4-Azafluorenone and α-Carboline Fluorophores with Green and Violet/Blue Emission

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Emission Characteristics
2.2. Surface Potential Measurement
2.3. Organic Field-Effect Transistors
2.4. Organic Light-Emitting Diodes
3. Materials and Methods
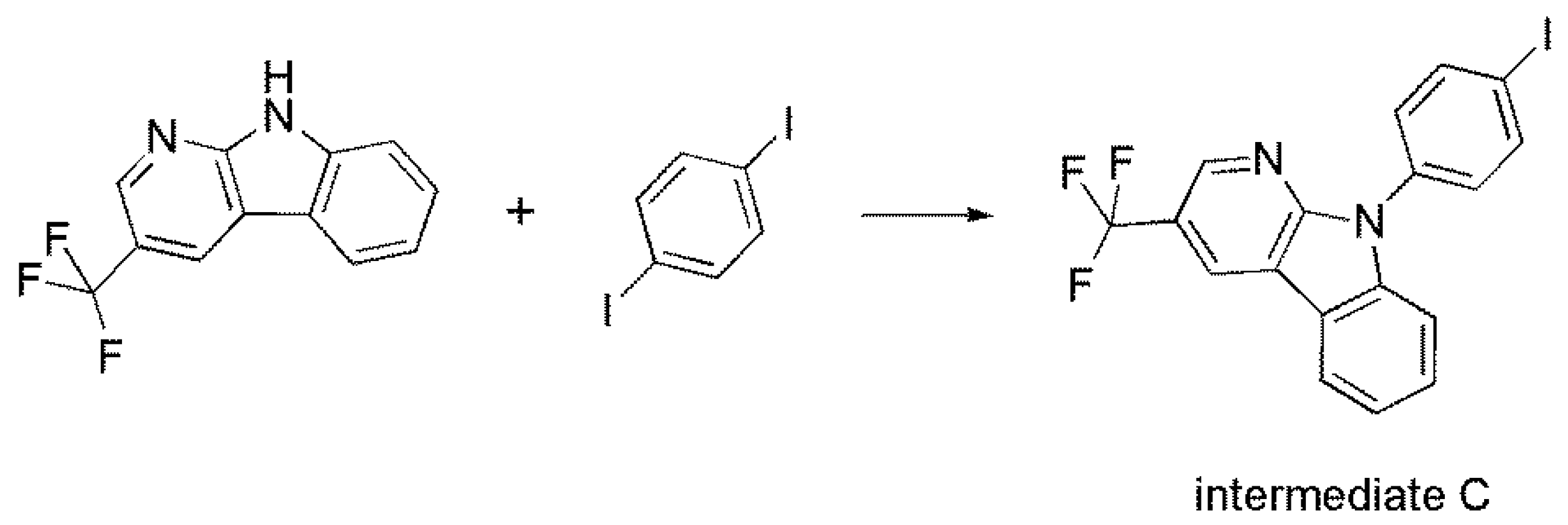
3.1. Synthesis
3.1.1. 4-Azafluorenones
3.1.2. α-Carbolines
3.2. Spectroscopic Measurements
3.3. Organic Thin Films and Thin Film Devices
3.4. Quantum-Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Hu, X.; Liu, J.; Li, B.; Leung, N.L.C.; Viglianti, L.; Cheung, T.S.; Sung, H.H.Y.; Kwok, R.T.K.; Williams, I.D.; et al. Rational design of red AIEgens with a new core structure from non-emissive heteroaromatics. Chem. Sci. 2018, 9, 7829–7834. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Zhuang, Z.; Zhao, Z.; Tang, B.Z. AIEgens based on main group heterocycles. J. Mater. Chem. C 2018, 6, 11835–11852. [Google Scholar] [CrossRef]
- Robb, M.J.; Li, W.; Gergely, R.C.R.; Matthews, C.C.; White, S.R.; Sottos, N.R.; Moore, J.S. A Robust Damage-Reporting Strategy for Polymeric Materials Enabled by Aggregation-Induced Emission. ACS Cent. Sci. 2016, 2, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Houk, K.N. Nitrogen-rich oligoacenes: Candidates for n-channel organic semiconductors. J. Am. Chem. Soc. 2007, 129, 1805–1815. [Google Scholar] [CrossRef]
- Li, G.; Duong, H.M.; Zhang, Z.; Xiao, J.; Liu, L.; Zhao, Y.; Zhang, H.; Huo, F.; Li, S.; Ma, J.; et al. Approaching a stable, green twisted heteroacene through “clean reaction” strategy. Chem. Commun. 2012, 48, 5974. [Google Scholar] [CrossRef]
- Wu, Y.; Yin, Z.; Xiao, J.; Liu, Y.; Wei, F.; Tan, K.J.; Kloc, C.; Huang, L.; Yan, Q.; Hu, F.; et al. Crystal Structure and Phototransistor Behavior of N-Substituted Heptacence. ACS Appl. Mater. Interfaces 2012, 4, 1883–1886. [Google Scholar] [CrossRef]
- Li, G.; Wu, Y.; Gao, J.; Wang, C.; Li, J.; Zhang, H.; Zhao, Y.; Zhao, Y.; Zhang, Q. Synthesis and Physical Properties of Four Hexazapentacene Derivatives. J. Am. Chem. Soc. 2012, 134, 20298–20301. [Google Scholar] [CrossRef]
- Bunz, U.H.F.; Engelhart, J.U.; Lindner, B.D.; Schaffroth, M. Large N-Heteroacenes: New Tricks for Very Old Dogs? Angew. Chemie Int. Ed. 2013, 52, 3810–3821. [Google Scholar] [CrossRef]
- Miao, Q. N-Heteropentacenes and N-Heteropentacenequinones: From Molecules to Semiconductors. Synlett 2012, 23, 326–336. [Google Scholar] [CrossRef]
