
Critical Appraisal of Kinetic Calculation Methods Applied to Overlapping Multistep Reactions




Abstract
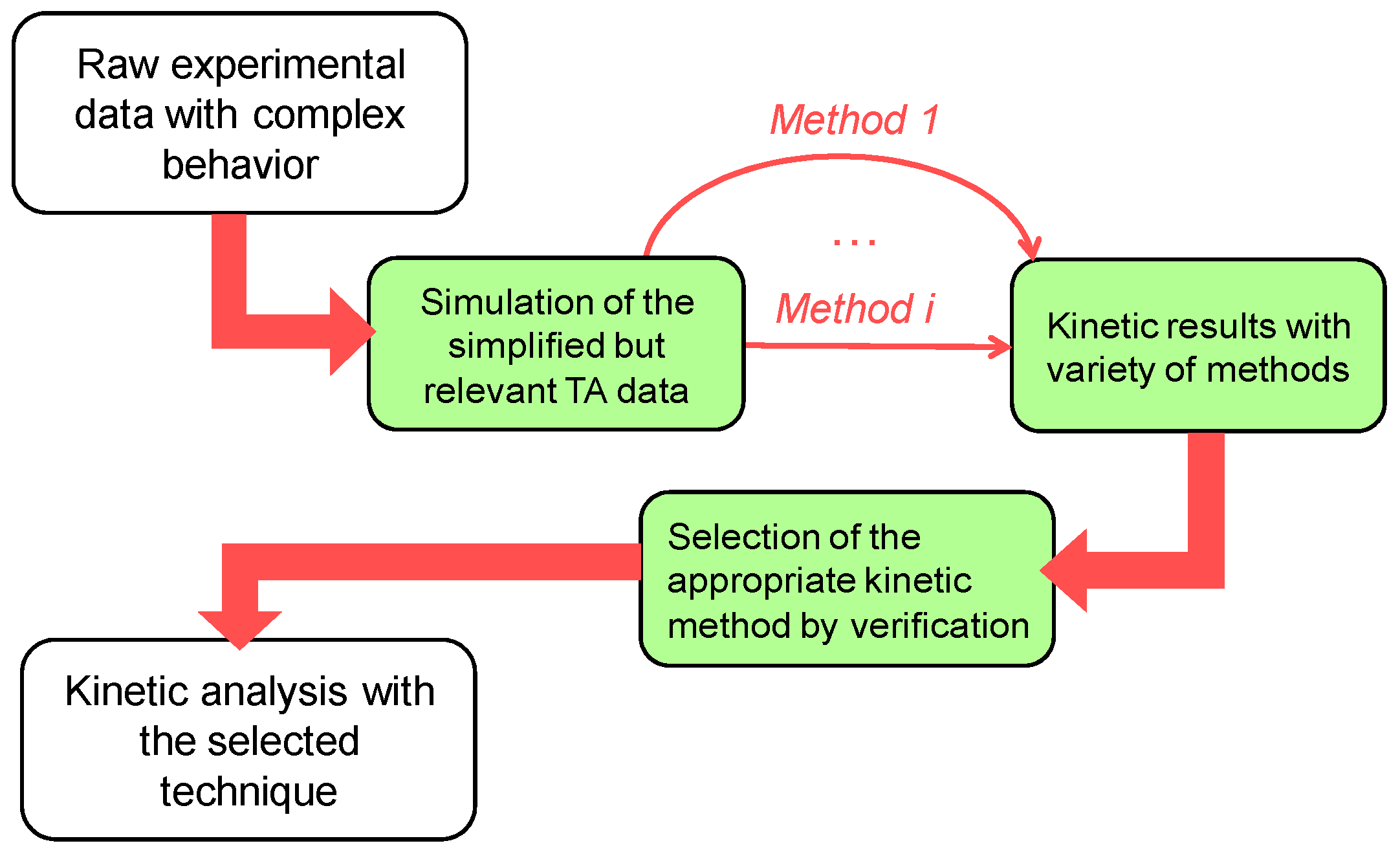
:1. Introduction
2. Theoretical
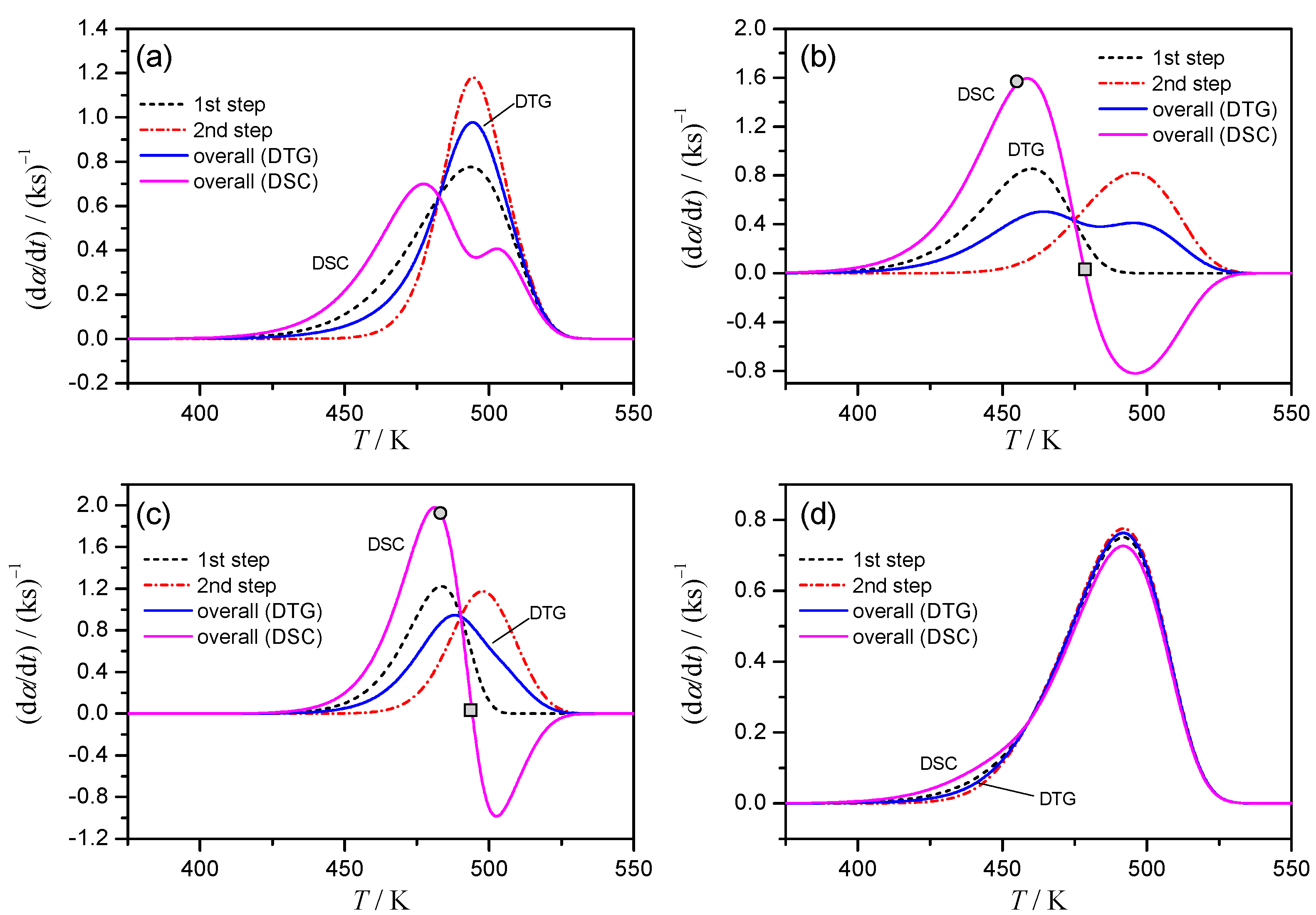
2.1. Simulation of Successive Reactions
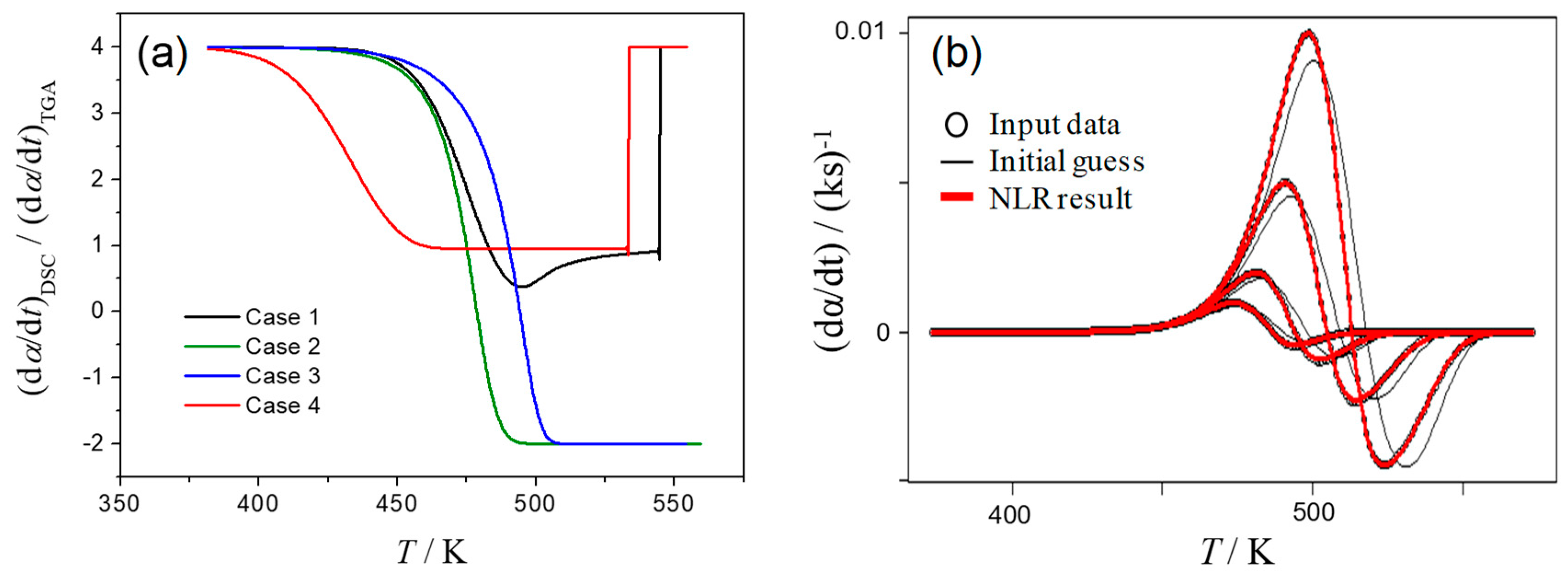
- Case 1: the rate constant for the first reaction, k1, is larger than that of the second reaction, k2, and this difference decreases with increasing temperature, T;
- Case 2: the value of k1 is larger than k2, and its ratio, k1/k2, is constant independent of T;
- Case 3: the value of k1 is smaller than k2, and this difference increases with T;
- Case 4: the value of k1 is smaller than k2, and k1/k2 is constant independent of T.
2.2. Kinetic Calculation Methods
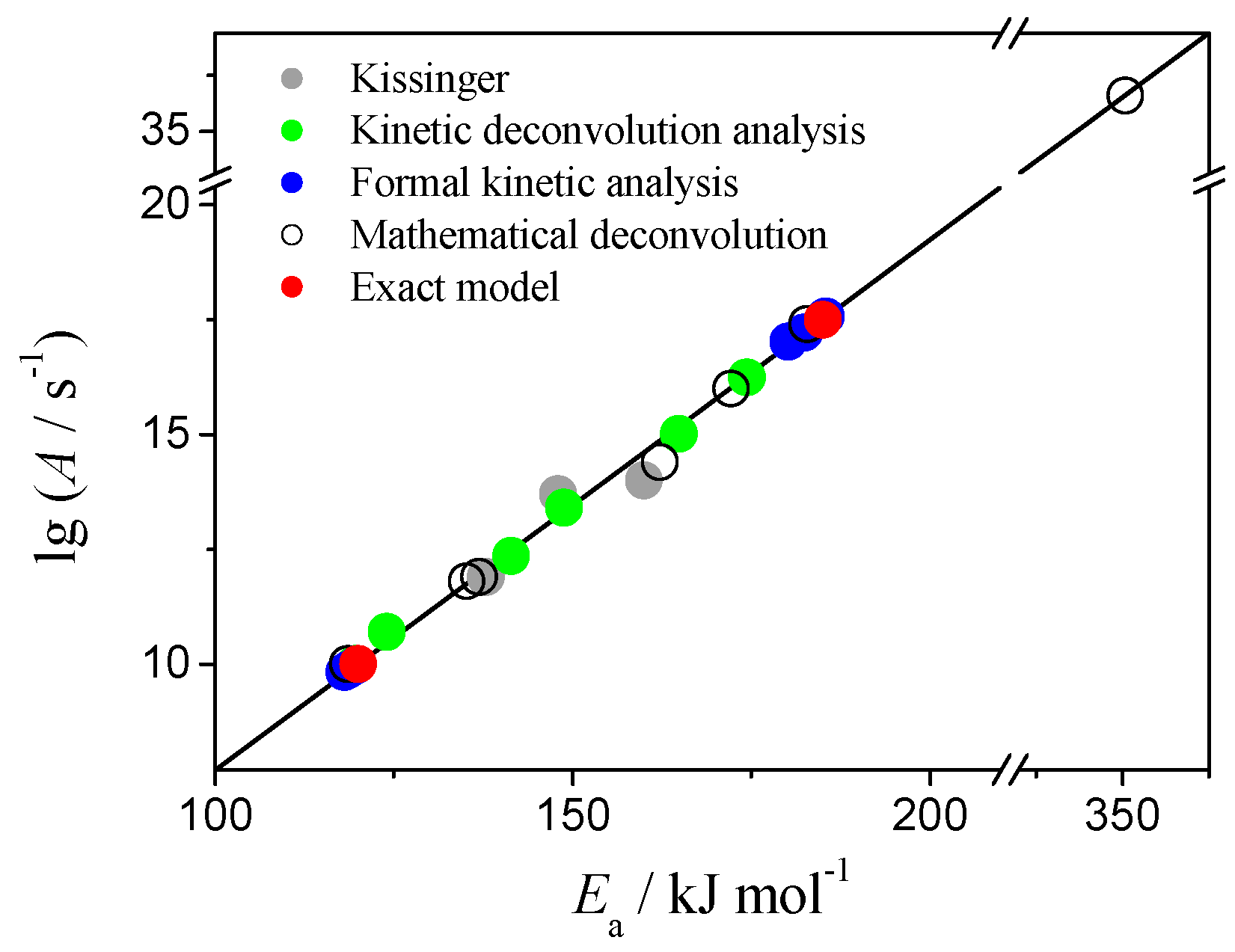
3. Results and Discussion
3.1. Apparent Features of the Simulated Kinetic Datas
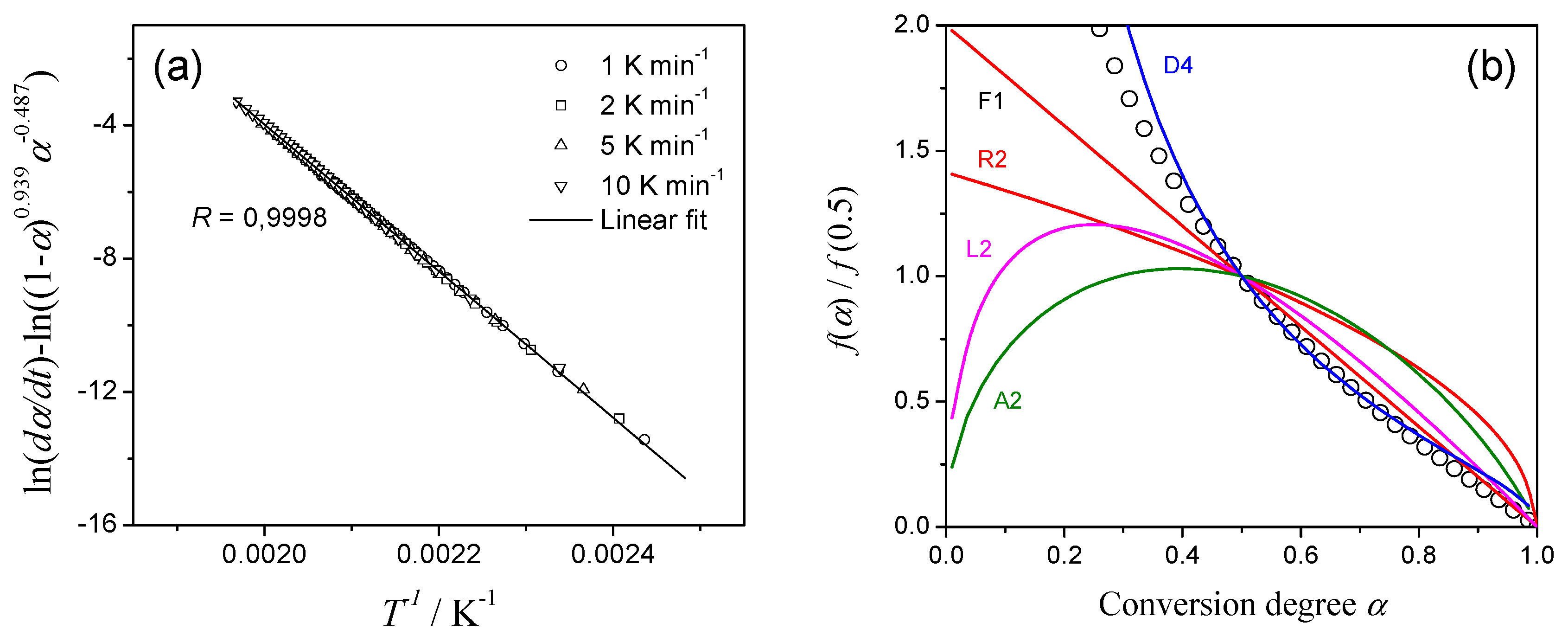
3.2. Kissinger Method
3.3. Isoconversional Methods
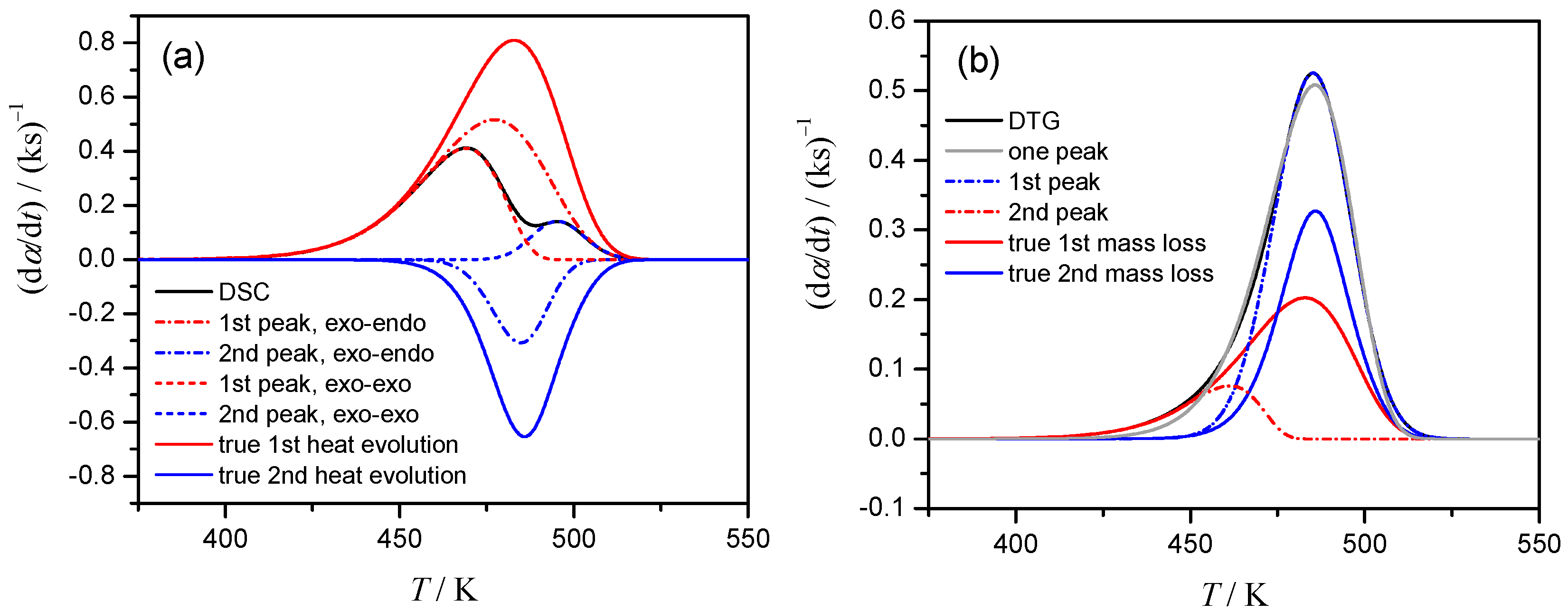
3.4. Mathematical Deconvolution and Subsequent Combined Kinetic Analysis
3.5. Kinetic Deconvolution Analysis
3.6. Formal Kinetic Analysis
4. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kaisersberger, E.; Opfermann, J. Kinetic evaluation of exothermal reactions measured by DSC. Thermochim. Acta 1991, 187, 151–158. [Google Scholar] [CrossRef]
- Burnham, A.K.; Weese, R.K.; Weeks, B.L. A Distributed Activation Energy Model of Thermodynamically Inhibited Nucleation and Growth Reactions and Its Application to the β−δ Phase Transition of HMX. J. Phys. Chem. B 2004, 108, 19432–19441. [Google Scholar] [CrossRef]
- Kitabayashi, S.; Koga, N. Thermal Decomposition of Tin(II) Oxyhydroxide and Subsequent Oxidation in Air: Kinetic Deconvolution of Overlapping Heterogeneous Processes. J. Phys. Chem. C 2015, 119, 16188–16199. [Google Scholar] [CrossRef] [Green Version]
