 128.58, 127.96, 127.47, 126.86, 80.52, 70.00, 52.71, 37.28, 28.24, 25.59.
Synthesis of Compound 5. To a stirred solution of phenylalanine methyl ester hydrochloride (5 g, 23.3 mmol) in dry THF (50 mL) was added trimethylamine (2.4 g, 23.3 mmol), dropwise, followed by addition of 4 (10.9 g, 23.3 mmol) in THF (20 mL). The mixture was stirred at room temperature for 5 h and then concentrated in vacuo. The residue was dissolved in EtOAc (100 mL) and washed with 5% NaHCO3 solution (100 mL × 3), 5% citric acid solution (100 mL × 3), brine, and dried over Na2SO4. Filtration followed by concentration in vacuo afforded 11.6 g of 5, as a white solid, in 94% yield. HRMS: m/z Calcd. 533.2652 For C31H37N2O6, Found 533.2647; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.40–7.42(d, 2H, J = 10.0 Hz), 7.35–7.38(t, 2H, J = 7.50 Hz), 7.31–7.32(d, 1H, J = 5.0 Hz), 7.22–7.25(t, 3H, J = 7.50 Hz), 7.09–7.10(d, 2H, J = 5.0 Hz), 6.97–6.99(d, 2H, J = 10.0 Hz), 6.88–6.89(d, 2H, J = 5.0 Hz), 6.27(s, 1H), 5.02(s, 2H), 4.95(s, 1H), 4.77(s, 1H), 4.82(s, 1H), 3.66(s, 3H), 2.96–3.05(m, 4H), 1.40(s, 9H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 171.36, 170.81, 157.89, 136.98, 135.65, 130.42, 129.22, 128.58, 128.54, 127.96, 127.42, 127.11, 115.03, 80.16, 70.01, 55.80, 53.26, 52.25, 37.99, 37.43, 28.26.
Synthesis of fragment C. To a stirred solution of 5 (6.0 g, 11.6 mmol) in CH2Cl2 (25 mL) was added trifluoroacetic acid (TFA) (8.6 mL, 116 mmol), dropwise, at 5 °C. The mixture was stirred at room temperature for 4 h and monitored by TLC. The solvent was evaporated off, after which 5.9 g of fragment C was obtained after recrystallization in ether (98% yield). HRMS: m/z Calcd. 433.2127 For C26H29N2O4, Found 433.2124; 1H-NMR(500 MHz, CDCl3) δ/ppm = 8.97–8.99(d, 1H, J = 10.0 Hz), 8.10(s, 3H), 7.44–7.45(d, 2H, J = 5.0 Hz), 7.38–7.41(t, 2H, J = 7.5 Hz), 7.29–7.34(m, 3H), 7.22–7.25(t, 3H, J = 7.5 Hz), 7.17–7.18(d, 2H, J = 5.0 Hz), 6.96–6.98(d, 2H, J = 10.0 Hz), 5.08(s, 2H), 4.55–4.60(q, 1H, J = 8.3 Hz), 3.98(s, 1H), 3.61(s, 3H), 3.02–3.09(m, 2H), 2.55–3.00(dd, 1H, J1 = 10.0, J2 = 15.0Hz), 2.85–2.89(dd, 1H, J1 = 5.0, J2 = 15Hz); 13C-NMR(125 MHz, CDCl3) δ/ppm = 171.64, 168.76, 158.08, 137.58, 137.14, 131.12, 129.53, 128.89, 128.86, 128.09, 127.21, 127.15, 115.27, 69.63, 54.27, 53.76, 52.50, 37.10, 36.51.
