Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone

 and
and
Abstract
:1. Introduction
2. Results and Discussion
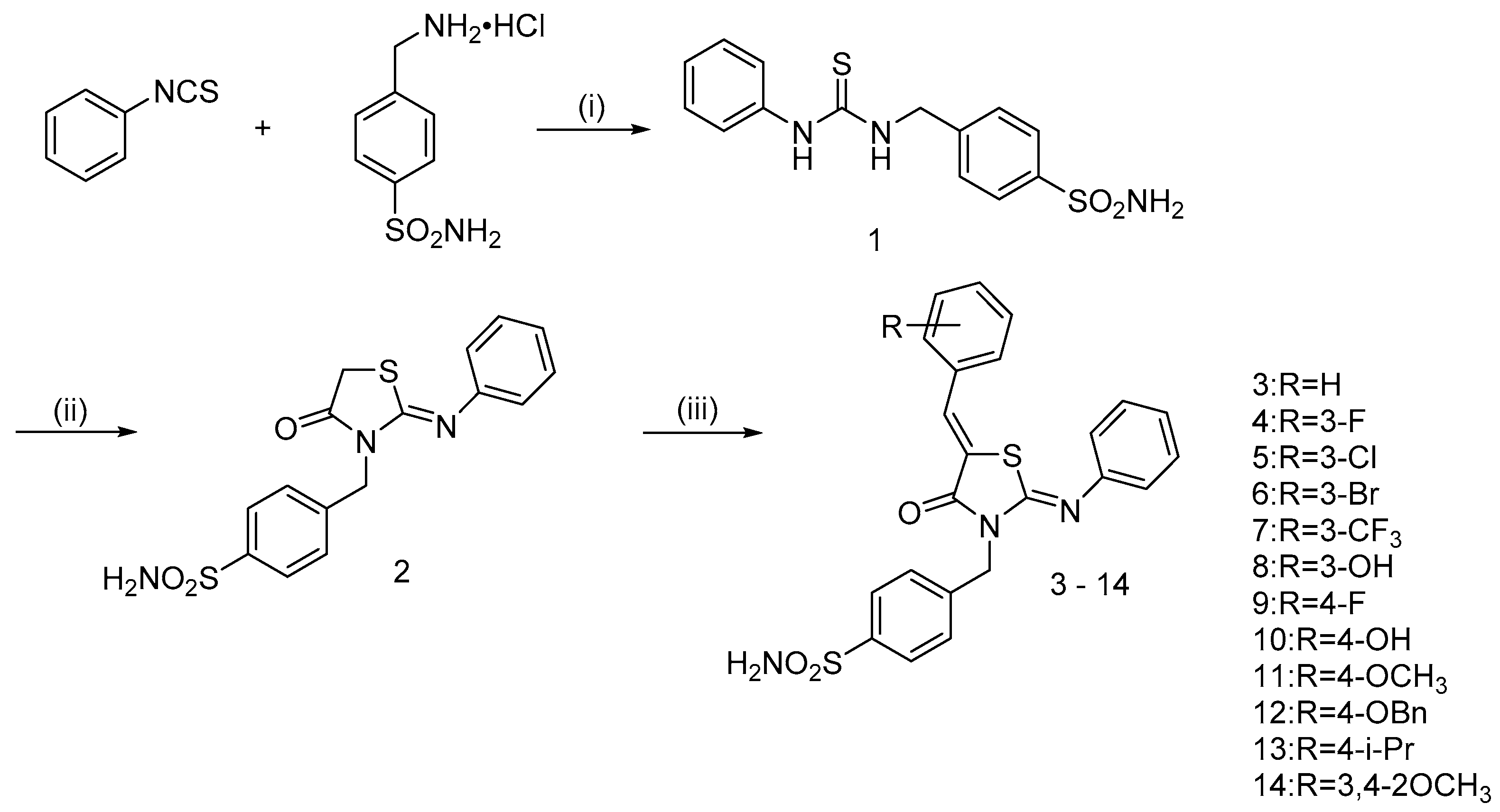
2.1. Chemistry
2.2. hCA Enzyme Inhibition Studies
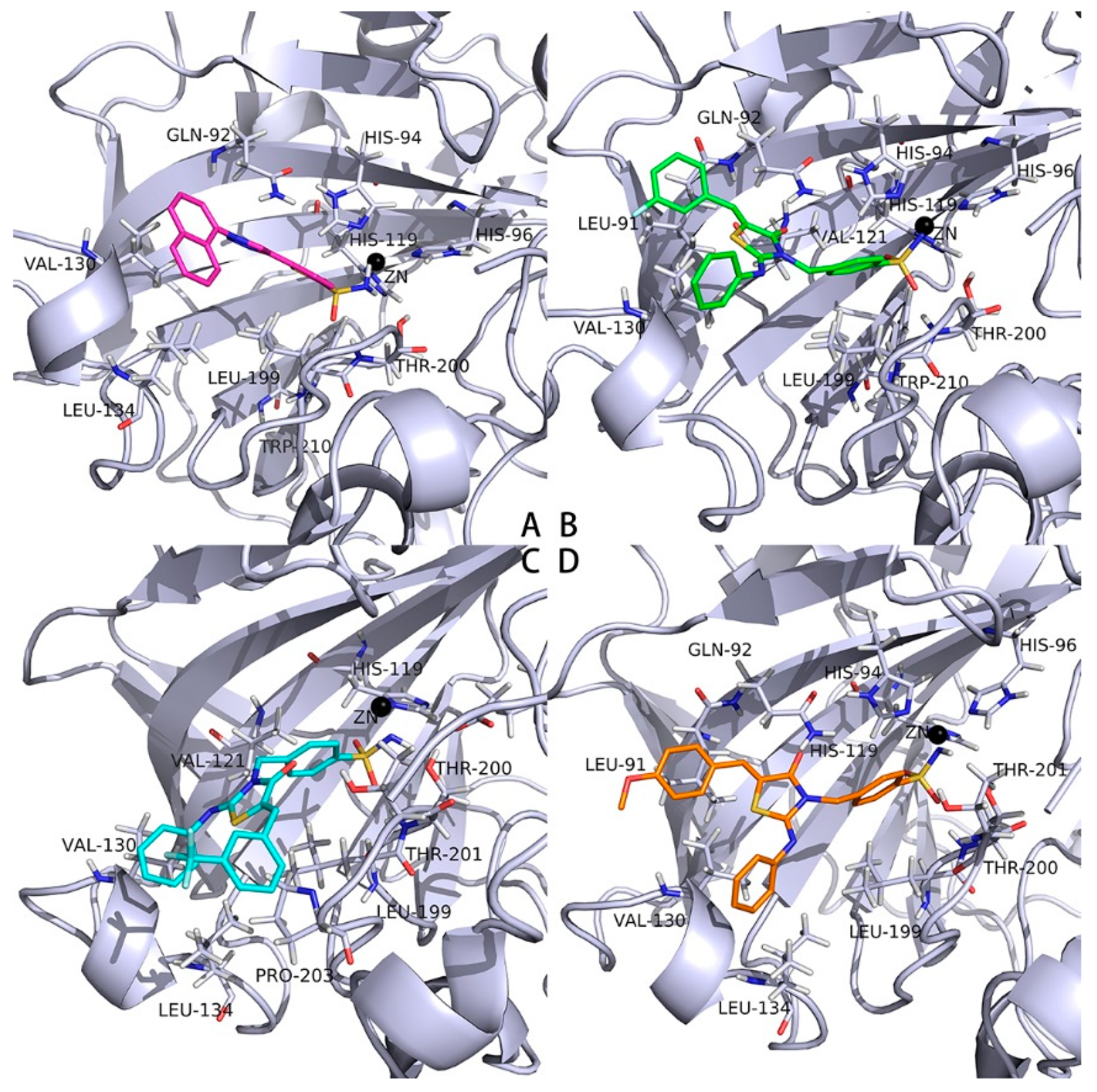
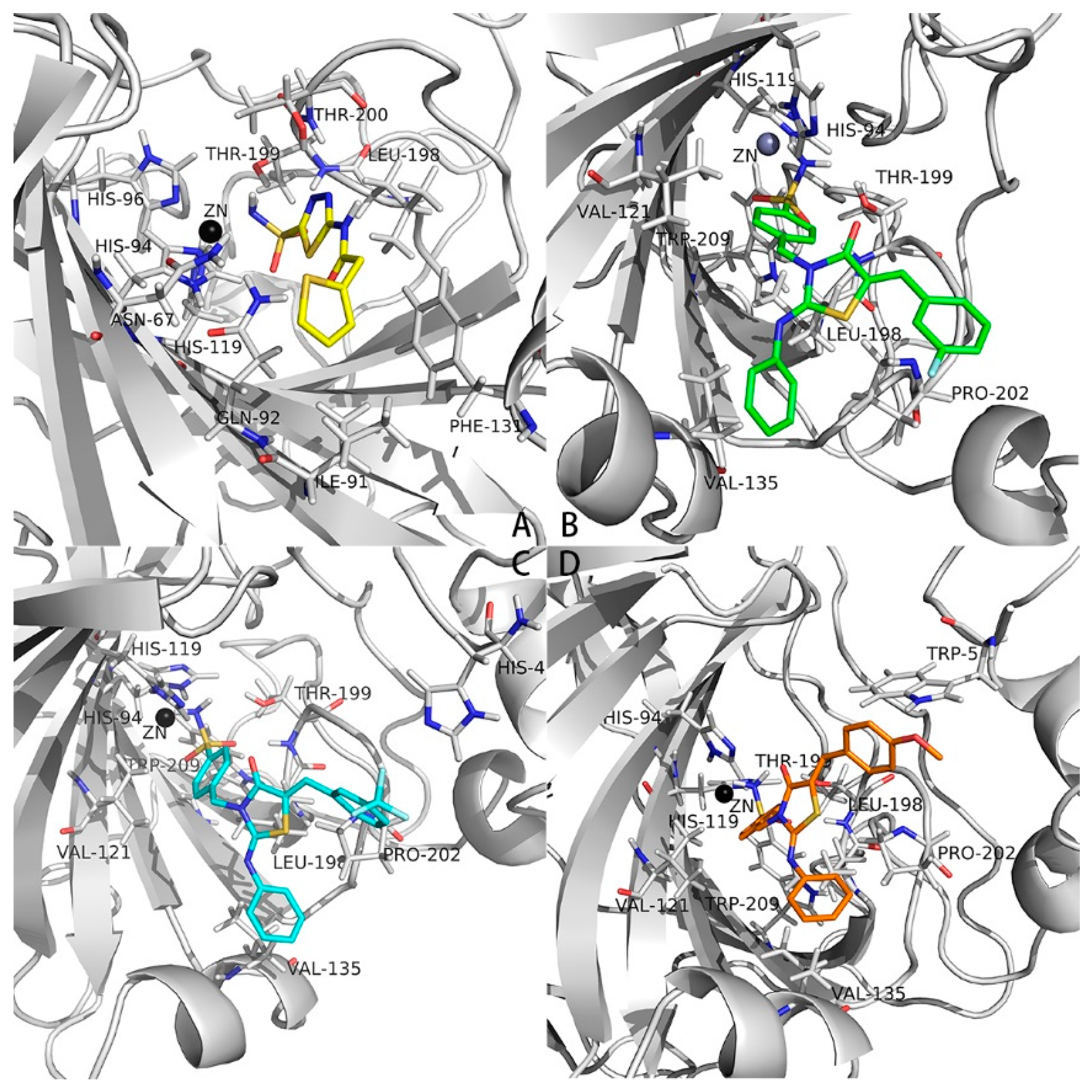
2.3. Docking Studies into the Active Site of hCAs
3. Materials and Methods
3.1. Chemistry
3.1.1. 4-((3-phenylthioureido)methyl)benzenesulfonamide (1)
3.1.2. (Z)-4-((4-oxo-2-(phenylimino)thiazolidin-3-yl)methyl)benzenesulfonamide (2)
3.1.3. General Procedure of Synthesis of 4-(((Z)-5-((Z)-benzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)methyl)benzenesulfonamide hybrids (3–14)
3.2. hCA Enzyme Inhibition Assays
3.3. Single-Crystal Structure
3.4. Preparation of Compound 2–14 for Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stadie, W.C.; O’Brien, H. The catalytic of the hydration of carbon dioxide and dehydration of carbonic acid by an enzyme isolated from red blood cells. J. Biol. Chem. 1933, 103, 521–529. [Google Scholar]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications, Subcellular Biochemistry; Frost, S.C.; McKenna, R. (Eds.) Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Švastová, E.; Hulíková, A.; Rafajová, M.; Zat’Ovičová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic Anhydrases: Role in pH Control and Cancer. Metabolites 2018, 8, 19. [Google Scholar] [Green Version]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A.; Harris, A.L.; Harris, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Boil. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T. Carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1377–1378. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 1–13. [Google Scholar] [CrossRef]
- A Phase I, Multi-center, Open-label, Study to Investigate the Safety, Tolerability and Pharmacokinetic of SLC-0111 in Subjects with Advanced Solid Tumours. 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02215850 (accessed on 15 September 2017).