- Galwey, A.K.; Brown, M.E. Thermal Decomposition of Ionic Solids: Chemical Properties and Reactivities of Ionic Crystalline Phases; Elsevier: Burlington, NJ, USA, 1999; ISBN 978-0-08-054279-9. [Google Scholar]
- Long, G.T.; Vyazovkin, S.; Brems, B.A.; Wight, C.A. Competitive Vaporization and Decomposition of Liquid RDX. J. Phys. Chem. B 2000, 104, 2570–2574. [Google Scholar] [CrossRef] [Green Version]
- Muravyev, N.V.; Monogarov, K.A.; Asachenko, A.F.; Nechaev, M.S.; Ananyev, I.V.; Fomenkov, I.V.; Kiselev, V.G.; Pivkina, A.N. Pursuing reliable thermal analysis techniques for energetic materials: Decomposition kinetics and thermal stability of dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (TKX-50). Phys. Chem. Chem. Phys. 2017, 19, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Koga, N.; Kameno, N.; Tsuboi, Y.; Fujiwara, T.; Nakano, M.; Nishikawa, K.; Iwasaki Murata, A. Multistep thermal decomposition of granular sodium perborate tetrahydrate: A kinetic approach to complex reactions in solid–gas systems. Phys. Chem. Chem. Phys. 2018, 20, 12557–12573. [Google Scholar] [CrossRef] [PubMed]
- Kameno, N.; Koga, N. Heterogeneous Kinetic Features of the Overlapping Thermal Dehydration and Melting of Thermal Energy Storage Material: Sodium Thiosulfate Pentahydrate. J. Phys. Chem. C 2018, 122, 8480–8490. [Google Scholar] [CrossRef]
- Opfermann, J.; Hädrich, W. Prediction of the thermal response of hazardous materials during storage using an improved technique. Thermochim. Acta 1995, 263, 29–50. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, P.E.; Perejón, A.; Criado, J.M.; Diánez, M.J.; Pérez-Maqueda, L.A. Kinetic model for thermal dehydrochlorination of poly(vinyl chloride). Polymer 2010, 51, 3998–4007. [Google Scholar] [CrossRef] [Green Version]