Synthesis of Compound 6. To a stirred solution of l-hydroxyproline methyl ester (10.0 g, 69.9 mmol) in 1,4-dioxane/H2O (v:v = 2:1) was added Na2CO3 (7.3 g, 69.9 mmol), slowly, followed by Fmoc N-hydroxysuccinimide ester (Fmoc-OSu) (23.6 g, 69.9 mmol). The resulting solution was stirred at room temperature for 10 h and then concentrated in vacuo. The residue was dissolved in EtOAc (100 mL), washed with brine, dried over Na2SO4, and concentrated to yield 6, as a colorless oil, in 94% yield. HRMS: m/z Calcd. 368.1498 For C21H22NO5, Found 368.1494; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.80–7.79(d, J = 4 Hz, 2H), 7.66–7.58(m, 2H), 7.45–7.42(t, J = 6.0 Hz, 2H), 7.36–7.34(t, J = 4.0 Hz, 2H), 4.59–4.44(m, 3H), 4.41–4.35(m, 1H), 4.31–4.15(m, 1H), 3.79(s, 2H), 3.70–3.60(m, 3H), 2.74(s, 1H), 2.41–2.32(m, 1H), 2.15–2.13(m, 1H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 173.16, 155.10, 144.09, 141.33, 127.74, 127.12, 125.15, 119.99, 70.08, 69.22, 67.64, 60.51, 57.98, 57.62, 55.31, 54.68, 52.38, 47.27, 39.33, 38.42.
Synthesis of Compound 7. To a stirred solution of triphosgene (3.2 g, 10.6 mmol) in dry THF (40 mL) was added 6 (6.5 g, 17.7 mmol) in THF (10 mL), slowly, and the mixture was stirred at room temperature for 2 h. Next, N-boc-ethylenediamine (14.2 g, 88.5 mmol) in THF (20 mL) and 4-dimethylaminopyridine (2.2 g, 17.7 mmol) was added. The resulting mixture was stirred at room temperature for 6 h. After the reaction was completed, the solvents were evaporated and then the residue was dissolved in EtOAc (100 mL), washed with 0.1 M HCl solution (100 mL × 3), brine (100 mL × 3), and dried over Na2SO4. The solvent was evaporated off, after which 6.7 g of 7 was obtained after recrystallization in EtOAc in 69% yield. HRMS: m/z Calcd. 576.2322 For C29H35N3O8Na, Found 576.2313; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.77(d, J = 8.0 Hz, 2H), 7.57(dd, J = 12.0, 4.0 Hz, 2H), 7.40(t, J = 8.0 Hz, 2H), 7.31(t, J = 6.0 Hz, 2H), 5.26(d, J = 20.0 Hz, 1H), 5.14(d, J = 20.0 Hz, 1H), 4.79(s, 1H), 4.51–4.47(m, 1H), 4.43–4.39(m, 1H), 4.34(t, J = 8.0 Hz, 1H), 4.29–4.15(m, 1H), 3.85–3.72(m, 4H), 3.66(s, 1H), 3.27(s, 4H), 2.46(dd, J = 12.0, 20.0 Hz, 2H), 2.22(s, 1H), 1.44(s, 9H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 172.53, 155.78, 154.75, 143.91, 143.59, 141.29, 127.73, 127.08, 125.12, 120.00, 79.75, 73.13, 72.23, 67.73, 60.40, 52.43, 47.27, 28.35, 21.04, 14.20.
Synthesis of fragment D. To a stirred solution of 7 (5.0 g, 9.0 mmol) in dry MeOH (30 mL) was added NaOH solution (18.0 mmol, 10 mL), dropwise, at 0 °C and the mixture was stirred at room temperature for 2 h. Solvents were removed by vacuo and the residue was resolved in 1, 4-dioxane followed by addition of Fmoc-OSu (3.0 g, 9.0 mmol). The resulting mixture was stirred at room temperature for 6 h and solvents were evaporated, after which 4.2 g of fragment D was obtained in 88% yield. HRMS: m/z Calcd. 562.5667 For C28H33N3O8Na, Found 562.2167. 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 7.93–7.89(t, J = 10.0 Hz, 1H)7.77–7.74(dd, J1 = 10.0, J2 = 45 Hz, 1H), 7.66(t, J = 7.5 Hz, 1H), 7.44(q, J = 6.6 Hz, 2H), 7.36(t, J = 5.0 Hz, 2H), 7.27(s, 1H), 6.85(s, 1H), 5.11(s, 1H), 4.28–4.19(m, 3H), 4.05(q, J = 10.0 Hz, 1H), 3.65–3.73(ddd, J = 27.8, 11.9, 5.3 Hz, 1H), 3.46–3.38(m, 1H), 3.43(s, 1H), 3.01(s, 4H), 2.25(s, 1H), 2.15(s, 1H), 1.39(s, 9H). 13C-NMR (125 MHz, DMSO-d6) δ/ppm = 171.00, 156.22, 155.12, 141.34, 140.04, 138.05, 129.57, 127.94, 122.05, 120.69, 78.25, 72.56, 67.37, 60.42, 52.99, 47.22, 28.87, 21.43, 14.74.
