New HSV-1 Anti-Viral 1′-Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Fragment as a Glycoside Moiety

 , ,
, ,  ,
,  ,
,
Abstract
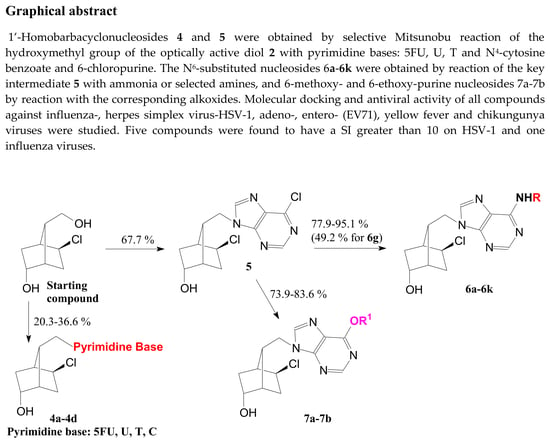
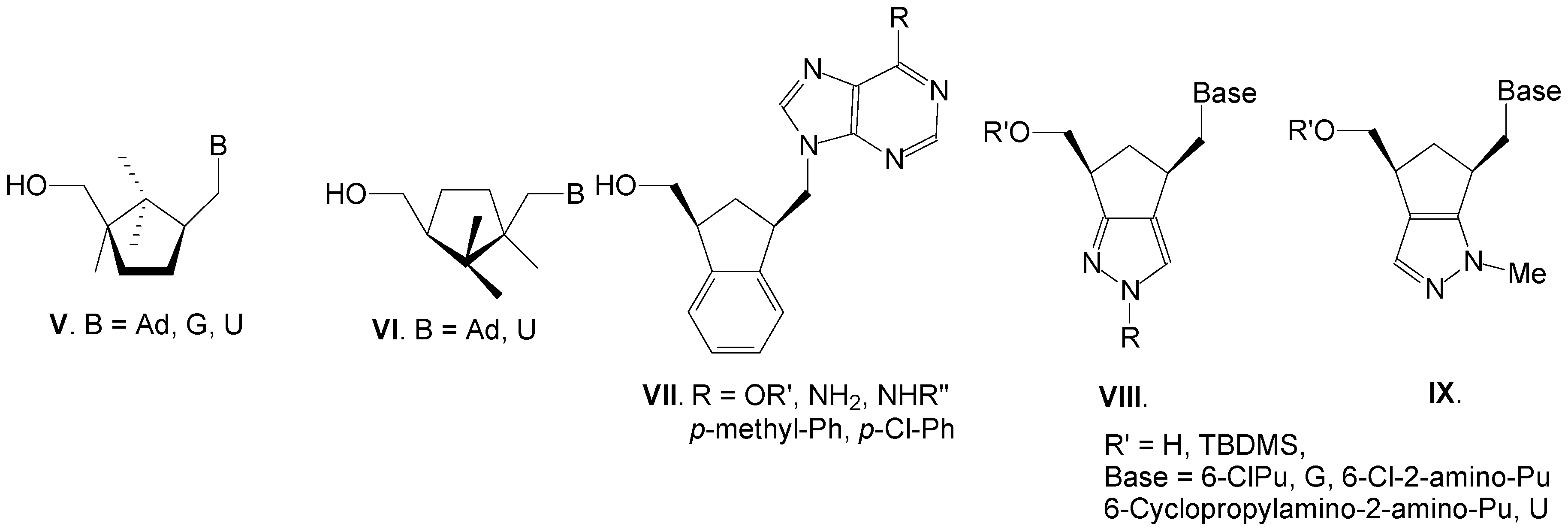
:
1. Introduction
- -
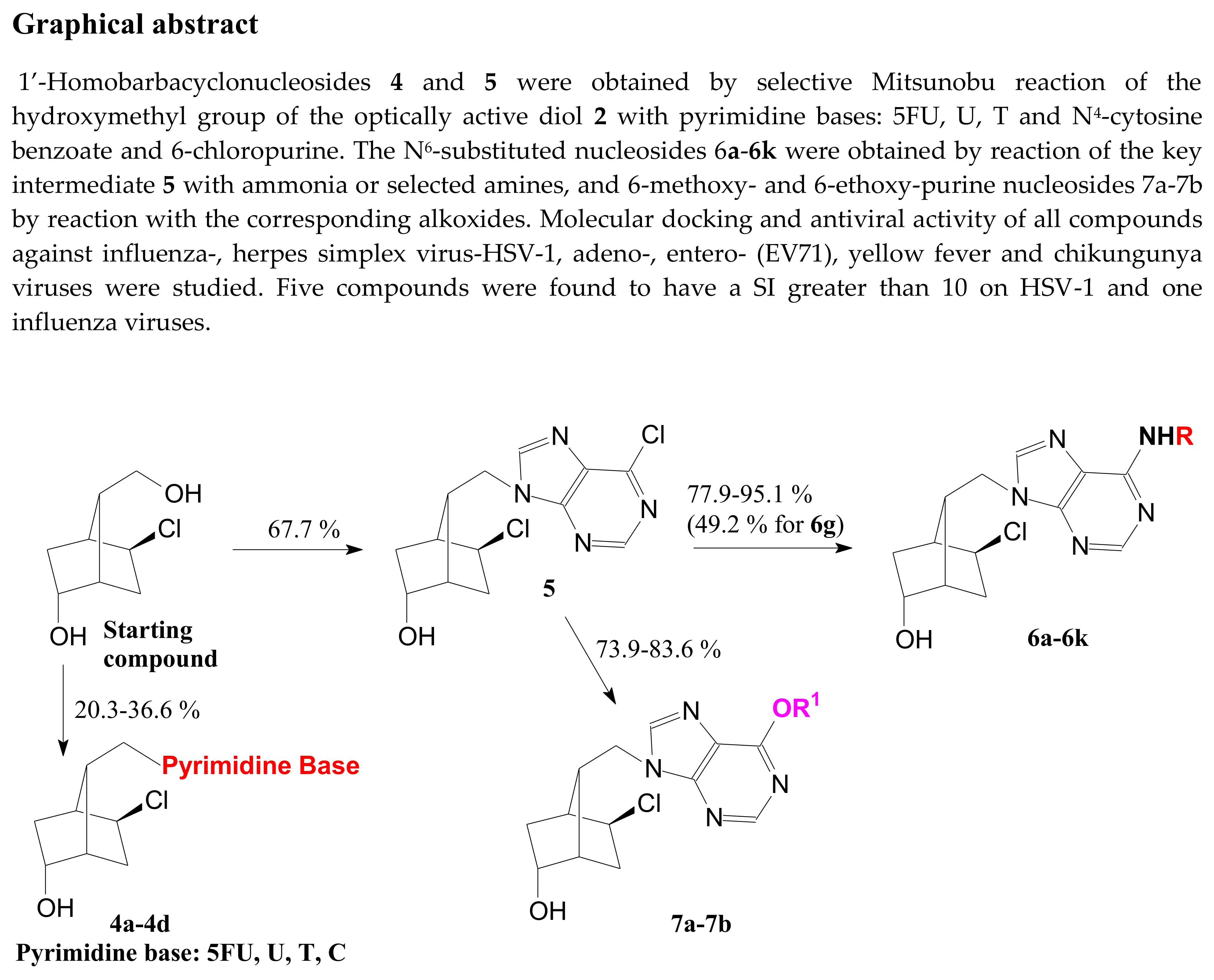
- V, with a 2,2,3-trimethylcyclopentanol, active against HIV-1 and HIV-2 at an EC50 = 4–14 µg/mL [25,26]. Some of these compounds exhibited considerable cytostatic activity, on Molt4/C8 (IC50 = 13.2 and 3.8 µg/mL with nucleobase 6-Cl-2-amino-purine and 2,6 diamino-purine) and L1210/0 (IC50 = 16.2 and 13.9 µg/mL with nucleobase 6-Cl-2-amino-purine and 2-chloro-purine) cancer lines [25,26]. It is interesting that compound VI with the opposite conformation of the cyclopentane ring is inactive (Figure 2) [27].