- Li, J.; Yan, F.; Gao, J.; Li, P.; Xiong, W.-W.; Zhao, Y.; Sun, X.W.; Zhang, Q. Synthesis, physical properties and OLED performance of azatetracenes. Dye. Pigment. 2015, 112, 93–98. [Google Scholar] [CrossRef]
- Zhang, Y.; Lai, S.-L.; Tong, Q.-X.; Lo, M.-F.; Ng, T.-W.; Chan, M.-Y.; Wen, Z.-C.; He, J.; Jeff, K.-S.; Tang, X.-L.; et al. High Efficiency Nondoped Deep-Blue Organic Light Emitting Devices Based on Imidazole-π-triphenylamine Derivatives. Chem. Mater. 2012, 24, 61–70. [Google Scholar] [CrossRef]
- Yang, X.; Xu, X.; Zhou, G. Recent advances of the emitters for high performance deep-blue organic light-emitting diodes. J. Mater. Chem. C 2015, 3, 913–944. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, S.; Ding, J.; Wang, L. Solution processible distyrylarylene-based fluorescent dendrimers: Tuning of carbazole-dendron generation leads to nondoped deep-blue electroluminescence. Org. Electron. 2018, 53, 43–49. [Google Scholar] [CrossRef]
- Liu, H.; Bai, Q.; Li, W.; Guo, Y.; Yao, L.; Gao, Y.; Li, J.; Lu, P.; Yang, B.; Ma, Y. Efficient deep-blue non-doped organic light-emitting diode with improved roll-off of efficiency based on hybrid local and charge-transfer excited state. RSC Adv. 2016, 6, 70085–70090. [Google Scholar] [CrossRef]
- Wang, Z.; Li, X.; Xue, K.; Li, H.; Zhang, X.; Liu, Y.; Yu, Z.; Lu, P.; Chen, P. Towards stable deep-blue emission and low efficiency roll-off in OLEDs based on phenanthroimidazole dimers. J. Mater. Chem. C 2016, 4, 1886–1894. [Google Scholar] [CrossRef]
- Chen, W.-C.; Yuan, Y.; Wu, G.-F.; Wei, H.-X.; Tang, L.; Tong, Q.-X.; Wong, F.-L.; Lee, C.-S. Staggered Face-to-Face Molecular Stacking as a Strategy for Designing Deep-Blue Electroluminescent Materials with High Carrier Mobility. Adv. Opt. Mater. 2014, 2, 626–631. [Google Scholar] [CrossRef]
- Sun, W.; Zhou, N.; Xiao, Y.; Wang, S.; Li, X. Novel carbazolyl-substituted spiro[acridine-9,9′-fluorene] derivatives as deep-blue emitting materials for OLED applications. Dye. Pigment. 2018, 154, 30–37. [Google Scholar] [CrossRef]
- Mucur, S.P.; Kök, C.; Bilgili, H.; Canımkurbey, B.; Koyuncu, S. Conventional and inverted organic light emitting diodes based on bright green emmisive polyfluorene derivatives. Polymer (Guildf). 2018, 151, 101–107. [Google Scholar] [CrossRef]