Synthesis of Compound 8. To a stirred solution of fragments B (3.5 g, 4.4 mmol) and C (1.4 g, 4.4 mmol) in dry THF (50 mL) was added TBTU (2.1 g, 6.6 mmol) and 4-methylmorpholine (0.9 g, 8.8 mmol), slowly. The mixture was stirred at room temperature for 6 h and solvents were removed by vacuo. The residue was dissolved in EtOAc (100 mL) and washed with 5% NaHCO3 solution (100 mL × 3), 5% citric acid solution (100 mL × 3), brine, and dried over Na2SO4. Solvents were evaporated off, after which 4.7 g of 8 was obtained after recrystallization in MeOH in 88% yield. HRMS: m/z Calcd. 1202.5603 For C71H76N7O11, Found 1202.5590; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 10.72(s, 1H), 8.64(d, J = 8.0 Hz, 1H), 8.27(d, J = 4.0 Hz, 1H), 8.17(d, J = 8.0 Hz, 1H), 7.97–7.91(dd, J = 16.0, 4.0 Hz, 1H), 7.85(d, J = 8.0 Hz, 2H), 7.74–7.68(dd, J = 16.0, 8.0 Hz, 2H), 7.53(d, J = 4.0 Hz, 1H), 7.37–7.33(m, 5H), 7.32–7.25(m, 7H), 7.23–7.17(m, 8H), 7.13(d, J = 8.0 Hz, 2H), 7.00(t, J = 6.0Hz, 1H), 6.93–6.89(m, 2H), 6.84(d, J = 8.0 Hz, 2H), 6.67(t, J = 6.0 Hz, 1H), 5.38(d, J = 8.0 Hz, 1H), 4.97(s, 2H), 4.48–4.45(m, 3H), 4.22–4.16(m, 3H), 4.09(s, 1H), 3.58–3.51(m, 1H), 3.47(s, 2H), 3.07–2.98(m, 3H), 2.94–2.86(m, 2H), 2.79–2.74(m, 3H), 1.48–1.43(m, 2H), 1.33(s, 9H), 1.20(s, 2H), 0.93(d, J = 8.0 Hz, 2H); 13C-NMR(125 MHz, DMSO-d6) δ/ppm = 171.88, 170.73, 157.52, 156.22, 144.66, 144.22, 141.30, 140.05, 138.97, 138.05, 137.77, 136.62, 130.88, 129.72, 129.61, 129.02, 128.90, 128.30, 127.98, 127.73, 127.62, 127.23, 126.20, 122.05, 120.70, 119.10, 118.83, 114.92, 111.83, 110.48, 110.00, 78.05, 69.71, 66.72, 58.40, 54.52, 53.49, 52.30, 47.17, 37.29, 35.75, 31.93, 29.87, 28.89, 27.19, 22.93.