- -
- -
- VIII and IX, with a cyclopenta[c]pyrazole moiety [30] or cyclopenta[d]pyrazole moiety and hydroxymethyl protected as TBDMS [31,32]. Compounds VIII, with adenine or 6-Cl-2-aminopurine as the base, are more active against VZV/TK- strain (EC50 = 1.5 and 2.1 µmol) than acyclovir, used as reference (EC50 = 27 µmol) [30]. The same compounds are as active as ganciclovir (EC50 = 0.25 and 0.40 µmol for both compounds) against cytomegalovirus AD169 and DAVIS 07/1 strains (EC50 = 0.50 and 0.50 µmol for adenine analog and EC50 = 0.44 and 0.39 µmol for 6-Cl-2-amino-purine analog). Compounds IX, with 6-Cl-Pu, 6-Cl-2-amino-Pu, Uracil, and with the opposite position of N-Me, present significant anticancer activity on L1210/0, Molt4/C8 and CEM cell lines (IC50 = 3.2–11 µg/mL) [8,31,32].
2. Results and Discussion
2.1. Molecular Design
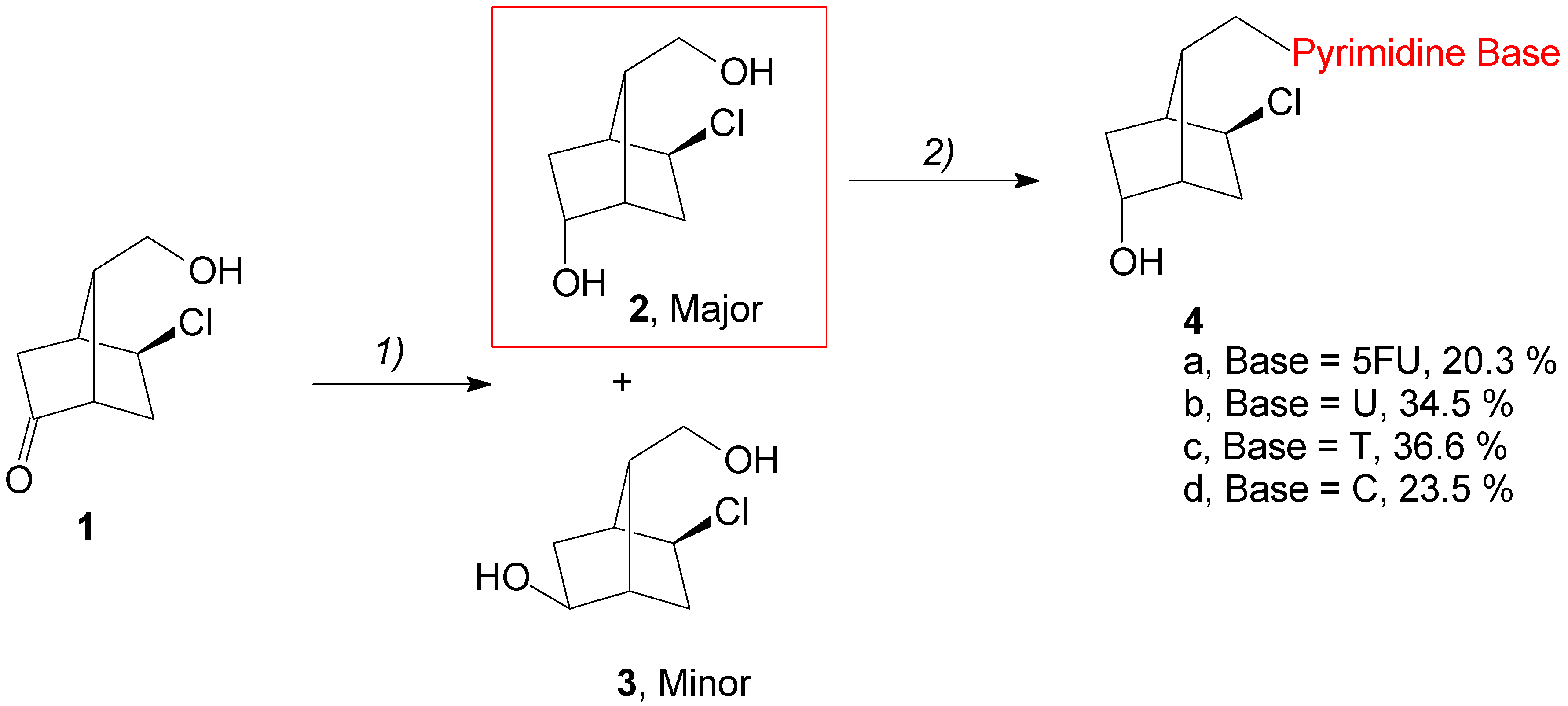
2.2. Chemistry
2.3. 1H, 13C-NMR, MS, Elemental Analysis and Optical Rotation
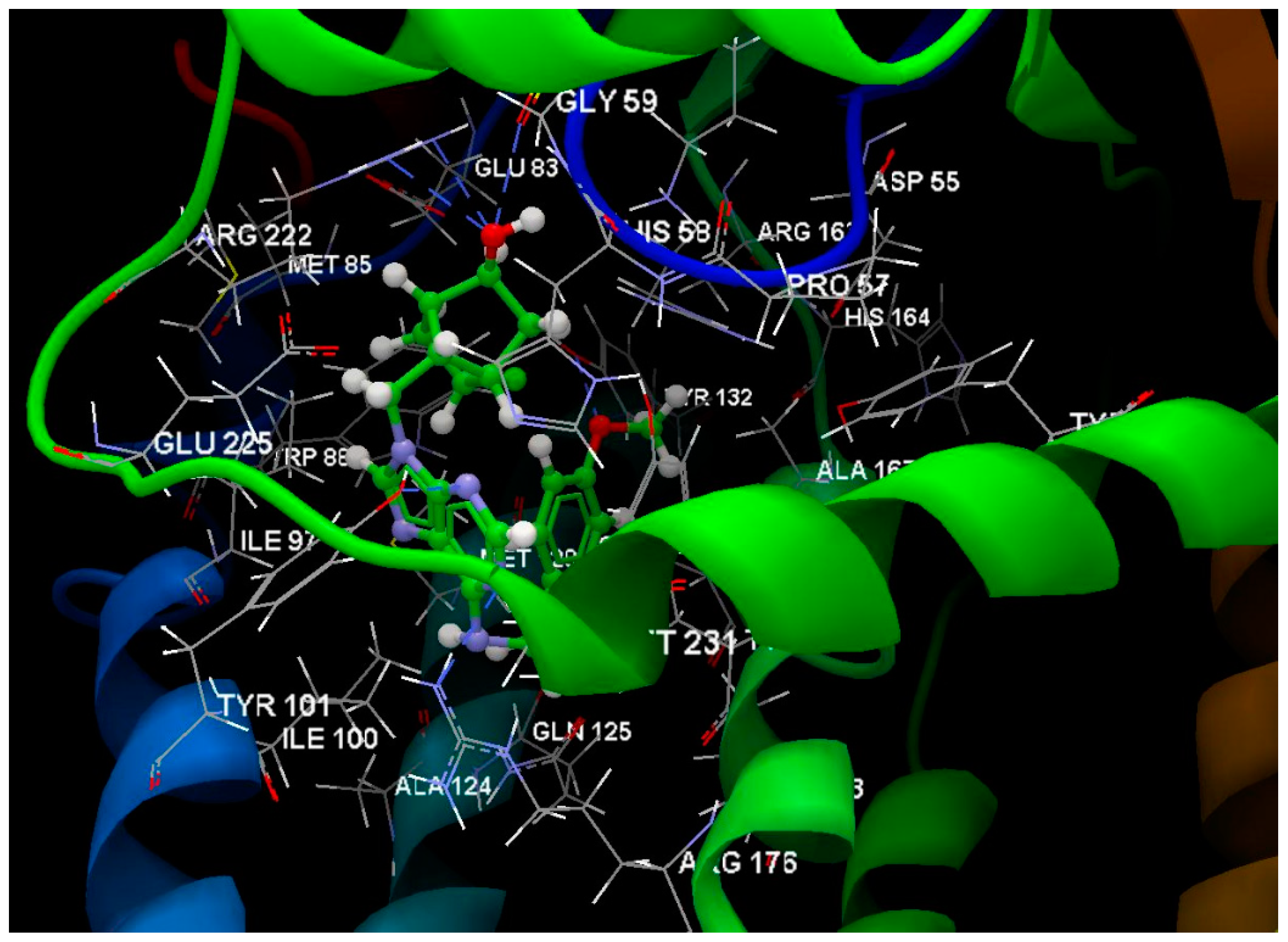
2.4. Antiviral Activity of the Compounds
- 1)
- adeno-, herpes- and influenza viruses, at Pasteur Institute of Epidemiology and Microbiology, Department of Virology, St. Petersburg, Russia, and
- 2)
- enteroviruses type: enterovirus 71 (EV71), yellow fever and Chikungunya viruses, at Rega Institute, Laboratory of Virology and Chemotherapy, Leuven-Belgium
3. Materials and Methods
3.1. Experimental-Chemistry
3.1.1. Synthesis of 1-(((1S,2S,4S,7R)-2-Chloro-5-hydroxybicyclo[2.2.1]heptan-7-yl)methyl)-5-fluoropyrimidine-2,4(1H,3H)-dione, 4a
3.1.2. Synthesis of 1-(((1S,2S,4S,5R,7R)-2-Chloro-5-hydroxybicyclo[2.2.1]heptan-7-yl)methyl)pyrimidine-2,4(1H,3H)-dione, 4b
3.1.3. Synthesis of 1-(((1S,2S,4S,7R)-2-Chloro-5-hydroxybicyclo[2.2.1]heptan-7-yl)methyl)-5-methylpyrimidine-2,4(1H,3H)-dione, 4c
3.1.4. Synthesis of 4-Amino-1-(((1S,2S,4S,7R)-2-chloro-5-hydroxybicyclo[2.2.1]heptan-7-yl)methyl)pyrimidin-2(1H)-one, 4d
3.1.5. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-chloro-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 5
General Procedure for Synthesis of Carbocyclic 1′-Homonucleosides 6a–7k
3.1.6. Synthesis of (1S,4S,5S,7R)-7-((6-Amino-9H-purin-9-yl)methyl)-5-chlorobicyclo[2.2.1]heptan-2-ol, 6a
3.1.7. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-(cyclopropylamino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6b
3.1.8. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-(cyclopentylamino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6c
3.1.9. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-(cyclohexylamino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6d
3.1.10. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-(((R)-1-hydroxy-3-phenylpropan-2-yl)amino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6e
3.1.11. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-(4-methylpiperazin-1-yl)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6f
3.1.12. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-((4-methylpiperazin-1-yl)amino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6g
3.1.13. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-morpholino-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6h
3.1.14. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-((4-hydroxyphenethyl)amino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6i
3.1.15. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-((4-hydroxyphenethyl)amino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6j
3.1.16. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-(phenethylamino)-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 6k
General Procedure for Obtaining the 6-Alkoxy-purine Analogs 7a and 7b
3.1.17. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-methoxy-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 7a