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Andreucci, E.; Ruzzolini, J.; Peppicelli, S.; Bianchini, F.; Laurenzana, A.; Carta, F.; Supuran, C.T.; Calorini, L. The carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells toconventional chemotherapy. J. Enzym. Inhib. Med. Chem. 2019, 8, 117–123. [Google Scholar]
- Romagnoli, R.; Baraldi, P.G.; Prencipe, F.; Oliva, P.; Baraldi, S.; Salvador, M.K.; Lopez-Carz, L.C.; Brancale, A.; Ferla, S.; Hamel, E.; et al. Synthesis and biological evaluation of 2-methyl-4, 5-disubstituted oxazoles as a novel class of highly potent antitubulin agents. Sci. Rep. 2017, 7, 1–19. Available online: https://www.nature.com/articles/srep46356. (accessed on 13 April 2017). [CrossRef] [PubMed]
- Li, Y.-S.; Hu, D.-K.; Zhao, D.-S.; Liu, X.-Y.; Jin, H.-W.; Song, G.-P.; Cui, Z.-N.; Zhang, L.-H. Design, synthesis and biological evaluation of 2,4-disubstituted oxazole derivatives as potential PDE4 inhibitors. Bioorg. Med. Chem. 2017, 25, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Sun, X.; Liu, Y.; Long, W.; Chen, B.; Shen, S.; Ma, H. Synthesis, crystal structure, biological activity and theoretical calculations of novel isoxazole derivatives. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 152, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Tomi, I.H.; Tomma, J.H.; Al-Daraji, A.H.; Al-Dujaili, A.H. Synthesis, characterization and comparative study the microbial activity of some heterocyclic compounds containing oxazole and benzothiazole moieties. J. Saudi Chem. Soc. 2015, 19, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Rouf, A.; Tanyeli, C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sato, M.; Kakinuma, H.; Miyata, N.; Taniguchi, K.; Bando, K.; Koda, A.; Kameo, K. Pyrazole and Isoxazole Derivatives as New, Potent, and Selective 20-Hydroxy-5,8,11,14-eicosatetraenoic Acid Synthase Inhibitors. J. Med. Chem. 2003, 46, 5416–5427. [Google Scholar] [CrossRef]
- Patel, N.B.; Shaikh, F.M. Synthesis and antimicrobial activity of new 4-thiazolidinone derivatives containing 2-amino-6-methoxybenzothiazole. Saudi Pharm. J. 2010, 18, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Shiradkar, M.R.; Ghodake, M.; Bothara, K.G.; Bhandari, S.V.; Nikalje, A.; Akula, K.C.; Desai, N.C.; Burange, P.J. Synthesis and anticonvulsant activity of clubbed thiazolidinoneebarbituric acid and thiazolidinoneetriazole derivatives. Arkivoc 2007, 14, 58–74. [Google Scholar]
- Ottana, R.; Maccari, R.; Giglio, M.; Del Corso, A.; Cappiello, M.; Mura, U.; Cosconati, S.; Marinelli, L.; Novellino, E.; Sartini, S.; et al. Identification of 5-arylidene-4-thiazolidinone derivatives endowed with dual activity as aldose reductase inhibitors and antioxidant agents for the treatment of diabetic complications. Eur. J. Med. Chem. 2011, 46, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K.; Agrawal, R.K. Recent developments and biological activities of thiazolidinone derivatives: A review. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar]
- Barreca, M.L.; Balzarini, J.; Chimirri, A.; De Clercq, E.; De Luca, L.; Höltje, H.D.; Höltje, M.; Monforte, A.M.; Monforte, P.; Pannecouque, C.; et al. Design, Synthesis, Structure−Activity Relationships, and Molecular Modeling Studies of 2,3-Diaryl-1,3-thiazolidin-4-ones as Potent Anti-HIV Agents. J. Med. Chem. 2002, 45, 5410–5413. [Google Scholar] [CrossRef] [PubMed]
- Takasu, K.; Pudhom, K.; Kaiser, M.; Brun, R.; Ihara, M. Synthesis and Antimalarial Efficacy of Aza-Fused Rhodacyanines in Vitro and in the P. berghei Mouse Model. J. Med. Chem. 2006, 49, 4795–4798. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Tan, O.; Ozadali, K.; Piskin, K.; Balkan, A. Molecular modeling, synthesis and screening of some new 4-thiazolidinone derivatives with promising selective COX-2 inhibitory activity. Eur. J. Med. Chem. 2012, 57, 59–64. [Google Scholar] [CrossRef]