- Koga, N.; Yamada, S.; Kimura, T. Thermal Decomposition of Silver Carbonate: Phenomenology and Physicogeometrical Kinetics. J. Phys. Chem. C 2013, 117, 326–336. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. General treatment of the thermogravimetry of polymers. J. Res. Natl. Bur. Stand. Phys. Chem. 1966, 70A, 487. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Linert, W. The Application of Isoconversional Methods for Analyzing Isokinetic Relationships Occurring at Thermal Decomposition of Solids. J. Solid State Chem. 1995, 114, 392–398. [Google Scholar] [CrossRef]
- Agrawal, R.K. Analysis of non-isothermal reaction kinetics: Part 2. Complex reactions. Thermochim. Acta 1992, 203, 111–125. [Google Scholar] [CrossRef]
- Svoboda, R.; Málek, J. Is the original Kissinger equation obsolete today? J. Therm. Anal. Calorim. 2014, 115, 1961–1967. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- ASTM. ASTM E698-05 Standard Test Method for Arrhenius Kinetic Constants for Thermally Unstable Materials; ASTM International: West Conshohocken, PA, USA, 2005. [Google Scholar]
- Vyazovkin, S.V.; Goryachko, V.I.; Lesnikovich, A.I. An approach to the solution of the inverse kinetic problem in the case of complex processes. Part III. Parallel independent reactions. Thermochim. Acta 1992, 197, 41–51. [Google Scholar] [CrossRef]
- Vyazovkin, S. Conversion dependence of activation energy for model DSC curves of consecutive reactions. Thermochim. Acta 1994, 236, 1–13. [Google Scholar] [CrossRef]
- Vyazovkin, S.V.; Lesnikovich, A.I. An approach to the solution of the inverse kinetic problem in the case of complex processes. Thermochim. Acta 1990, 165, 273–280. [Google Scholar] [CrossRef]
- Criado, J.M.; Sánchez-Jiménez, P.E.; Pérez-Maqueda, L.A. Critical study of the isoconversional methods of kinetic analysis. J. Therm. Anal. Calorim. 2008, 92, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Wu, W.; Liu, R. Isoconversional Kinetic Analysis of Complex Solid-State Processes: Parallel and Successive Reactions. Ind. Eng. Chem. Res. 2012, 51, 16157–16161. [Google Scholar] [CrossRef]