3.1.18. Synthesis of (1S,4S,5S,7R)-5-Chloro-7-((6-ethoxy-9H-purin-9-yl)methyl)bicyclo[2.2.1]heptan-2-ol, 7b
3.2. Anti-Viral Testing of the Compounds Viruses and Cells
3.2.1. Cytotoxicity Assay.
3.2.2. Virus Titration.
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Galmarini, C.M.; Popowycz, F.; Joseph, B. Cytotoxic nucleoside analogues: Different strategies to improve their clinical efficacy. Curr. Med. Chem. 2008, 15, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Analysis of FDA Approved Anticancer Drugs Reveals the Future of Cancer Therapy. Cell Cycle 2004, 3, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Highlights in the discovery of antiviral drugs: A personal retrospective. J. Med. Chem. 2010, 53, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Deval, J. Antimicrobial strategies: Inhibition of viral polymerases by 3′-hydroxyl nucleosides. Drugs 2009, 69, 151–166. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004, 30, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Wroblewski, A.E.; Glowacka, I.E.; Piotrowska, D.G. 1′-homonucleosides and their structural analogues: A Review. Eur. J. Med. Chem. 2016, 118, 121–142. [Google Scholar] [CrossRef]
- Lecerclé, D.; Clouet, A.; Al-Dabbagh, B.; Crouvoisier, M.; Bouhss, A.; Gravier-Pelletier, C.; Le Merrer, Y. Bacterial transferase MraY inhibitors: Synthesis and biological evaluation. Bioorg. Med. Chem. 2010, 18, 4560–4569. [Google Scholar] [CrossRef]
- Lee, H.W.; Choi, W.J.; Jacobson, K.A.; Jeong, L.S. Synthesis and Binding Affinity of Homologated Adenosine Analogues as A3 Adenosine Receptor Ligands. Bull. Korean Chem. Soc. 2011, 32, 961–964. [Google Scholar] [CrossRef]
- Rinaldo-Matthis, A.; Wing, C.; Ghanem, M.; Deng, H.; Wu, P.; Tyler, P.C.; Evans, G.B.; Furneaux, R.H.; Almo, S.C.; Wang, C.C.; et al. Inhibition and structure of Trichomonas vaginalis purine nucleoside phosphorylase with picomolar transition state analogues. Biochemistry 2007, 46, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Kim, H.O.; Choi, W.J.; Choi, S.; Lee, J.H.; Park, S.; Loo, L.; Jacobson, K.A.; Jeong, L.S. Design, synthesis, and binding of homologated truncated 4′-thioadenosine derivatives at the human A3 adenosine receptors. Bioorg. Med. Chem. 2010, 18, 7015–7021. [Google Scholar] [CrossRef] [PubMed]
- Sekiyama, T.; Hatsuya, S.; Tanaka, Y.; Uchiyama, M.; Ono, N.; Iwayama, S.; Oikawa, M.; Suzuki, K.; Okunishi, H.; Tsuji, T. Synthesis and Antiviral Activity of Novel Acyclic Nucleosides: Discovery of a Cyclopropyl Nucleoside with Potent Inhibitory Activity against Herpesviruses. J. Med. Chem. 1998, 41, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Onishi, T.; Mukai, C.; Nakagawa, R.; Sekiyama, T.; Aoki, M.; Suzuki, K.; Nakazawa, H.; Ono, N.; Ohmura, Y.; Iwayama, S.; et al. Synthesis and Antiviral Activity of Novel Anti-VZV 5-Substituted Uracil Nucleosides with a Cyclopropane Sugar Moiety. J. Med. Chem. 2000, 43, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.L.; Ksebati, M.B.; Ptak, R.G.; Fan, B.Y.; Breitenbach, J.M.; Lin, J.S.; Cheng, Y.C.; Kern, E.R.; Drach, J.C.; Zemlicka, J. (Z)- and (E)-2-((hydroxymethyl)cyclo-propylidene)methyladenine and -guanine. New nucleoside analogues with a broad- spectrum antiviral activity. J. Med. Chem. 1998, 41, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Baldanti, F.; Sarasini, A.; Drach, J.C.; Zemlicka, J.; Gerna, G. Z-isomers of 2-hy-droxymethylcyclopropylidenemethyl adenine (synadenol) and guanine (synguanol) are active against ganciclovir- and foscarnet-resistant human cytomegalovirus UL97 mutants. Antivir. Res. 2002, 56, 273–278. [Google Scholar] [CrossRef]