- Ansari, M.F.; Idrees, D.; Hassan, M.I.; Ahmad, K.; Avecilla, F.; Azam, A. Design, synthesis and biological evaluation of novel pyridine-thiazolidinone derivatives as anticancer agents: Targeting human carbonic anhydrase IX. Eur. J. Med. Chem. 2018, 144, 544–556. [Google Scholar]
- Güzel-Akdemir, Ö.; Angeli, A.; Demir, K.; Supuran, C.T.; Akdemir, A. Novel thiazolidinone-containing compounds, without the well-known sulphonamide zinc-binding group acting as human carbonic anhydrase IX inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 1299–1308. [Google Scholar] [Green Version]
- Li, F.-R.; Fan, Z.-F.; Qi, S.-J.; Wang, Y.-S.; Wang, J.; Liu, Y.; Cheng, M.-S. Design, Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase IX Inhibitory Evaluations of Novel N-Substituted-β-d-Glucosamine Derivatives that Incorporate Benzenesulfonamides. Molecules 2017, 22, 785. [Google Scholar]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Boil. Chem. 1967, 242, 4221–4229. [Google Scholar]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient Expression and Crystallization System of Cancer-Associated Carbonic Anhydrase Isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef]
- Biswas, S.; McKenna, R.; Supuran, C.T. Effect of incorporating a thiophene tail in the scaffold of acetazolamide on the inhibition of human carbonic anhydraseisoforms I, II, IX and XII. Bioorg. Med. Chem. Lett. 2013, 23, 5646–5649. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An Improved AutoDock Force Field for Small-Molecule Docking to Zinc Metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar]
- Discovery Studio User Manual; Accelrys Inc.: San Diego, CA, USA, 2008.
Sample Availability: Samples of the compounds (2)–(14) are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | hCA II (IC50, nM) | hCA IX (IC50, nM) | Docking Scores (Kcal/mol) |
|---|---|---|---|---|
| 2 | / | 266.2 | 242.1 | −5.16 |
| 3 | H | 166.3 | 182.9 | −6.14 |
| 4 | 3-F | 146.5 | 130.5 | −6.68 |
| 5 | 3-Cl | 270.3 | 292.3 | −5.00 |
| 6 | 3-Br | 148.0 | 154.2 | −6.18 |
| 7 | 3-CF3 | 335.0 | 544.4 | −3.24 |
| 8 | 3-OH | 151.3 | 140.9 | −6.90 |
| 9 | 4-F | 216.7 | 246.1 | −5.02 |
| 10 | 4-OH | 160.8 | 144.9 | −6.30 |
| 11 | 4-OCH3 | 115.4 | 117.2 | −6.79 |
| 12 | 4-OBn | 1725 | >10,000 | 0.70 |
| 13 | 4-i-Pr | 692.1 | 1246 | −2.15 |
| 14 | 3,4-2OCH3 | 121.3 | 146.5 | −6.51 |
| Mafenide hydrochloride | 8912 | >30,000 | NA a | |
| AcetazolaMide (AZM) | 30.47 | 88.1 | −5.46 | |
| SLC-0111 | 137.8 | 180.7 | −6.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-P.; Yin, Z.-F.; Li, J.-Y.; Wang, Z.-P.; Wu, Q.-J.; Wang, J.; Liu, Y.; Cheng, M.-S. Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone. Molecules 2019, 24, 2418. https://doi.org/10.3390/molecules24132418
Zhang Z-P, Yin Z-F, Li J-Y, Wang Z-P, Wu Q-J, Wang J, Liu Y, Cheng M-S. Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone. Molecules. 2019; 24(13):2418. https://doi.org/10.3390/molecules24132418
Chicago/Turabian StyleZhang, Zuo-Peng, Ze-Fa Yin, Jia-Yue Li, Zhi-Peng Wang, Qian-Jie Wu, Jian Wang, Yang Liu, and Mao-Sheng Cheng. 2019. "Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone" Molecules 24, no. 13: 2418. https://doi.org/10.3390/molecules24132418
APA StyleZhang, Z. -P., Yin, Z. -F., Li, J. -Y., Wang, Z. -P., Wu, Q. -J., Wang, J., Liu, Y., & Cheng, M. -S. (2019). Synthesis, Molecular Docking Analysis, and Carbonic Anhydrase Inhibitory Evaluations of Benzenesulfonamide Derivatives Containing Thiazolidinone. Molecules, 24(13), 2418. https://doi.org/10.3390/molecules24132418