- Gan, L.; Li, X.; Cai, X.; Liu, K.; Li, W.; Su, S.-J. D–A–D-type orange-light emitting thermally activated delayed fluorescence (TADF) materials based on a fluorenone unit: simulation, photoluminescence and electroluminescence studies. Beilstein J. Org. Chem. 2018, 14, 672–681. [Google Scholar] [CrossRef]
- Ni, F.; Zhu, Z.; Tong, X.; Xie, M.; Zhao, Q.; Zhong, C.; Zou, Y.; Yang, C. Organic emitter integrating aggregation-induced delayed fluorescence and room-temperature phosphorescence characteristics, and its application in time-resolved luminescence imaging. Chem. Sci. 2018, 9, 6150–6155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Einzinger, M.; Zhu, T.; Chae, H.S.; Jeon, S.; Ihn, S.-G.; Sim, M.; Kim, S.; Su, M.; Teverovskiy, G.; et al. Molecular Design of Deep Blue Thermally Activated Delayed Fluorescence Materials Employing a Homoconjugative Triptycene Scaffold and Dihedral Angle Tuning. Chem. Mater. 2018, 30, 1462–1466. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Yuan, Y.; Wang, Q.; Hu, Y.; Khan, A.; Jiang, Z.-Q.; Liao, L.-S. Highly efficient non-doped deep-blue organic light-emitting diodes by employing a highly rigid skeleton. Dye. Pigment. 2018, 158, 396–401. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Zhang, Y.-J.; Fei, X.; Ran, Q.; Fung, M.-K.; Fan, J. Diazaspirocycles: novel platforms for efficient phosphorescent organic light-emitting diodes. J. Mater. Chem. C 2019, 7, 1370–1378. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, Y.J.; Son, Y.H.; Hur, J.A.; Um, H.A.; Shin, J.; Lee, T.W.; Cho, M.J.; Kim, J.K.; Joo, S.; et al. High-efficiency blue phosphorescent organic light-emitting diodes using a carbazole and carboline-based host material. Chem. Commun. 2013, 49, 6788. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Im, Y.; Seo, J.-A.; Lee, J.Y. Carboline derivatives with an ortho-linked terphenyl core for high quantum efficiency in blue phosphorescent organic light-emitting diodes. Chem. Commun. 2013, 49, 9860. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Sølling, T.I.; Diau, E.W.-G. Revisiting the photophysics of 9-fluorenone: Ultrafast time-resolved fluorescence and theoretical studies. Chem. Phys. Lett. 2017, 686, 218–222. [Google Scholar] [CrossRef]
- Ghosh, I.; Mukhopadhyay, A.; Koner, A.L.; Samanta, S.; Nau, W.M.; Moorthy, J.N. Excited-state properties of fluorenones: influence of substituents, solvent and macrocyclic encapsulation. Phys. Chem. Chem. Phys. 2014, 16, 16436–16445. [Google Scholar] [CrossRef]