- Danappe, S.; Pal, A.; Alexandre, C.; Aubertin, A.-M.; Bourgougnon, N.; Huet, F. Synthesis of new nucleoside analogues comprising a methylenecyclobutane unit. Tetrahedron 2005, 61, 5782–5787. [Google Scholar] [CrossRef]
- Santana, L.; Teijeira, M.; Uriarte, E.; Teran, C.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E. Synthesis and Biological Evaluation of 1,2-Disubstituted Carbonucleosides of 6-Substituted Purine and 8-Azapurine. Nucl. Nucl. 1999, 18, 733–734. [Google Scholar] [CrossRef] [PubMed]
- Balo, M.C.; Fernandez, F.; Lens, E.; Lopez, C.; De Clercq, E.; Andrei, G.; Snoeck, R.; Balzarini, J. Synthesis and Antiviral Activities of Some Novel Carbocyclic Nucleosides. Nucl. Nucl. 1996, 15, 1335–1346. [Google Scholar] [CrossRef]
- Balo, M.C.; Blanco, J.M.; Fernandez, F.; Lens, E.; Lopez, C. Synthesis of novel carbocyclic nucleosides with a cyclopentenyl ring: Homocarbovir and analogues. Tetrahedron 1998, 54, 2833–2842. [Google Scholar] [CrossRef]
- Rajapan, V.P.; Yin, X.; Schneller, S.W. A flexible synthesis of carbanucleosides and 5′-nor-1′-homo carbanucleosides from a common precursor. Tetrahedron 2002, 58, 9889–9895. [Google Scholar] [CrossRef]
- Kim, A.; Hong, J.H. Synthesis and Anti-HCMV Activity of Novel 5′-Norcarboacyclic Nucleosides. Arch. Pharm. Chem. Life Sci. 2005, 338, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Celona, D.; Di Rita, S.; Guiso, M.; Melchioni, C.; Umani, F. Chiral Synthons from the Iridoid Glucoside Antirrhinoside−Synthesis of a Carbocyclic Homonucleoside Analogue. Eur. J. Org. Chem. 2001, 21, 4061–4066. [Google Scholar] [CrossRef]
- Franzyk, H.; Rasmussen, J.H.; Mazzei, R.A.; Jensen, S.R. Synthesis of Carbocyclic Homo-N-Nucleosides from Iridoids. Eur. J. Org. Chem. 1998, 12, 2931–2935. [Google Scholar] [CrossRef]
- Blanco, J.M.; Caamano, O.; Fernandez, F.; Gomez, M.; Nieto, M.I.; Balzarini, J.; Padalko, E.; De Clerq, E. Synthesis and Antiviral and Cytostatic Activities of Carbocyclic Nucleosides Incorporating a Modified Cyclopentane Ring. I: Guanosine Analogues. Nucl. Nucl. 1997, 16, 159–171. [Google Scholar] [CrossRef]
- Nieto, M.J.; Blanco, J.M.; Caamano, O.; Fernandez, F.; Garcia-Mera, X.; Balzarini, J.; Padalko, E.; Neyts, J.; De Clerq, E. Synthesis, Antiviral and Cytostatic Activities of Carbocyclic Nucleosides Incorporating a Modified Cyclopentane Ring. Part 2:1 Adenosine and Uridine Analogues. Nucl. Nucl. 1998, 17, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.J.; Caamano, O.; Fernandez, F.; Gomez, M.; Balzarini, J.; De Clerq, E. Synthesis, Antiviral and Cytostatic Activities, of Carbocyclic Nucleosides Incorporating a Modified Cyclopentane Ring. IV. Adenosine and Uridine Analogues. Nucl. Nucl. Nucleic Acids 2002, 21, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, F.; Garcia-Mera, X.; Morales, M.; Rodriguez-Borges, De Clerq, E. Synthesis and Cytostatic Activities of New 6-Substituted Purinylcarbonucleosides Derived from Indan. Synthesis 2002, 21, 1084–1090. [Google Scholar] [CrossRef]
- Yao, S.-W.; Lopez, V.H.C.; Fernandez, F.; Garcia-Mera, X.; Morales, M.; Rodriguez-Borges, J.E.; Cordeiro, M.N. Synthesis and QSAR study of the anticancer activity of some novel indane carbocyclic nucleosides. Bioorg. Med. Chem. 2003, 11, 4999–5006. [Google Scholar] [CrossRef]
- Garcia, M.D.; Caamano, O.; Fernandez, F.; Lopez, M.C.; De Clerck, E. Synthesis of Purinyl homo-Carbonucleoside Derivatives of 2-Benzylcyclopenta[c]pyrazol. Synthesis 2005, 925–932. [Google Scholar]