Synthesis of Compound 9. To a stirred solution of 8 (4.0 g, 3.3 mmol) in dry THF (40 mL) was added piperidine (16.5 mL, 165 mmol), dropwise, at 0 °C and the mixture was stirred at room temperature for 40 min. Solvents were evaporated off, after which 3.0 g of amine intermediate was obtained after recrystallization in MeOH in 93% yield. HRMS: m/z Calcd. 980.4922 For C56H66N7O9, Found 980.4932. To a stirred solution of the above amine (1.7 g, 3.1 mmol) and fragment D (3.0 g, 3.1 mmol) in dry THF (50 mL) was added TBTU (1.5 g, 4.7 mmol) and 4-methylmorpholine (0.9 g, 9.3 mmol), slowly. The mixture was stirred at room temperature for 6 h and solvents were removed by vacuo. The residue was dissolved in EtOAc (100 mL) and washed with 5% NaHCO3 solution (100 mL × 3), 5% citric acid solution (100 mL × 3), brine, and dried over Na2SO4. Solvents were evaporated off, after which 3.96 g of 9 was obtained after recrystallization in MeOH in 85% yield. HRMS: m/z Calcd. 1501.7084 For C84H97N10O16, Found 1501.7087; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 10.69(dd, J = 27.1, 6.2 Hz, 1H), 8.54(dd, J = 42.4, 12.0 Hz, 2H), 8.36(dd, J = 21.2, 8.6 Hz, 1H), 8.08(dd, J = 19.8, 9.3 Hz, 1H), 7.96–7.90(m, 1H), 7.87(d, J = 5.0 Hz, 2H), 7.59(d, J = 7.8 Hz, 2H), 7.56–7.51(m, 1H), 7.42–7.11(m, 26H), 6.88(m, 2H), 5.58(dd, J = 39.8, 8.0 Hz, 1H), 4.96(m, 2H), 4.89–4.70(m, 1H), 4.49(s, 2H), 4.23(d, J = 9.8 Hz, 2H), 4.15(s, 1H), 4.08–4.00(m, 1H), 3.90(s, 1H), 3.80(t, J = 14.3 Hz, 1H), 3.52(d, J = 4.9 Hz, 3H), 3.29(s, 2H), 3.07–2.93(m, 9H), 2.92–2.82(m, 4H), 2.28–2.14(m, 1H),2.02(dd, J = 15.3, 9.7 Hz, 1H), 1.55–1.40(m, 2H), 1.35(s, 18H), 1.30–1.23(m, 2H), 0.97(m, J = 16.8, 10.8 Hz, 2H); 13C-NMR (125 MHz, DMSO-d6) δ/ppm = 172.25, 171.84, 171.77, 171.37, 170.22, 157.60, 156.14, 154.48, 144.47, 141.36, 138.60, 137.58, 130.90, 129.70, 129.00, 128.90, 128.39, 128.27, 128.09, 127.77, 127.32, 127.21, 125.70, 124.82, 122.92, 120.65, 120.12, 116.62, 114.99, 77.93, 73.23, 67.42, 62.14, 58.56, 58.06, 54.54, 54.25, 53.15, 52.42, 47.13, 37.79, 37.35, 36.43, 32.33, 31.43, 29.82, 28.88, 28.36, 23.02,
Synthesis of Compound 10. To a stirred solution of 9 (2.0 g, 1.3 mmol) in dry THF (20 mL) was added piperidine (6.5 mL, 65 mmol), dropwise, at 0 °C and the mixture was stirred at room temperature for 30 min. Solvents were evaporated off and residue was re-dissolved in dry THF (20 mL). Next, LiBr (0.5 g, 6.5 mmol) in H2O (1 mL) was added, dropwise, to the above solution at 0 °C followed by addition of 1M NaOH solution (6.5 mL, 6.5 mmol) over a period of 3 h. The mixture was stirred at room temperature for 12 h and concentrated in vacuo to yield 1.2 g of 10, as a white solid, in 72% yield. HRMS: m/z Calcd. 