- Moukhina, E. Determination of kinetic mechanisms for reactions measured with thermoanalytical instruments. J. Therm. Anal. Calorim. 2012, 109, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Burnham, A.K. Application of the Šesták-Berggren Equation to Organic and Inorganic Materials of Practical Interest. J. Therm. Anal. Calorim. 2000, 60, 895–908. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Criado, J.M.; Sánchez-Jiménez, P.E. Combined Kinetic Analysis of Solid-State Reactions: A Powerful Tool for the Simultaneous Determination of Kinetic Parameters and the Kinetic Model without Previous Assumptions on the Reaction Mechanism. J. Phys. Chem. A 2006, 110, 12456–12462. [Google Scholar] [CrossRef]
- Perejón, A.; Sánchez-Jiménez, P.E.; Criado, J.M.; Pérez-Maqueda, L.A. Kinetic Analysis of Complex Solid-State Reactions. A New Deconvolution Procedure. J. Phys. Chem. B 2011, 115, 1780–1791. [Google Scholar] [CrossRef] [Green Version]
- Noda, Y.; Koga, N. Phenomenological Kinetics of the Carbonation Reaction of Lithium Hydroxide Monohydrate: Role of Surface Product Layer and Possible Existence of a Liquid Phase. J. Phys. Chem. C 2014, 118, 5424–5436. [Google Scholar] [CrossRef]
- Koga, N.; Kodani, S. Thermally induced carbonation of Ca(OH)2 in a CO2 atmosphere: Kinetic simulation of overlapping mass-loss and mass-gain processes in a solid–gas system. Phys. Chem. Chem. Phys. 2018, 20, 26173–26189. [Google Scholar] [CrossRef]
- Schmid, H.; Eisenreich, N.; Krause, C.; Pfeil, A. Analysis of complex thermoanalytical curves: The thermodynamic and kinetic parameters of isopropylammonium nitrate. J. Therm. Anal. Calorim. 1989, 35, 569–576. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Koga, N.; Meerov, D.B.; Pivkina, A.N. Kinetic analysis of overlapping multistep thermal decomposition comprising exothermic and endothermic processes: Thermolysis of ammonium dinitramide. Phys. Chem. Chem. Phys. 2017, 19, 3254–3264. [Google Scholar] [CrossRef]
- Nakano, M.; Wada, T.; Koga, N. Exothermic Behavior of Thermal Decomposition of Sodium Percarbonate: Kinetic Deconvolution of Successive Endothermic and Exothermic Processes. J. Phys. Chem. A 2015, 119, 9761–9769. [Google Scholar] [CrossRef] [Green Version]