- Garcia, M.D.; Caamano, O.; Fernandez, F.; Abeijon, P.; Blanco, J.M. Synthesis of New 1′(N)-Homocarbanucleosides Based on 1-Methylcyclopenta[c]pyrazole Scaffold. Synthesis 2006, 73–80. [Google Scholar]
- Garcia, M.D.; Caamano, O.; Fernandez, F.; Garcia-Mera, X.; Perez-Castro, I. Synthesis of Purinyl and Pyrimidinyl 1′(N)-Homocarbanucleosides Based on a 1-Methylcyclopenta[c]pyrazole Scaffold; Part 2. Synthesis 2006, 3967–3972. [Google Scholar]
- Tănase, C.; Drăghici, C.; Căproiu, M.T.; Shova, S.; Mathe, C.; Cocu, F.G.; Enache, C.; Maganu, M. New carbocyclic nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; synthesis, X-ray crystallography and anticancer activity. Bioorg. Med. Chem. 2014, 22, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Tănase, C.; Drăghici, C.; Cojocaru, A.; Galochkina, A.V.; Orshanskaya, J.R.; Zarubaev, V.V.; Shova, S.; Enache, C.; Maganu, M. New carbocyclic N6-substituted adenine and pyrimidine nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; synthesis, antiviral, anticancer activity and X-ray crystallography. Bioorg. Med. Chem. 2015, 23, 6346–6354. [Google Scholar] [CrossRef] [PubMed]
- Tănase, C.; Cocu, F.G.; Neagu, M.; Manda, G. New Bicyclo[2.2.1]heptane Nucleoside Analogues as Antitumor Agents. Rev. Chim. (Bucharest) 2009, 60, 147–151. [Google Scholar]
- Tănase, C.; Cojocaru, A.; Drăghici, C. New 1′-homocarbonucleosides analogs with a constrained bicyclo[2.2.1]heptane fragment. Patent RO 141479 A2, 13 May 2015. [Google Scholar]
- Tănase, C.; Drăghici, C.; Neguț, C.; Pintilie, L. New constrained Amines in a Bicyclo[2.2.1]heptane Skeleton. Rev. Chim. (Bucharest) 2018, 69, 2448–2453. [Google Scholar]
- Tănase, C.; Drăghici, C.; Neguț, C.; Pintilie, L. Bicyclo[2.2.1]heptane Amines Protected at the Hydroxyl Group. Patent RO 133248 A2, 30 April 2019. [Google Scholar]
- Bennett, M.S.; Wien, F.; Champness, J.N.; Batuwangala, T.; Rutherford, T.; Summers, W.C.; Sun, H.; Wright, G.; Sanderson, M.R. Structure to 1.9 Å resolution of a complex with herpex simplex virus type-1 thymidine kinase of a novel, non-substrate inhibitor: X-ray crystallographic comparison with binding of acyclovir. FEBS Lett. 1999, 443, 121–125. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Alswaidan, I.; Ghabbour, H.A.; Ezzeldin, E.; Elaasser, M.; Marzouk, M. Docking and antiherpetic activity of 2-aminobenzo[de]-isoquinoline-1,3-diones. Molecule 2015, 20, 5099–5111. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with plants. J. Chem. Inf. Model 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Tănase, C.; Căproiu, M.T.; Drăghici, C.; Enache, C.; Cocu, F.G.; Manda, G. Secondary Compounds and Intermediates in Deprotection of the Benzoate Groups of O2,O4- and N1,O4-Nucleoside Analogues With Optically Active Bicyclo[2.2.1]Heptane Sugar Moieties. Rev. Chim. (Bucharest) 2011, 62, 380–385. [Google Scholar]
- Hamon, N.; Kaci, M.; Uttaro, J.-P.; Perigaud, C.; Mathe, C. Synthesis of 3′-halo-5′-norcarbocyclic nucleoside phosphonates as potential anti-HIV agents. Eur. J. Med. Chem. 2018, 150, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, K. Antiviral Drugs Against Alphaherpesvirus. Adv. Exp. Med. Biol. 2018, 1045, 103–122. [Google Scholar] [PubMed]