- Breton, G.W.; Vang, X. Photodimerization of Anthracene. J. Chem. Educ. 1998, 75, 81. [Google Scholar] [CrossRef]
- Becker, H.-D.; Andersson, K. Photochemical Diels—Alder dimerization of 9-phenylethylnylanthracene. J. Photochem. 1984, 26, 75–77. [Google Scholar] [CrossRef]
- Tu, M.; Reinsch, H.; Rodríguez-Hermida, S.; Verbeke, R.; Stassin, T.; Egger, W.; Dickmann, M.; Dieu, B.; Hofkens, J.; Vankelecom, I.; et al. Reversible Optical Writing and Data Storage in an Anthracene-Loaded Metal-Organic Framework. Angew. Chemie 2018, 58, 2423–2427. [Google Scholar] [CrossRef] [PubMed]
- Zdobinsky, T.; Sankar Maiti, P.; Klajn, R. Support Curvature and Conformational Freedom Control Chemical Reactivity of Immobilized Species. J. Am. Chem. Soc. 2014, 136, 2711–2714. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Hohman, J.N.; Cao, Y.; Houk, K.N.; Ma, H.; Jen, A.K.-Y.; Weiss, P.S. Creating favorable geometries for directing organic photoreactions in alkanethiolate monolayers. Science 2011, 331, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.H.; Kaddah, A.M.; El-Abbady, S.A.; Gado, S.H. Cyclocondensation of Cyanoacetamide and N-Substituted Cyanoacetamides with 2-Arylmethylene indan-1-one and -indan-1,3-diones. J. Prakt. Chemie 1982, 324, 1045–1051. [Google Scholar] [CrossRef]
- Shrestha, A.; Park, S.; Jang, H.J.; Katila, P.; Shrestha, R.; Kwon, Y.; Lee, E.-S. A new phenolic series of indenopyridinone as topoisomerase inhibitors: Design, synthesis, and structure-activity relationships. Bioorg. Med. Chem. 2018, 26, 5212–5223. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Z.; Zhang, X.; Fan, X. Synthesis of Functionalized Pyridines via Cu(II)-Catalyzed One-Pot Cascade Reactions of Inactivated Saturated Ketones with Electron-Deficient Enamines. J. Org. Chem. 2017, 82, 11230–11237. [Google Scholar] [CrossRef] [PubMed]
- Hostyn, S.; Van Baelen, G.; Lemière, G.L.F.; Maes, B.U.W. Synthesis of α-Carbolines Starting from 2,3-Dichloropyridines and Substituted Anilines. Adv. Synth. Catal. 2008, 350, 2653–2660. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | λA (nm) | λE (nm) | ΦF (%) | αAIE | |
|---|---|---|---|---|---|
| Soln | Soln | Soln | Film | ||
| 1a | 386 | 475 | 1.0 | 11.2 | 11.2 |
| 1b | 388 | 477 | 0.5 | 75.0 | 150.0 |
| 1c | 376 | 476 | 1.0 | 26.1 | 26.1 |
| Cpd | λA (nm) | λF (nm) | |||
|---|---|---|---|---|---|
| CHCl3 | MeOH | CHCl3 | MeOH | Film | |
| 2a | 368 | 365 | 425/415 * | 408/420 * | 450 |
| 2b | 340 | 342 | 407 | 407 | 455 |