1265.6247 For C68H85N10O14, Found 1265.6262; 1H-NMR(600 MHz, DMSO-d6) δ/ppm = 10.72(s, 1H), 8.95(s, 1H), 8.74–8.63(m, 1H), 8.16(t, J = 9.4 Hz, 1H), 8.06(d, J = 7.3 Hz, 1H), 7.87(dd, J = 11.8, 8.0 Hz, 1H), 7.54(d, J = 7.9 Hz, 1H), 7.39(d, J = 7.2 Hz, 2H), 7.37–7.32(m, 2H), 7.32–7.28(m, 1H), 7.29–7.14(m, 10H), 7.14–6.98(m, 4H), 6.92–6.88(m, 1H), 6.85(d, J = 8.3 Hz, 2H), 6.79(dd, J = 18.0, 7.3 Hz, 1H), 6.68(t, J = 5.7 Hz, 1H), 5.56(d, J = 40.3, 8.0 Hz, 1H), 5.00(s, 2H), 4.54(dq, J = 35.6, 7.4 Hz, 1H), 4.46–4.34(m, 2H), 4.07(s, 2H), 3.09–2.88(m, 9H), 2.74(dt, J = 31.7, 10.3 Hz, 3H), 2.64(d, J = 15.3 Hz, 1H), 2.55(d, J = 15.3 Hz, 1H), 2.26(d, J = 44.3 Hz, 1H), 1.94–1.83(m, 1H), 1.46–1.37(m, 1H), 1.36–1.31(m, 18H), 1.27(t, J = 7.6 Hz, 1H), 1.19(dt, J = 17.5, 8.5 Hz, 2H), 0.98–0.79(m, 2H); 13C-NMR(150 MHz, DMSO-d6) δ/ppm = 173.02, 171.60, 171.16, 169.77, 157.28, 156.09, 139.56, 139.27, 138.16, 137.58, 136.65, 136.48, 130.59, 130.15, 130.05, 128.81, 128.72, 128.12, 128.03, 127.81, 127.06, 125.98, 124.16, 121.24, 118.99, 118.65, 114.75, 111.70, 110.77, 110.38, 77.77, 76.08, 69.51, 59.74, 55.92, 55.35, 55.10, 53.90, 53.07, 38.35, 37.75, 37.13, 32.22, 31.65, 29.59, 28.68, 27.97, 23.02.
Synthesis of intermediate A. To a stirred solution of 10 (1.0 g, 0.8 mmol) in dry DMF (250 mL) was added HATU (0.8 g, 2.0 mmol) and anhydrous HOBt (0.3 g, 2.0 mmol), slowly, and the mixture was stirred at −5 °C for 2 h. After the reaction was completed, H2O (20 mL) was added to the above solution and white solid precipitated out. The mixture was filtered through celite and the solid residue was obtained to give 0.86 g of intermediate A in 87% yield. HRMS: m/z Calcd. 1247.6141 For C68H83N10O13, Found 1247.6098; 1H-NMR(600 MHz, DMSO-d6) δ/ppm = 10.85(s, 1H), 9.34(d, J = 7.4 Hz, 1H), 8.33(d, J = 6.8 Hz, 1H), 7.90(d, J = 12.6 Hz, 1H), 7.71(d, J = 9.8 Hz 1H), 7.55(d, J = 16.2 Hz, 1H), 7.43(d, J = 18.4 Hz, 2H), 7.33(ddt, J = 36.4, 21.3, 7.4 Hz, 10H), 7.22(dd, J = 15.1, 7.6 Hz, 3H), 7.19–7.14(m,1H), 7.06–6.91(m, 3H), 6.88(d, J = 6.5 Hz, 1H), 6.83–6.74(m, 3H), 6.72–6.61(m, 1H), 5.39(d, J = 8.3 Hz, 1H), 5.18(s, 1H), 5.05–4.92(m, 3H), 4.60–4.46(m, 1H), 4.49–4.32(m, 2H), 3.69(dd, J = 34.6, 7.6 Hz, 1H), 3.62–3.51(m, 1H), 3.04–2.85(m, 7H), 2.83–2.55(m, 5H), 2.15(d, J = 7.0 Hz, 1H), 2.08–1.89(m, 1H), 1.44(s, 1H), 1.33(s, 18H), 1.26–1.08(m, 3H), 1.03–0.86(m, 2H); 13C-NMR(150 MHz, DMSO-d6) δ/ppm = 172.60, 171.70, 171.17, 169.87, 157.41, 156.06, 139.95, 137.64, 137.56, 136.76, 136.45, 136.41, 131.19, 130.67, 130.28, 129.73, 129.31, 129.16, 128.85, 128.50, 128.24, 128.12, 127.43, 126.28, 124.21, 121.27, 118.62, 114.50, 111.65, 109.67, 78.22, 77.89, 69.58, 59.91, 57.41, 54.89, 53.76, 51.74, 51.11, 36.62, 36.29, 31.70, 30.95, 29.54, 28.69, 27.95, 22.98.