- Lacroix, C.; Querol-Audi, J.; Roche, M.; Franco, D.; Froen, M.; Guerra, P.; Terme, T.; Vanelle, P.; Verdaguer, N.; Neyts, J.; et al. A novel benzonitrile analogue inhibits rhinovirus replication. J. Antimicrob. Chemother. 2014, 69, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Saudi, M.; Zmurko, J.; Kaptein, S.; Rozenski, J.; Neyts, J.; Van Aerchot, A. Syntesis and evaluation of imidazole-4,5- and pyrazine-2,3-dicarboxamides targeting dengue and yellow fever virus. Eur. J. Med. Chem. 2014, 87, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Li, C.; Tas, A.; Querat, G.; Albulescu, I.C.; De Burghgraeve, T.; Segura Guerrero, N.A.; Gigante, A.; Piorkowski, G.; Decroly, E.; et al. The viral capping enzyme nsP1: A novel target for the inhibition of chikungunya virus infection. Sci. Rep. 2016, 6, 31819. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4, 5, 6 and 7 (with exception of 4d and 6g) are available in amount of 5-to 10 mg from the authors, for a year. |








{kind=link}
{kind=link}
{kind=link}
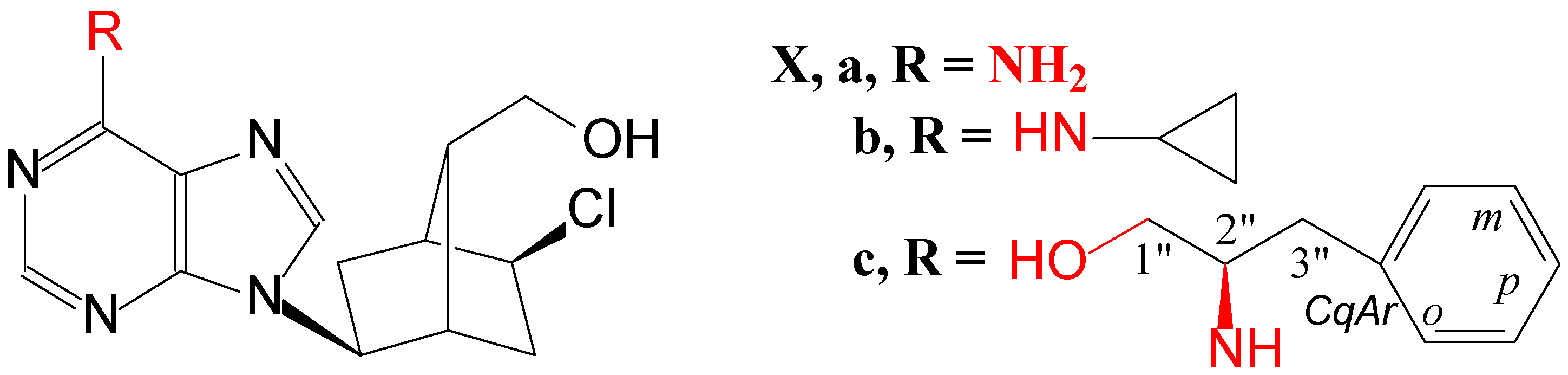{kind=link}
{kind=link}
{kind=link}
{kind=link}
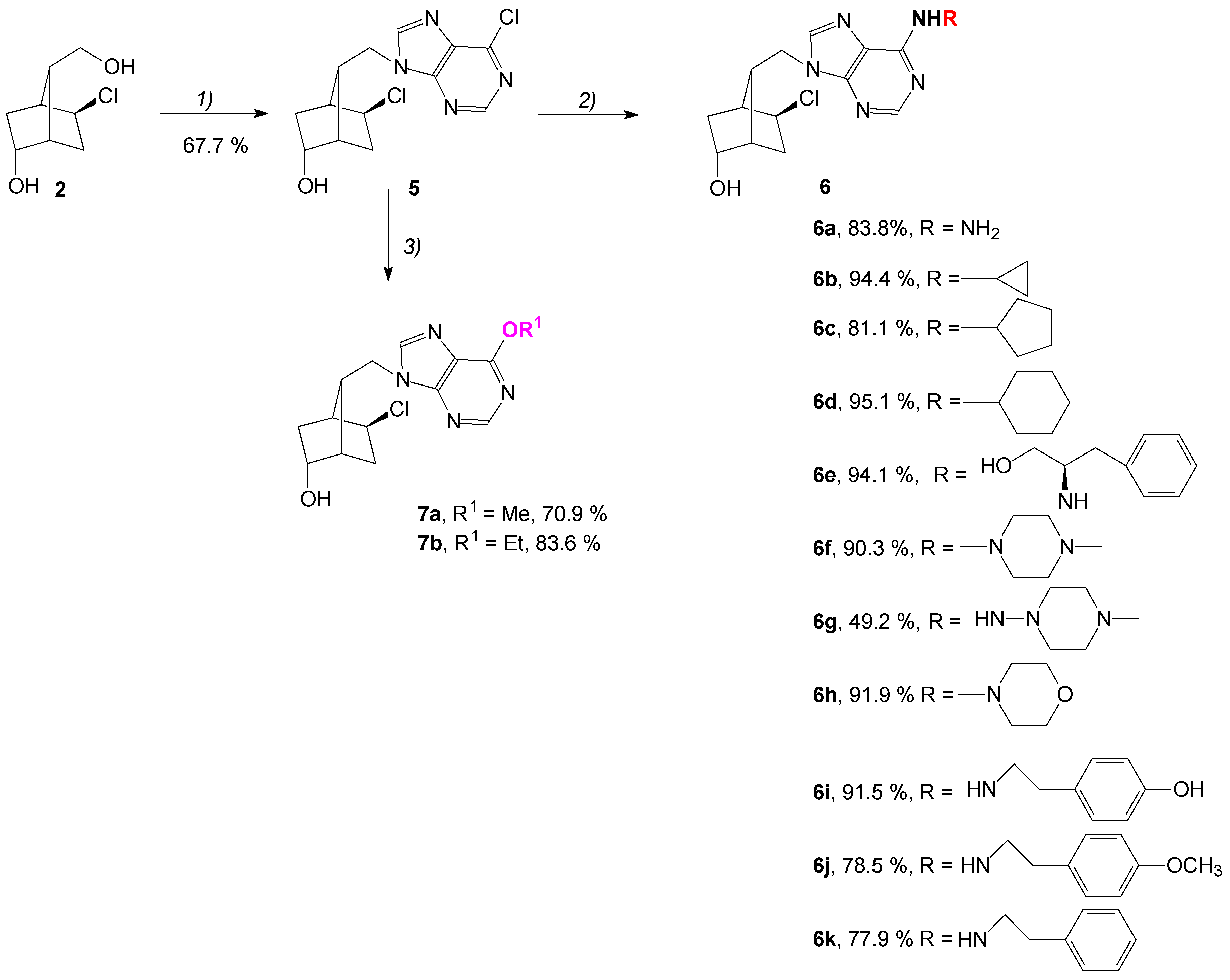{kind=link}
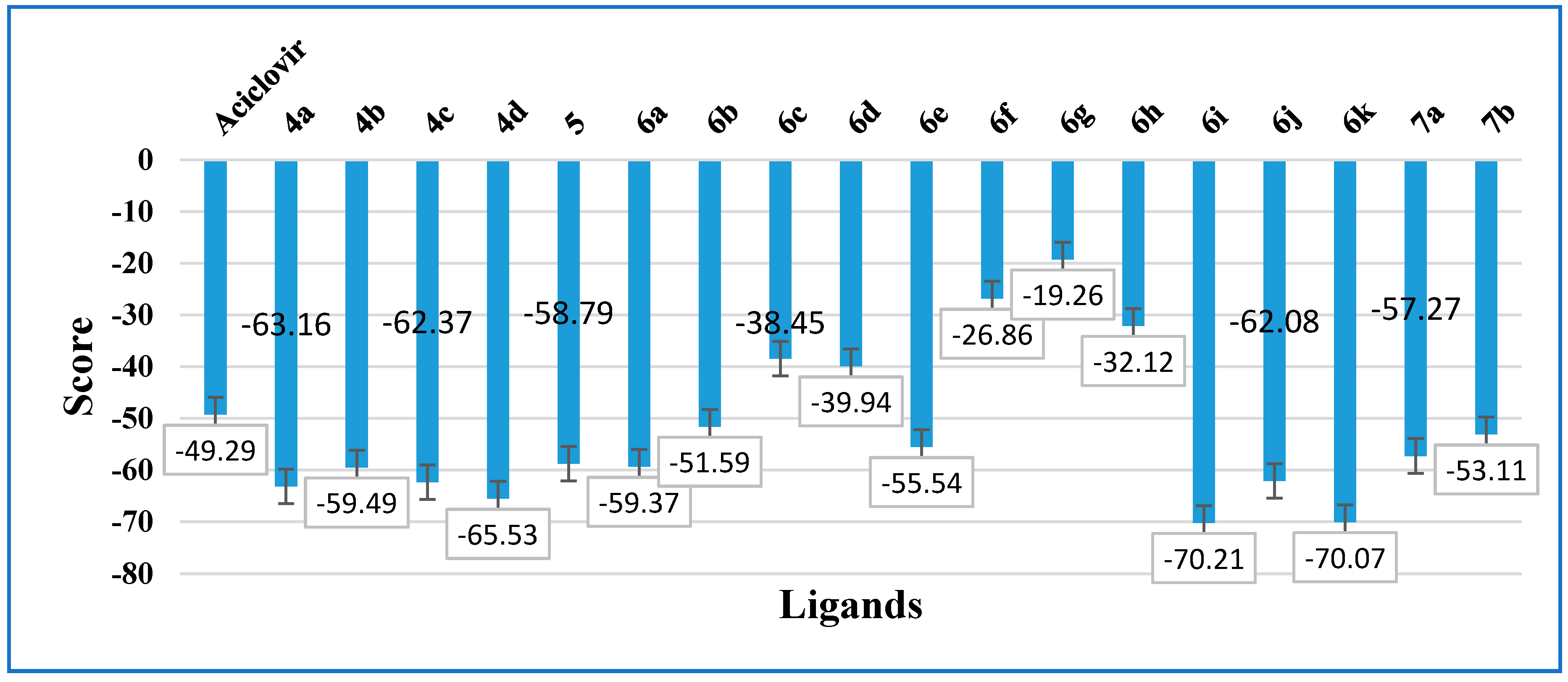
| Compound | Atoms | Weight [Daltons] | Score | RMSD (Å) | Flexible Bonds | Lipinski Violations | Hydrogen Donors | Hydrogen Acceptors | Log P |
|---|---|---|---|---|---|---|---|---|---|
| Acyclovir | 26 | 224.20 | −49.29 | 0.71 | 4 | 0 | 3 | 8 | 0.50 |
| 4a | 33 | 288.70 | −63.16 | 0.01 | 2 | 0 | 2 | 5 | 2.68 |
| 4b | 33 | 270.71 | −59.49 | 0.008 | 2 | 0 | 2 | 5 | 2.58 |
| 4c | 36 | 284.74 | −62.37 | 0.02 | 2 | 0 | 2 | 5 | 2.94 |
| 4d | 34 | 269.73 | −65.53 | 0.01 | 2 | 0 | 4 | 5 | 2.76 |
| 5 | 34 | 313.18 | −58.79 | 0.03 | 2 | 0 | 3 | 6 | 2.02 |
| 6a | 36 | 293.75 | −59.37 | 0.02 | 2 | 0 | 3 | 6 | 0.71 |
| 6b | 43 | 333.82 | −51.59 | 0.06 | 4 | 0 | 2 | 6 | 1.93 |
| 6c | 49 | 361.87 | −38.45 | 0.02 | 4 | 0 | 2 | 6 | 2.64 |
| 6d | 52 | 375.90 | −39.94 | 0.13 | 4 | 0 | 2 | 6 | 3.19 |
| 6e | 56 | 427.93 | −55.54 | 0.29 | 7 | 0 | 3 | 7 | 2.71 |
| 6f | 51 | 376.88 | −26.86 | 0.02 | 3 | 0 | 1 | 7 | 1.33 |
| 6g | 53 | 391.90 | −19.26 | 0.02 | 4 | 0 | 2 | 8 | 1.42 |
| 6h | 47 | 363.84 | −32.12 | 0.47 | 3 | 0 | 1 | 7 | 1.14 |
| 6i | 53 | 413.90 | −70.21 | 0.09 | 6 | 0 | 3 | 7 | 2.97 |
| 6j | 56 | 427.93 | −62.08 | 1.57 | 7 | 0 | 2 | 7 | 3.30 |
| 6k | 52 | 397.90 | −70.07 | 0.07 | 6 | 0 | 2 | 6 | 3.33 |
| 7a | 38 | 308.76 | −57.27 | 0.02 | 3 | 0 | 1 | 6 | 1.36 |
| 7b | 41 | 322.79 | −53.11 | 0.55 | 4 | 0 | 1 | 6 | 1.73 |
| Cmpd | Mw | Anti-Viral Activity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Influenza Virus | Herpes Simplex Virus Type 1 | Human Adenovirus Type 5 | ||||||||
| CC50, µM a | IC50, µM b | SI c | CC50, µM | IC50, µM | SI | CC50, µM | IC50, µM | SI | ||
| 4a | 288.71 | >1039 | 173 ± 21 | 6 | >1039 | >1039 | 1 | >1039 | >1039 | 1 |
| 4b | 270.72 | >1108 | 369 ± 26 | 3 | 1108 | 705 ± 81 | 2 | >1108 | >1108 | 1 |
| 4c | 284.74 | >1054 | >1054 | 1 | >1054 | >1054 | 1 | >1054 | >1054 | 1 |
| 4d | 269.73 | >1112 | 102 ± 12 | 11 | >1112 | 727 ± 77 | 2 | >1112 | >1112 | 1 |
| 6a | 293.76 | >1021 | 328 ± 29 | 3 | 975 ± 64 | 102 ± 11 | 10 | 977 ± 103 | >340 | 3 |
| 6b | 333.82 | 752 ± 44 | 419 ± 51 | 2 | >773 | 599 ± 64 | 1 | >899 | >899 | 1 |
| 6c | 361.87 | 227 ± 19 | >83 | 3 | 605 ± 42 | 47 ± 6 | 13 | 824 ± 91 | >276 | 3 |
| 6d | 375.90 | 42 ± 3 | >27 | 2 | 521 ± 48 | 21 ± 4 | 25 | 697 ± 75 | 123 ± 14 | 6 |
| 6e | 427.93 | 188 ± 22 | >26 | 7 | >608 | 380 ± 46 | 2 | >701 | >701 | 1 |
| 6f | 376.89 | >796 | 241 ± 30 | 3 | >719 | 28 ± 4 | 26 | >796 | 105 ± 11 | 8 |
| 6g | 391.90 | >766 | >766 | 1 | >766 | 540 ± 32 | 1 | >766 | >766 | 1 |
| 6h | 500.00 | >600 | 196 ± 15 | 3 | >600 | 341 ± 40 | 2 | >600 | >600 | 1 |
| 6i | 413.91 | 124 ± 9 | >72 | 2 | 447 ± 31 | 155 ± 13 | 3 | 217 ± 17 | >72 | 3 |