| 2c | 376 | 372 | 422/433 * | 427/413 * | 460 |
| 2d | 403 | 399 | 437 | 432 | 450 |
| 2e | 285 | 294 | 401 | 414 | 420 |
| Cpd | ΦF CHCl3 | ΦF CH3OH | ΦF ss | τ(CHCl3) (ns) | τ(CH3OH) (ns) | τ(ss) (ns) |
|---|---|---|---|---|---|---|
| 2a | 0.58 | 0.29 | 0.49 | τ = 3.52 χ2 = 1.181 | τ = 3.47 χ2 = 1.161 | τ1 = 1.58 (23.0%) τ2 = 4.07 (77.0%) χ2 = 1.194 |
| 2b | 0.48 | 0.53 | 0.41 | τ = 2.91 χ2 = 1.149 | τ = 5.09 χ2 = 1.163 | τ1 = 0.64 (65.1%) τ2 = 2.58 (29.0%) τ3 = 13.1 (5.9%) χ2 = 1.124 |
| 2c | 0.94 | 0.70 | 0.23 | τ = 5.05 χ2 = 1.172 | τ = 5.11 χ2 = 1.159 | τ1 = 1.30 (48.7%) τ2 = 3.86 (51.3%) χ2 = 1.113 |
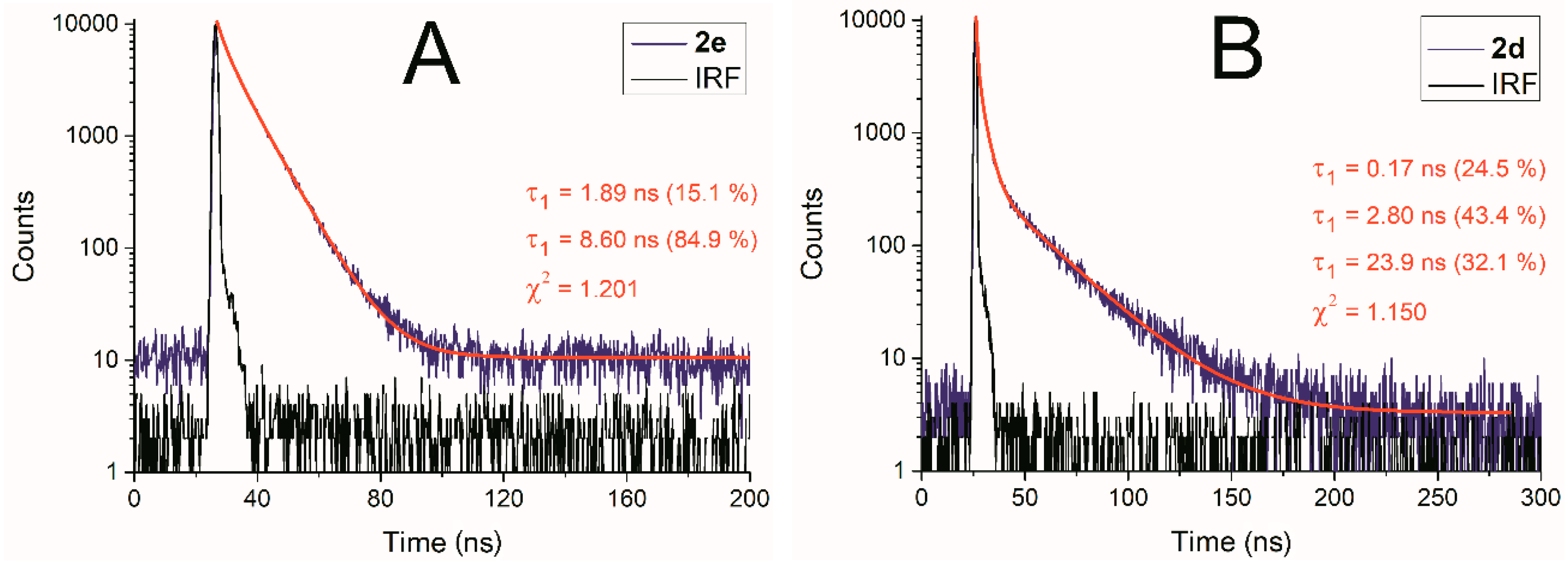
| 2d | 0.68 | 0.74 | 1.00 | τ = 2.95 χ2 = 1.155 | τ = 3.49 χ2 = 1.135 | τ1 = 0.17 (24.5%) τ2 = 2.80 (43.4%) τ3 = 23.9 (32.1%) χ2 = 1.150 |
| 2e | 0.07 | 0.07 | 0.52 | Low signal intensity | Low signal intensity | τ1 = 1.89 (15.1%) τ2 = 8.60 (84.9%) χ2 = 1.201 |
| Cpd | Eg,opt (eV) | EL (eV) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|
| 2a | 3.08 | 3.25; 2.45 | −5.7 | −2.6 |
| 2b | 3.26 | 2.55 | −5.7 | −2.4 |
| 2c | 3.05 | 2.85; 2.60 | −5.0 | −2.0 |
| 2d | 2.82 | 2.30 | −4.5 | −1.7 |
| 2e | 3.08 | 2.95; 2.60; 2.00 | −4.7 | −1.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cigáň, M.; Danko, P.; Brath, H.; Čakurda, M.; Fišera, R.; Donovalová, J.; Filo, J.; Weis, M.; Jakabovič, J.; Novota, M.; et al. 4-Azafluorenone and α-Carboline Fluorophores with Green and Violet/Blue Emission. Molecules 2019, 24, 2378. https://doi.org/10.3390/molecules24132378
Cigáň M, Danko P, Brath H, Čakurda M, Fišera R, Donovalová J, Filo J, Weis M, Jakabovič J, Novota M, et al. 4-Azafluorenone and α-Carboline Fluorophores with Green and Violet/Blue Emission. Molecules. 2019; 24(13):2378. https://doi.org/10.3390/molecules24132378
Chicago/Turabian StyleCigáň, Marek, Peter Danko, Henrich Brath, Matúš Čakurda, Roman Fišera, Jana Donovalová, Juraj Filo, Martin Weis, Ján Jakabovič, Miroslav Novota, and et al. 2019. "4-Azafluorenone and α-Carboline Fluorophores with Green and Violet/Blue Emission" Molecules 24, no. 13: 2378. https://doi.org/10.3390/molecules24132378
APA StyleCigáň, M., Danko, P., Brath, H., Čakurda, M., Fišera, R., Donovalová, J., Filo, J., Weis, M., Jakabovič, J., Novota, M., & Gáplovský, A. (2019). 4-Azafluorenone and α-Carboline Fluorophores with Green and Violet/Blue Emission. Molecules, 24(13), 2378. https://doi.org/10.3390/molecules24132378