- Heym, F.; Etzold, B.J.M.; Kern, C.; Jess, A. Analysis of evaporation and thermal decomposition of ionic liquids by thermogravimetrical analysis at ambient pressure and high vacuum. Green Chem. 2011, 13, 1453. [Google Scholar] [CrossRef]
- Skala, D.; Sokić, M.; Tomić, J.; Kopsch, H. Kinetic analysis of consecutive reactions using TG and DSC techniques: Theory and application. J. Therm. Anal. Calorim. 1989, 35, 1441–1458. [Google Scholar] [CrossRef]
- Friedman, H.L. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J. Polym. Sci. Part C Polym. Symp. 1964, 6, 183–195. [Google Scholar] [CrossRef]
- Ozawa, T. A New Method of Analyzing Thermogravimetric Data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef] [Green Version]
- Vyazovkin, S. Isoconversional Kinetics of Thermally Stimulated Processes; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-14174-9. [Google Scholar]
- Sánchez-Jiménez, P.E.; Pérez-Maqueda, L.A.; Perejón, A.; Criado, J.M. A new model for the kinetic analysis of thermal degradation of polymers driven by random scission. Polym. Degrad. Stab. 2010, 95, 733–739. [Google Scholar] [CrossRef]
- Opfermann, J. Kinetic Analysis Using Multivariate Non-linear Regression. I. Basic concepts. J. Therm. Anal. Calorim. 2000, 60, 641–658. [Google Scholar] [CrossRef]
- Burnham, A.K.; Dinh, L.N. A comparison of isoconversional and model-fitting approaches to kinetic parameter estimation and application predictions. J. Therm. Anal. Calorim. 2007, 89, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Budrugeac, P.; Homentcovschi, D.; Segal, E. Critical Considerations on the Isoconversional Methods. III. On the evaluation of the activation energy from non-isothermal data. J. Therm. Anal. Calorim. 2001, 66, 557–565. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Criado, J.M. The Accuracy of Senum and Yang’s Approximations to the Arrhenius Integral. J. Therm. Anal. Calorim. 2000, 60, 909–915. [Google Scholar] [CrossRef]