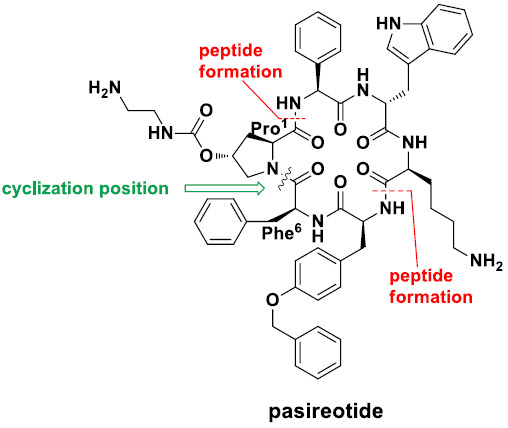
Synthesis of pasireotide. TFA (6.0 mL) was added to intermediate A (0.5 g, 0.4 mmol) and the mixture was stirred at 0 °C for 2 h. After the reaction was completed, excess TFA was evaporated off. Diethyl ether was added to the residue and a white solid was precipitated off to give 0.45 g of pasireotide after filtration (89% yield). ESI-MS(m/z): 524.04[M/2 + H−CF3COOH]+; 1047.86[M + H−2CF3COOH]+; 1069.81[M + Na−2CF3COOH]+; HRMS: m/z Calcd. 1047.5092 For C58H67N10O9, Found 1047.5107; 1H-NMR(600 MHz, DMSO-d6) δ/ppm = 10.87(s, 1H), 7.82(d, J = 42 Hz, 10H), 7.42–7.38(m, 5H), 7.36–7.25(m, 10H), 7.23–7.21(t, J = 6.0 Hz, 3H), 7.07–7.04(t, J = 9.0 Hz, 1H), 7.0(s, 1H), 6.96–6.93(t, J = 9.0 Hz, 3H), 6.82(d, J = 12 Hz, 2H), 5.48(d, J = 6.0 Hz, 1H), 5.22(s, 1H), 5.01(s, 2H), 4.59(d, J = 6.0 Hz, 1H), 4.46–4.43(dd, J = 10.2, 7.0 Hz, 1H), 4.41–4.39(m, 1H), 3.98(s, 1H), 3.78(d, J = 12.0 Hz, 1H), 3.70(q, J = 6.0 Hz, 1H), 3.60(d, J = 6.0 Hz, 1H), 3.28–3.24(m, 2H), 3.22–3.16(m, 2H), 3.03–3.01(m, 2H), 2.90–2.84(m, 4H), 2.69(d, J = 12.0 Hz, 2H), 2.20(d, J = 6.0 Hz, 1H), 2.05–2.01(t, J = 12.0 Hz, 1H), 1.43–1.27(m, 2H), 1.22–1.17(m, 2H), 0.92(s, 1H), 0.72(s, 1H); 13C-NMR (150 MHz, DMSO-d6) δ/ppm = 172.57, 170.58, 170.30, 170.10, 168.90, 157.33, 156.03, 138.32, 137.61, 137.01, 136.43, 130.50, 130.32, 130.27, 128.92, 128.88, 128.50, 128.26, 128.20, 128.15, 128.08, 127.04, 124.29, 121.30, 120.68, 118.88, 118.70, 116.71, 114.80, 111.70, 109.77, 74.13, 69.60, 59.97, 57.72, 57.55, 56.71, 56.14, 54.79, 53.11, 40.52, 39.12, 38.96, 38.45, 37.54, 36.25, 34.49, 31.09, 26.93, 22.22.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}