| 6j | 427.93 | 467 ± 28 | >234 | 2 | 205 ± 19 | 15 ± 2 | 14 | 241 ± 22 | >70 | 3 |
| 6k | 397.91 | 254 ± 21 | 74 ± 8 | 3 | 185 ± 16 | 48 ± 6 | 4 | 245 ± 18 | >75 | 3 |
| 5 | 313.19 | >958 | 254 ± 14 | 4 | >830 | 69 ± 9 | 12 | >958 | >958 | 1 |
| 7a | 308.77 | >972 | 233 ± 33 | 4 | >972 | >972 | 1 | >972 | >972 | 1 |
| 7b | 322.78 | >929 | >929 | 1 | >929 | 524 ± 63 | 2 | >929 | >929 | 1 |
| Acyclovir | N/T | >1131 | 28 ± 4 | 40 | N/T | |||||
| Oseltamivir | >100 | 0.3 ± 0.1 | >333 | N/T | N/T | |||||
| Cmpd | Mw | Anti-Viral Activity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Yellow Fever Virus | Chikungunya Virus | Enterovirus 71 | ||||||||
| CC50, µM a | IC50, µM b | SI c | CC50, µM | IC50, µM | SI | CC50, µM | IC50, µM | SI | ||
| 4a | 288.71 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 4b | 270.72 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 4c | 284.74 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 4d | 269.73 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 6a | 293.76 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | 71 | 1 |
| 6b | 333.82 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 6c | 361.87 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | 28 ± 16 | 4 |
| 6d | 375.90 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | 28 ± 13 | 4 |
| 6e | 427.93 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 6f | 376.89 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 6g | 391.90 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 6h | 500.00 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 6i | 413.91 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | 79 | 1 |
| 6j | 427.93 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | 42 ± 3.2 | 2 |
| 6k | 397.91 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | 25 ± 5.6 | 4 |
| 5 | 313.19 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 7a | 308.77 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
| 7b | 322.78 | >100 | >100 | 1 | >100 | >100 | 1 | >100 | >100 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tănase, C.I.; Drăghici, C.; Hanganu, A.; Pintilie, L.; Maganu, M.; Volobueva, A.; Sinegubova, E.; Zarubaev, V.V.; Neyts, J.; Jochmans, D.; et al. New HSV-1 Anti-Viral 1′-Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Fragment as a Glycoside Moiety. Molecules 2019, 24, 2446. https://doi.org/10.3390/molecules24132446
Tănase CI, Drăghici C, Hanganu A, Pintilie L, Maganu M, Volobueva A, Sinegubova E, Zarubaev VV, Neyts J, Jochmans D, et al. New HSV-1 Anti-Viral 1′-Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Fragment as a Glycoside Moiety. Molecules. 2019; 24(13):2446. https://doi.org/10.3390/molecules24132446
Chicago/Turabian StyleTănase, Constantin I., Constantin Drăghici, Anamaria Hanganu, Lucia Pintilie, Maria Maganu, Alexandrina Volobueva, Ekaterina Sinegubova, Vladimir V. Zarubaev, Johan Neyts, Dirk Jochmans, and et al. 2019. "New HSV-1 Anti-Viral 1′-Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Fragment as a Glycoside Moiety" Molecules 24, no. 13: 2446. https://doi.org/10.3390/molecules24132446
APA StyleTănase, C. I., Drăghici, C., Hanganu, A., Pintilie, L., Maganu, M., Volobueva, A., Sinegubova, E., Zarubaev, V. V., Neyts, J., Jochmans, D., & Slita, A. V. (2019). New HSV-1 Anti-Viral 1′-Homocarbocyclic Nucleoside Analogs with an Optically Active Substituted Bicyclo[2.2.1]Heptane Fragment as a Glycoside Moiety. Molecules, 24(13), 2446. https://doi.org/10.3390/molecules24132446