- Muravyev, N.V. THINKS—Thermokinetic Software, Moscow, 2016.
- Starink, M.J. The determination of activation energy from linear heating rate experiments: A comparison of the accuracy of isoconversion methods. Thermochim. Acta 2003, 404, 163–176. [Google Scholar] [CrossRef]
- Vyazovkin, S. Modification of the integral isoconversional method to account for variation in the activation energy. J. Comput. Chem. 2001, 22, 178–183. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Monogarov, K.A.; Bragin, A.A.; Fomenkov, I.V.; Pivkina, A.N. HP-DSC study of energetic materials. Part I. Overview of pressure influence on thermal behavior. Thermochim. Acta 2016, 631, 1–7. [Google Scholar] [CrossRef]
- Roduit, B.; Borgeat, C.; Berger, B.; Folly, P.; Alonso, B.; Aebischer, J.N. The prediction of thermal stability of self-reactive chemicals: From milligrams to tons. J. Therm. Anal. Calorim. 2005, 80, 91–102. [Google Scholar] [CrossRef]
- Koga, N.; Goshi, Y.; Yamada, S.; Pérez-Maqueda, L.A. Kinetic approach to partially overlapped thermal decomposition processes: Co-precipitated zinc carbonates. J. Therm. Anal. Calorim. 2013, 111, 1463–1474. [Google Scholar] [CrossRef]
- Yan, Q.-L.; Zeman, S.; Zhang, J.-G.; Qi, X.-F.; Li, T.; Musil, T. Multistep Thermolysis Mechanisms of Azido-s-triazine Derivatives and Kinetic Compensation Effects for the Rate-Limiting Processes. J. Phys. Chem. C 2015, 119, 14861–14872. [Google Scholar] [CrossRef]
- Svoboda, R.; Málek, J. Applicability of Fraser–Suzuki function in kinetic analysis of complex crystallization processes. J. Therm. Anal. Calorim. 2013, 111, 1045–1056. [Google Scholar] [CrossRef]
- Šesták, J.; Berggren, G. Study of the kinetics of the mechanism of solid-state reactions at increasing temperatures. Thermochim. Acta 1971, 3, 1–12. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Pivkina, A.N. New concept of thermokinetic analysis with artificial neural networks. Thermochim. Acta 2016, 637, 69–73. [Google Scholar] [CrossRef]
- Bohn, M.A. Assessment of description quality of models by information theoretical criteria based on Akaike and Schwarz-Bayes applied with stability data of energetic materials. In Proceedings of the 46th International Annual Conference of the Fraunhofer ICT, Karlsruhe, Germany, 23–26 June 2015; pp. 6.1–6.23. [Google Scholar]
- Roduit, B.; Hartmann, M.; Folly, P.; Sarbach, A.; Baltensperger, R. Prediction of thermal stability of materials by modified kinetic and model selection approaches based on limited amount of experimental points. Thermochim. Acta 2014, 579, 31–39. [Google Scholar] [CrossRef]
- Garn, P.D. An examination of the kinetic compensation effect. J. Therm. Anal. Calorim. 1975, 7, 475–478. [Google Scholar] [CrossRef]
- Agrawal, R.K. On the compensation effect. J. Therm. Anal. Calorim. 1986, 31, 73–86. [Google Scholar] [CrossRef]
- Koga, N. A review of the mutual dependence of Arrhenius parameters evaluated by the thermoanalytical study of solid-state reactions: The kinetic compensation effect. Thermochim. Acta 1994, 244, 1–20. [Google Scholar] [CrossRef]
- Koga, N.; Šesták, J. Kinetic compensation effect as a mathematical consequence of the exponential rate constant. Thermochim. Acta 1991, 182, 201–208. [Google Scholar] [CrossRef]
- Brown, M.E.; Galwey, A.K. The significance of “compensation effects” appearing in data published in “computational aspects of kinetic analysis”: ICTAC project, 2000. Thermochim. Acta 2002, 387, 173–183. [Google Scholar] [CrossRef]
- Koga, N.; Šesták, J.; Málek, J. Distortion of the Arrhenius parameters by the inappropriate kinetic model function. Thermochim. Acta 1991, 188, 333–336. [Google Scholar] [CrossRef]
- Koga, N.; Šesták, J. Further aspects of the kinetic compensation effect. J. Therm. Anal. Calorim. 1991, 37, 1103–1108. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Step | Assumed Kinetic Parameters 1 | Ea/kJ mol−12 | lg(A/s−1) 2 | Δαp 3 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ea/kJ mol−1 | lg(A/s−1) | DTG | DSC | DTG | DSC | DTG | DSC | ||
| 1 | 1 | 120 | 10 | 138 ± 4 | 148 ± 1 | 12.9 ± 0.4 | 13.7 ± 0.1 | 0.06 | 0.19 |
| 2 | 185 | 17.5 | – | 160 ± 10 | – | 14.0 ± 1.1 | – | 0.19 | |
| 2 | 1 | 115 | 10.4 | 115 ± 1 | 115 ± 1 | 10.3 ± 0.1 | 10.4 ± 0.1 | 0.002 | 0.007 |
| 2 | 115 | 9.4 | 115 ± 1 | 115 ± 1 | 9.4 ± 0.01 | 9.4 ± 0.1 | 0.002 | 0.003 | |
| 3 | 1 | 185 | 17.5 | 203 ± 4 | 177 ± 2 | 19.3 ± 0.4 | 16.7 ± 0.3 | 0.11 | 0.11 |
| 2 | 120 | 10 | – | 159 ± 3 | – | 14.0 ± 0.3 | – | 0.16 | |
| 4 | 1 | 115 | 9.5 | 116 ± 3 | 116 ± 2 | 9.6 ± 0.3 | 9.6 ± 0.2 | 0.03 | 0.02 |
| 2 | 115 | 11 | |||||||
| Kinetic Scheme | DSC Data | DTG Data | DSC+DTG Data |
|---|---|---|---|
| Single-step reaction | −4671 | −6339 | −23010 |
| Two parallel reactions | −4888 | −5355 | −18186 |
| Two consecutive reactions | −9873 | −10733 | −32343 |
| Two independent reactions (KDA) | −6791 | −7611 | −24676 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muravyev, N.V.; Pivkina, A.N.; Koga, N. Critical Appraisal of Kinetic Calculation Methods Applied to Overlapping Multistep Reactions. Molecules 2019, 24, 2298. https://doi.org/10.3390/molecules24122298
Muravyev NV, Pivkina AN, Koga N. Critical Appraisal of Kinetic Calculation Methods Applied to Overlapping Multistep Reactions. Molecules. 2019; 24(12):2298. https://doi.org/10.3390/molecules24122298
Chicago/Turabian StyleMuravyev, Nikita V., Alla N. Pivkina, and Nobuyoshi Koga. 2019. "Critical Appraisal of Kinetic Calculation Methods Applied to Overlapping Multistep Reactions" Molecules 24, no. 12: 2298. https://doi.org/10.3390/molecules24122298
APA StyleMuravyev, N. V., Pivkina, A. N., & Koga, N. (2019). Critical Appraisal of Kinetic Calculation Methods Applied to Overlapping Multistep Reactions. Molecules, 24(12), 2298. https://doi.org/10.3390/molecules24122298